

Abstract
Inflammatory pain is thought to be mediated in part through the action of inflammatory mediators on membrane receptors of peripheral nerve terminals, however, the downstream signaling events which lead to pain are poorly understood. In this study we investigated the nociceptive pathways induced by activation of protease-activated receptor 2 (PAR-2) in damage-sensing (nociceptive) neurons from rat dorsal root ganglion (DRG). We found that activation of PAR-2 in these cells strongly inhibited M-type potassium currents (conducted by Kv7 potassium channels). Such inhibition caused depolarization of the neuronal resting membrane potential leading, ultimately, to nociception. Consistent with this mechanism, injection of the specific M channel blocker XE991 into rat paw induced nociception in a concentration-dependent manner. Injection of a PAR-2 agonist peptide also induced nociception but coinjection of XE991 and the PAR-2 agonist did not result in summation of nociception, suggesting that the action of both agents may share a similar mechanism. We also studied the signaling pathway of M current inhibition by PAR-2 using patch-clamp and fluorescence imaging of DRG neurons. These experiments revealed that the PAR-2 effect was mediated by phospholipase C (PLC). Further experiments demonstrated that M current inhibition required concurrent rises in cytosolic Ca2+ concentration and depletion of membrane phosphatidylinositol 4,5-bisphosphate (PIP2). We propose that PLC- and Ca2+/PIP2-mediated inhibition of M current in sensory neurons may represent one of the general mechanisms underlying pain produced by inflammatory mediators, and may therefore open up a new therapeutic window for treatment of this major clinical problem.
Keywords: M current, PAR-2, KCNQ, inflammatory pain, PLC, sensory neuron
Introduction
Inflammation causes spontaneous pain and exaggerated perception of chemical, thermal, and mechanical stimuli (hyperalgesia, allodynia). The common cause of these uncomfortable sensations is the action of inflammatory mediators, such as prostaglandins, leukotrienes, bradykinin, histamine, ATP, and proteases, on the peripheral terminals of damage-sensing (nociceptive) neurons (McMahon et al., 2006). However, the mechanism(s) by which inflammatory mediators induce spontaneous pain and hyperalgesia are diverse and poorly understood.
Inflammatory mediators act by activating their specific receptors, and many of these belong to the G-protein-coupled receptors (GPCRs) that use Gq or G11 types of Gα and activate phospholipase C (PLC) (McMahon et al., 2006). One such receptor type, which is abundantly expressed in nociceptors, is the protease-activated receptor 2 (PAR-2) (Amadesi et al., 2004; Dai et al., 2004). Protease-activated receptors (PAR1–4) are a novel class of GPCRs with a unique activation mechanism: exogenous proteases cleave the receptor's N-terminal fragment unveiling a more proximal domain which acts as a tethered ligand and activates the receptor (Macfarlane et al., 2001). During inflammation, PAR-2 can be activated by tryptase, trypsin, and a few related serine proteases released by macrophages and mast cells (Luo et al., 2007). In in vivo studies, injection of PAR-2 agonists induced both spontaneous nociception and hyperalgesia (Vergnolle et al., 2001; Paszcuk et al., 2008), whereas a PAR-2 antagonist produced anti-nociceptive effects in an inflammatory pain model (Cenac et al., 2007). The exact molecular mechanisms of these effects are poorly understood, however it has been suggested that a group of pathways underlying inflammatory hyperalgesia/allodynia act via thermosensitive and/or mechanosensitive transient receptor potential (TRP) channels. Indeed, it has been demonstrated that activation of PAR-2 produces sensitization of native and overexpressed TRPV1 (Amadesi et al., 2004; Dai et al., 2004), TRPA1 (Dai et al., 2007), and TRPV4 (Grant et al., 2007; Sipe et al., 2008). This sensitization could ultimately lead to inflammatory hyperalgesia. However, the potential role of these channels in spontaneous inflammatory pain (that is inflammatory pain without any external mechanical or thermal stimulation) is less certain.
Here, we propose a novel mechanism for acute, spontaneous inflammatory pain: depolarization and hyperexcitability of nociceptors via Gq/11-coupled receptor-induced inhibition of the M current. M current conducted by the Kv7 (KCNQ) potassium channels is a mechanism used throughout the nervous system to maintain neurons at the resting membrane potential and to resist depolarizing inputs (Delmas and Brown, 2005). Numerous studies have now demonstrated that genetic or pharmacological inhibition of M current leads to increased neuronal excitability, whereas M channel openers suppress action potential firing (Biervert et al., 1998; Charlier et al., 1998; Passmore et al., 2003; Gamper et al., 2006; Zaika et al., 2006; Peretz et al., 2007). It is well established that M channels can be effectively inhibited via the Gq/11-PLC pathway, thus the origin of “M” in M current is from its inhibition by Gq/11-coupled muscarinic acetylcholine receptors (Brown and Adams, 1980, for review, see Delmas and Brown, 2005). However, the role of receptor-mediated regulation of M current in nociception has not been explored.
Materials and Methods
Cell cultures, transfections, and cDNA constructs.
Dorsal root ganglia were extracted from 7- to 10-d-old rats from all spinal levels. Ganglia were enzymatically dissociated in HBSS supplemented with collagenase type 1A (1.5 mg/ml) and dispase (15 mg/ml) (Invitrogen) at 37°C for 15–20 min. Cells were then mechanically triturated, washed twice by centrifugation (800 rpm for 5 min), resuspended in 800 μl of growth medium, and plated onto glass coverslips coated with poly-d-lysine and laminin. DRG neurons were cultured for 2–5 d in a humidified incubator (37°C, 5% CO2) in DMEM supplemented with GlutaMAX I (Invitrogen), 10% fetal bovine serum, penicillin (50 U/ml), and streptomycin (50 μg/ml); no nerve growth factor was added to the culture media. CHO cells were handled as described recently (Gamper et al., 2005a). Plasmids encoding human Kv7.2 and human Kv7.3 (GenBank accession numbers, AF110020 and AAC96101, respectively) were given to us by David McKinnon (State University of New York at Stony Brook, Stony Brook, NY) and Thomas Jentsch (Zentrum fur Molekulare Neurobiologie, Hamburg, Germany) and subcloned into pcDNA3.1 (Invitrogen). The PLCδ-PH-GFP and EGFP-Lyn-PH-PP constructs were kind gifts from Tobias Meyer (Stanford University, Palo Alto, CA). Human PAR-2 (GenBank accession number BT009856) was from Missouri S&T cDNA Resource Center. CHO cells were transfected using FuGENE HD transfection reagent (Roche) according to the instructions of the manufacturer. DRG neurons were transfected using Amaxa Nucleofector Device in combination with rat DRG transfection kit and O-03 voltage protocol. PAR-2 activating peptide (2-Furoyl-Leu-Ile-Gly-Arg-Leu-Orn-NH2) and edelfosine were from Calbiochem (VWR International) (except for behavioral experiments, see below); XE991 (Tocris), Xestospongin C (Cayman Europe); Fluo-4 AM (Invitrogen), diC8-PIP2 (Echelon Bioscience). All other chemicals were from Sigma unless otherwise stated.
Electrophysiology.
In patch-clamp experiments the standard bath solution contained (in mm) NaCl (160), KCl (2.5), CaCl2 (2), MgCl2 (1), HEPES (10), pH 7.4 adjusted with NaOH. In perforated-patch experiments the patch pipette contained K-acetate (90), KCl (20), CaCl2 (1), MgCl2 (3), EGTA (3), HEPES (40), amphotericin B (400 μg/ml), pH 7.4 adjusted with NaOH. In conventional whole-cell experiments the pipette solution contained K-acetate (120–140), NaCl (5), KCl (15), Mg-ATP (3), GTP (0.1), HEPES (10), pH 7.3 adjusted with KOH. The calcium concentration in this pipette solution was fixed with varying concentrations of BAPTA and CaCl2 as indicated in the text. Currents were amplified using an EPC-10 patch-clamp amplifier (HEKA) and recorded using Patchmaster software (v2.2. HEKA). The current signal was sampled at 1 kHz and filtered online at 500 Hz using a software based Bessel filter. Patch pipettes were fabricated from borosilicate glass (Harvard Apparatus) using a horizontal puller (DMZ-universal puller; Zeitz-Instrumente) and heat polished to a resistance of 2–4 MΩ. Cells were mounted on an inverted microscope (TE-2000; Nikon) in a low profile perfusion chamber fed by a gravity perfusion system flowing at ∼2 ml/min resulting in a bath exchange time of ∼15 s. Series resistance was corrected online by up to 70% using the Patchmaster software and liquid junction potentials were corrected. The magnitude of the neuronal M current was measured from the deactivation current when stepping the membrane voltage from −30 mV to −60 mV and was calculated as difference between current amplitude at 10 ms into the voltage pulse and that at the end of the pulse. This analysis method was designed to minimize any contribution from other K+ currents and capacitance artifacts. All analyses of patch-clamp data were conducted using FITMASTER software (v2.11, HEKA).
Fluorescence imaging.
For studies of cytosolic Ca2+ signals, cells were loaded with Fluo-4 AM (2 μm) in combination with pluronic F-127 (0.02%) and probenecid (2 mm) in standard bath solution at 37°C for 45 min. Cells were then mounted on an inverted microscope (TE-2000E, Nikon) illuminated with a halogen lamp filtered through a GFP bandpass filter (450–480 nm excitation wavelength) and imaged using an electron multiplying CCD camera (DQC-FS; Nikon) through a Plan Fluor 10× objective [10×/0.30 DIC, working distance (wd) 16 mm; Nikon]. Drugs were applied using a gravity fed perfusion system. In experiments in which bath Ca2+ was reduced to 0, CaCl2 was removed from the standard bath solution and replaced with 0.2 mm EGTA. Neurons were positively identified by perfusing the cells with a bath solution in which 30 mm NaCl had been replaced with KCl. The perimeter of each neuron was outlined using NIS Elements imaging software (Nikon) and the total pixel intensity within the defined area (F) normalized to the basal total pixel intensity (F0) and plotted against time. For measuring PIP2 hydrolysis, freshly isolated DRG neurons were transfected with 2 μg of PLCδ-PH-GFP using Nucleofector device (Amaxa) as per the manufacturer's instructions. Cells were imaged 2–5 d later using a 100× oil-immersion lens (Apo TIRF, 1.49 numerical aperture, 0.13–0.2 mm wd; Nikon) on a Nikon swept field confocal scanner, attached to an inverted microscope (TE-2000E; Nikon). Cells were illuminated at 488 nm using an argon laser, and the image was amplified and recorded using an electron multiplying CCD camera (DQC-FS, Nikon). The confocal was set to a slit diameter of 50 μm and cells exposed to the laser light for 50–100 ms every 1 s for the duration of the experiment. Analysis of imaging data were conducted using NIS Elements imaging software (Nikon).
Behavioral assays.
On the day of the experiment, PAR-2 activating peptide (Ser-Leu-Ile-Gly-Arg-Leu-NH2, Tocris, 50 μg) and XE991 (2–200 μm) were dissolved in saline (pH 7.4). The right hindpaw of the animals received an intraplantar injection (50 μl) of a selected concentration of drugs and the duration that the animal spent grooming and flinching the injected hindpaw over a 45 min period was collected. Thermal withdrawal latencies were measured using method described before (Hargreaves et al., 1988). After habituation and collection of basal withdrawal latencies, rats were injected with PAR-2 activating peptide or XE991. The drug-induced changes in withdrawal latency were measured 10 min after injection and normalized for baseline readings. To record withdrawal thresholds for mechanical stimulation rats were habituated for 45–60 min and then the baseline readings (three readings per animal) were taken on the right hindpaws using the Dynamic Plantar Aesthesiometer (Ugo Basile). The instrument applies constant ramp of mechanical pressure to the animal right hindpaw (from 0 to 50 g in 10 s) and the reading provides withdrawal threshold in grams. The test drugs (50 μl) were injected in the intraplantar region of the right hindpaws of the animals, and the readings were taken 10 min after injection. All drug treatments were blinded for the observer.
Statistics.
Data are presented as mean ± SEM and were statistically compared using paired or unpaired t test or one-way ANOVA with Tukey's posttest as appropriate.
Results
M current in small DRG neurons is inhibited by PAR-2
Recent research has established that the majority of neurons in rat DRG (both small and large) express robust M-type potassium currents conducted by Kv7.2, Kv7.3, and Kv7.5 subunits (Passmore et al., 2003), and some 90% of small-diameter nociceptive DRG neurons express PAR-2 receptors (Amadesi et al., 2004; Dai et al., 2004). We therefore reasoned there must be coexpression of PAR-2 and M channels in the majority of small nociceptive neurons. Our patch-clamp experiments confirmed this assumption. Whole-cell currents were measured from small-diameter (mean whole-cell capacitance of 20.8 ± 1.1 pF, n = 44), predominantly TRPV1-positive (73% or 51 of 70 of such cells responded to 1 μm capsaicin) DRG neurons. M current was measured using a standard voltage protocol (Fig. 1A, inset). Application of the hyperpolarizing test pulse resulted in a slowly deactivating whole-cell current, characteristic of M current (for time constants, see supplemental Table 1, available at www.jneurosci.org as supplemental material), which was partially inhibited by the specific M channel blocker XE991 (3–10 μm) (Fig. 1C) (57 ± 4% inhibition of deactivation current, n = 7). In subsequent voltage-clamp experiments, we termed IM the XE991-sensitive component of the slowly deactivating current produced by stepping the membrane voltage from −30 to −60 mV. Addition of a specific peptide agonist of PAR-2 (2f-LIGRLO-amide, 10 μm; PAR2-AP) to the bath solution resulted in a dramatic and sustained reduction in the deactivation current from 4.3 ± 0.4 to 2.5 ± 0.3 pA/pF (24/27 neurons, p < 0.0001) (Fig. 1B,C) which amounted to 80.5 ± 10.6% of IM. When applied after XE991, PAR2-AP had no further effect on the deactivation current (deactivation current in the presence of XE991 alone was 1.52 ± 0.32 pA/pF; and after subsequent coapplication of XE991 + PAR2-AP became 1.38 ± 0.30 pA/pF; n = 7, paired t test) (Fig. 1C). These data indicate that the fraction of the deactivation current inhibited by PAR2-AP is a classical M current. Inhibition of M current by PAR-2AP or XE991 was accompanied by a substantial depolarization of the membrane potential by 8.2 ± 1.9 mV (from −65.6 ± 1.6 mV to −57.4 ± 1.7 mV; n = 20, p < 0.001) and 12.8 ± 2.1 mV (from −61.5 ± 3.2 mV to −48.8 ± 2.7 mV; n = 8, p < 0.001) respectively (Fig. 1D). In current-clamp experiments, we found that IM inhibition by PAR2-AP lowered the threshold for action potential firing in 5/12 tested neurons (data not shown). The effect on excitability of DRG neurons in culture was most likely underestimated because of a slight depolarization causing partial inactivation of voltage-gated Na+ channels (Zimmermann et al., 2007). As we shall see later, behavioral experiments support a strong excitatory effect of M channel block.
Figure 1.

PAR-2 inhibits M current in DRG neurons. M current was recorded in small-diameter DRG neurons using the perforated-patch technique. A, Exemplar experiment showing whole-cell current elicited by the voltage protocol depicted in the inset above (values in mV). Drugs were added to the bath solution sequentially, protease activated receptor-2 activating peptide (PAR2-AP) (10 μm) followed by XE991 (XE, 10 μm). Zero current is indicated by the dotted line. B, Representative time course of M current inhibition by PAR2-AP. Plotted is the deactivation current measured as the difference between the current 10 ms into the voltage pulse from −30 mV to −60 mV and the steady-state current at −60 mV and normalized to cell capacitance (Ideac density). Drugs were applied during the periods indicated by the bars. C, D, Summary of the effects of PAR2-AP and XE991 on M current (C) and resting membrane potential (Vm) (D). Bars are mean ± SEM; number of experiments given above bars in parentheses. Significant difference from basal (*) and from PAR2-AP (†); NS, no significant difference.
To verify that the observed effects of PAR-2 on M current were not an artifact of culture conditions, we developed a method of perforated-patch recording from acute DRG “slices” (see supplemental Methods, available at www.jneurosci.org as supplemental material). These recordings (supplemental Fig. 1) were consistent with the experiments on cultured neurons validating both the expression of M current in native DRG neurons and its functional coupling to PAR-2 signaling.
DRG neurons express a wide variety of voltage-gated K+ channels, most of which are activated at membrane potentials more depolarized than M channels. We therefore investigated whether these were modulated by PAR-2 activation by measuring the total outward current at + 60 mV (termed IK) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). XE991 (10 μm) induced 16 ± 4% (n = 6) inhibition of IK, consistent with inhibition of M current. Subsequent application of PAR2-AP induced a relatively minor change in IK: an initial transient increase in the current (12.4 ± 4.5%, n = 6) followed by a steady-state inhibition of 6.3 ± 2.3% (n = 6). These results, together with previous observations that activation of PAR-2 does not affect TTX-resistant voltage-gated sodium channels (Kayssi et al., 2007) and H current (Alier et al., 2008) in DRG neurons, suggest that the PAR-2 action on the voltage-gated channels in DRG is specific to M channels.
We were also able to model PAR-2 inhibition of M current in CHO cells by cotransfection of human PAR-2 with hKv7.2 and hKv7.3 channels (Fig. 2A). In such cells, PAR2-AP induced robust inhibition of the voltage-gated K+ current at 0 mV [which is solely conducted by Kv7.2/7.3 because endogenous K+ conductance in CHO cells at this voltage is negligible (Gamper et al., 2005a)]. In both DRG and CHO cells, the effect of PAR2-AP was partially reversible after washout (Fig. 2A–C) (DRG: 60.5 ± 9.2% recovery, n = 12; CHO: 96.0 ± 19.6% recovery, n = 8). The recovery after inhibition appeared more complete in CHO cells than that in DRGs, however this was statistically insignificant (unpaired t test, p = 0.09). The recovery after PAR2-AP inhibition took 173 ± 25 s for DRG neurons and 392 ± 111 s (p = 0.03) for CHO cells. Importantly, the effect of PAR2-AP on IM could be mimicked by trypsin (20 nm, 51.6 ± 10.6% inhibition, n = 6) which is known to activate PAR-2 through the more physiological mechanism of N-terminal cleavage (Macfarlane et al., 2001), although as expected, the trypsin effect was not reversible after washout (Fig. 2C) (4.4 ± 15.0% recovery), reflecting the fact that in this case the endogenous ligand is tethered to the receptor and cannot be washed away. Inhibition of IM by trypsin in DRGs was somewhat smaller than that by PAR2-AP (51.6 ± 10.6% vs 80.5 ± 10.6%, respectively); however, the difference did not reach statistical significance (unpaired t test, p = 0.24).
Figure 2.
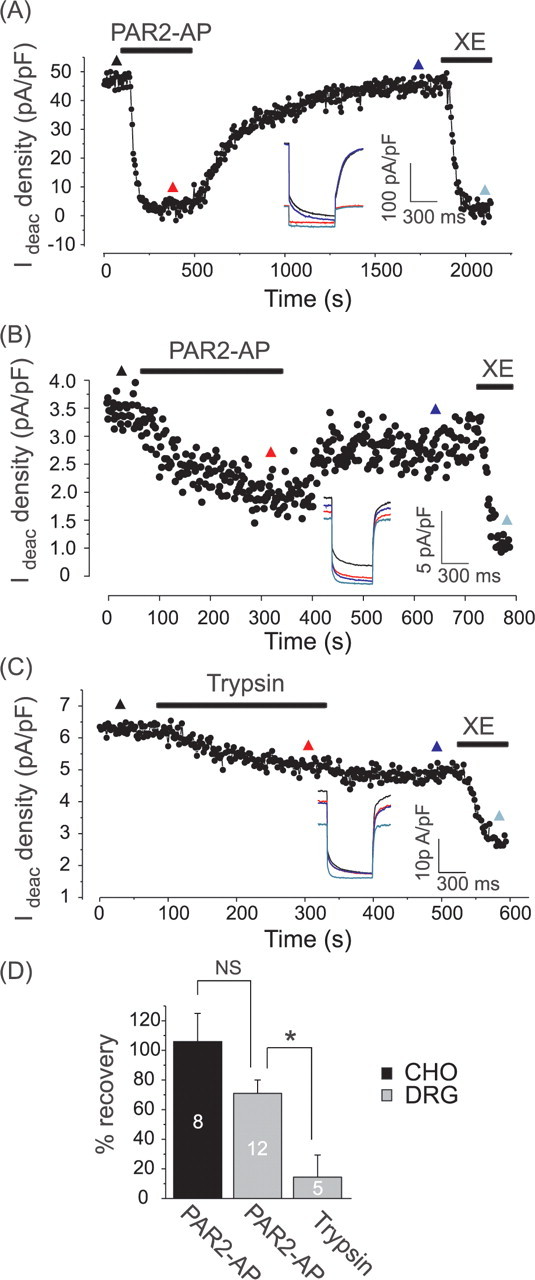
Recovery of M current from PAR-2 induced inhibition. A, Reconstitution of PAR-2 inhibition of M current in CHO cells. Cells were transfected with hKv7.2, hKv7.3, and hPAR-2 cDNA, and current was recorded in perforated-patch mode. Deactivation current when stepping the membrane voltage from 0 mV to −60 mV is normalized to cell capacitance. Drugs (PAR2-AP, 10 μm; XE991, 3 μm) were applied during the periods indicated by the bars. Inset shows current traces taken at the time points indicated by colored triangles. B, Recovery of M current in DRG neurons. Recording conditions as in Figure 1A,B. C, Recording similar to that shown in B, but trypsin (20 nm) is applied instead of PAR2-AP. D, Summary of recovery of Kv7.2/7.3 current (CHO) or native M current (DRG) after inhibition by PAR2-AP or trypsin. Data are represented as percentage recovery of the XE991-sensitive deactivation current fraction (IM).
PAR-2-mediated inhibition of M current requires PLC and Ca2+ rise
We next sought to unravel the intracellular signaling cascade linking PAR-2 and M current in DRG. In many cell types, PAR-2 receptors are coupled to Gq/11 pathways (Bushell et al., 2006), resulting in activation of PLC which hydrolyzes membrane PIP2 to form DAG and IP3. If IP3 release is strong enough to reach IP3 receptors in the endoplasmic reticulum (ER), then Ca2+ is released from the ER stores which, in turn, may trigger multiple secondary Ca2+-sensitive pathways (Berridge and Irvine, 1989). Does PAR2-AP activate PLC in cultured DRG neurons? We transfected DRG culture (using an Amaxa Nucleofector) with an optical probe for PIP2, PLCδ-PH-GFP (Stauffer et al., 1998). The probe contained the pleckstrin homology (PH) domain of PLCδ that binds with high affinity to PIP2 and IP3. After transfection, we imaged PAR2-AP induced membrane-to-cytosol translocation of the probe using a swept field confocal microscope (Fig. 3A,B). Under basal conditions, PLCδ-PH-GFP was predominantly localized to the plasma membrane as shown by an intensity line plot. Application of PAR2-AP caused a robust and reversible translocation of PLCδ-PH-GFP from the plasma membrane to the cytosol in 7/12 small DRG neurons which was manifested in a marked rise in cytosolic fluorescence intensity of 1.6 ± 0.2-fold (n = 7) and a consequent loss of fluorescence at the plasma membrane. This result is consistent with activation of PLC, hydrolysis of membrane PIP2, and generation of IP3 [for which the probe has a higher affinity (Várnai and Balla, 1998; Hirose et al., 1999)].
Figure 3.
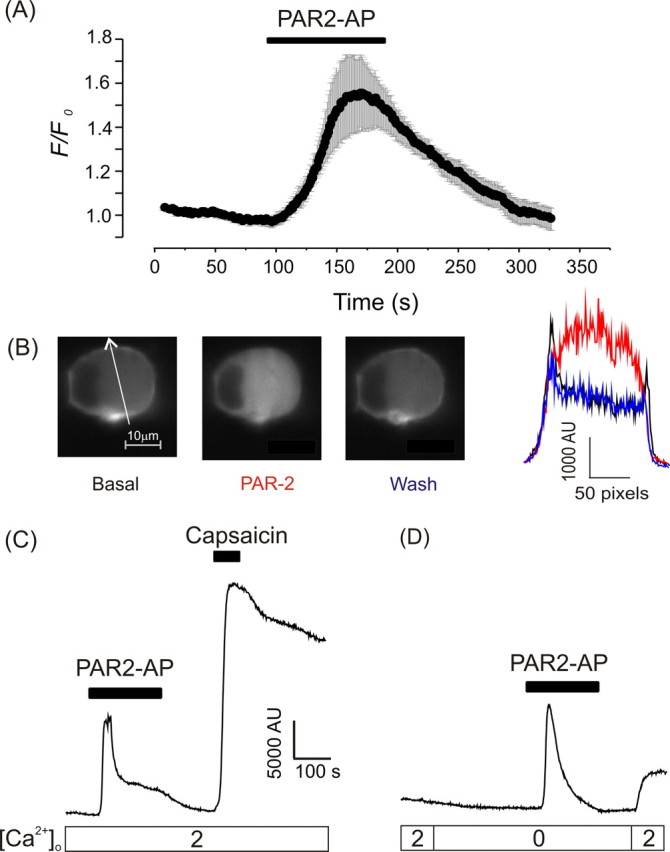
PAR-2 stimulates PIP2 hydrolysis and a global Ca2+ increase in DRG neurons. A, DRG neurons were transfected with the optical probe for PIP2/IP3 (PLCδ-PH-GFP) and imaged using a swept field confocal microscope. Time course of changes in cytosolic fluorescence intensity (F) normalized to basal fluorescence levels (F0) (mean ± SEM, n = 7). Only cells that displayed translocation of the probe were included (7 of 12). PAR2-AP was added to the bath solution as indicated by the black bar. B, An exemplar cell imaged as in A. Images before application (basal), during application (PAR-2), and after the wash-out of PAR2-AP (wash) are shown on the left. White arrow indicates line used for intensity profiles shown on the right. Note the two fluorescence intensity peaks under basal conditions which correspond to the plasma membrane and are lost after PAR-2 activation. C, D, Calcium imaging from individual DRG neurons loaded with Fluo-4 AM. Lower bars indicate [Ca2+]o (mm). Drugs (PAR2-AP, 10 μm; capsaicin, 1 μm) were applied during the periods indicated by the bars.
Because M channels can be inhibited by both PIP2 depletion (Suh and Hille, 2002; Zhang et al., 2003; Li et al., 2005) and cytosolic Ca2+ (Selyanko and Brown, 1996; Gamper and Shapiro, 2003; Gamper et al., 2005b), we asked whether PAR2-AP produces Ca2+ transients in DRG neurons in culture. Such cells were loaded with the calcium-sensitive fluorescent dye Fluo-4 AM and neurons identified morphologically and by their response to depolarization induced by bath application of 30 mm KCl. In line with recent reports (Steinhoff et al., 2000; Amadesi et al., 2004), PAR2-AP (10 μm) induced strong calcium rises in 77/169 (45%) of neurons in culture (Fig. 3C). Subsequent application of the TRPV1 agonist capsaicin (1 μm), a reliable biomarker for nociceptive neurons (Caterina et al., 1997; Binshtok et al., 2007), produced a large rise in [Ca2+]i in the majority of PAR-2 responsive neurons studied (59/77), further supporting the observations that expression of PAR-2 and TRPV1 largely overlap (Amadesi et al., 2004; Dai et al., 2004). PAR-2 activation in the absence of extracellular Ca2+ (0 Ca2+ and 0.2 mm EGTA) failed to prevent the response to PAR2-AP (Fig. 3D). It is of note that when PAR2-AP was applied in the presence of extracellular Ca2+ (2 mm), a smaller sustained phase of [Ca2+]i elevation was often observed after the main spike (Fig. 3B; supplemental Fig. 3, available at www.jneurosci.org as supplemental material). This second phase of Ca2+ response was absent when this experiment was repeated in Ca2+-free buffer (Fig. 3D) suggesting a role for secondary Ca2+ entry pathways in the integral Ca2+ signal induced by PAR-2 in DRG. Removal of Na+ from the extracellular solution failed to prevent PAR2-AP induced Ca2+ transients (6 of 14 neurons responded to PAR2-AP in Na+-free solution, the same proportion as for the experiments in standard extracellular solution), whereas experiments with the PLC blocker edelfosine (10 μm) abolished them (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), further supporting a major role for PLC-IP3 signaling in the generation of these Ca2+ transients.
Next we investigated which second messengers were required for M current inhibition in DRG. We first confirmed the requirement of PLC for M current inhibition. Pretreatment of DRG neurons with the selective PLC inhibitor edelfosine (10 μm) severely attenuated PAR2-AP induced inhibition of IM (Fig. 4). Next we tested whether PIP2 depletion by PLC is the main pathway of M current inhibition in DRG [as suggested for sympathetic neurons and expression systems (Suh and Hille, 2002; Ford et al., 2003; Zhang et al., 2003)]. We thought to prevent such PIP2 depletion by dialyzing 100 μm of the soluble PIP2 analog, diC-8 PIP2, into the cytosol via the patch pipette. To our surprise, buffering of plasma membrane PIP2 only slightly attenuated PAR2-AP inhibition of IM in DRG. With 100 μm diC-8 PIP2 in the pipette PAR2-AP still produced strong (69.2 ± 2.9%, n = 9) inhibition of IM (this value is however significantly lower than 82.7 ± 3.3% observed in control neurons in fast whole-cell mode, n = 9, p < 0.005) (Fig. 4). Supplemental Figure 4 (available at www.jneurosci.org as supplemental material) shows control experiments confirming that diC8-PIP2 was indeed effectively delivered to the inner leaflet of the plasma membrane when applied through the patch pipette.
Figure 4.

PAR-2 inhibits M current in DRG through a Ca2+ and PIP2-dependent mechanism downstream of PLC activation. A, Buffering of the cytosolic Ca2+ and membrane PIP2 through the whole-cell pipette. Representative recordings from DRG neurons temporally aligned for the onset of the PAR2-AP application. Data are normalized to the basal level of IM (I0). PAR2-AP was added to the bath solution 60 s after establishing a stable current level as indicated by the black bar. Pipette solutions contain: 0.2 mm BAPTA, no added Ca2+ (control, black circles); 0.2 mm BAPTA, 100 μm diC8-PIP2 (PIP2, red circles); 10 mm BAPTA, free [Ca2+]i buffered to 75 nm with CaCl2 (BAPTA, blue circles); 10 mm BAPTA, 100 μm diC8-PIP2 (BAPTA+PIP2, green circles). Recordings were performed in whole-cell mode. Individual traces were fit with a generic sigmoid function for better comparison. B, Mean data of the experiments shown in A. Number of experiments is given within bars. *Difference from control, †difference from PIP2. C, Perforated patch experiments similar to those in A. Black circles depict control experiments similar to that of Figure 1B; red circles, PLC inhibitor edelfosine (10 μm) added 10 min before commencing experiment and remained throughout; blue circles, as before, but specific IP3 receptor blocker Xestospongin C (1 μm) is used instead of edelfosine. D, Mean data of the experiments shown in C. *Difference from control, †difference from edelfosine.
The next step was to test whether disruption of the Ca2+ signal would block M current inhibition by PAR-2. With the cytosolic free Ca2+ concentration buffered to 75 nm using 10 mm of the fast Ca2+ buffer BAPTA intracellularly, the PAR2-AP induced inhibition of IM was reduced to 34.3 ± 5.6% (n = 14) (Fig. 4). Similarly, pretreatment with the IP3 receptor blocker Xestospongin C (1 μm) resulted in a reduction of the PAR2-AP induced inhibition of IM to 31.3 ± 10.0% (n = 6) (Fig. 4). In control Ca2+ imaging experiments, pretreatment with Xestospongin C diminished PAR2-AP induced Ca2+ transients in DRG neurons (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). In further experiments we combined 100 μm diC-8 PIP2 and 10 mm BAPTA in the whole-cell pipette to buffer both PIP2 and [Ca2+]i. Under such conditions PAR2-AP produced only a minor inhibition (24.5 ± 5.8%) of IM (n = 14). These experiments suggest that both pathways (PIP2 depletion and Ca2+/CaM) coincide in the PAR2-mediated inhibition of M current in DRGs with the Ca2+ signal probably playing a dominant role.
Pharmacological inhibition of M current induces acute nociception
The experiments described above establish that PAR-2 triggering induces strong inhibition of M current and depolarization in nociceptive DRG neurons. We next sought to establish whether pharmacological blockade of M current can induce nociception in the whole animal and also whether nociception induced by PAR-2 activation could be attributable to the M current inhibition. Intraplantar injection of the M channel blocker XE991 into the hindpaw of rats induced prominent, concentration-dependent, nocifensive behavior (quantified as time spent grooming and flinching during 45 min and compared with the vehicle control) (Fig. 5A). In such behavioral tests, drugs have to be used at 10–100 times higher concentrations than those used in pharmacological experiments on isolated cells because of drug absorption by the tissues and by dilution/absorption in blood and plasma. Our data indicate that injection of 20 μm XE991, a concentration that is still reasonably selective to M channels in expression system/primary neuron electrophysiological experiments (Elmedyb et al., 2007), induced spontaneous nociception (Fig. 5A), suggesting that this effect of XE991 is specifically attributable to the inhibition of peripheral M channels. Injection of 200 μm XE991 resulted in significantly elevated nocifensive behavior compared with 20 μm (Fig. 5A), indicating that the effect was concentration dependent.
Figure 5.

M channel blocker and PAR2-AP induce nocifensive behavior in rats. A, XE991 induces acute pain in a concentration-dependent manner. Solutions (50 μl) of different concentrations (as indicated below the bars) were injected into the hindpaw of adult rats. Nocifensive behavior was quantified by observing time spent grooming and flinching during 45 min, n = 6 for each experiment. *Difference with the vehicle control; †difference from XE991 (2 μm); ‡difference from XE991 (20 μm). B, Preapplication of XE991 does not significantly increase nocifensive behavior induced by PA2-AP. Drugs were injected as follows: Vh1, vehicle (50 μl) control; PAR2-AP, PAR2-AP, 50 μm; XE, XE991, 200 μm; Vh2, double injection of the vehicle (15 min apart); PAR2-AP + XE, 200 μm XE991 was preinjected 15 min before the injection of 50 μm PAR2-AP; n = 6 for all groups. C, PAR2-AP induces thermal hyperalgesia in rats. 50 μl of PAR2-AP (50 μm) or XE991 (200 μm) were injected into the hindpaw of adult rats. Hindpaw withdrawal latency was measured using Hargreaves apparatus as described previously (Hargreaves et al., 1988). Negative numbers indicate hyperalgesia, and positive numbers indicate anti-nociception. Readings were taken from the ipsilateral paw 10 min after injection (n = 6–9). **Significant difference from the vehicle control (one-way ANOVA with Tukey posttest). D, PAR2-AP induces mechanical hyperalgesia in rats. The mean of three baseline readings for mechanical threshold for paw withdrawal was taken for each animal on the right hindpaw intraplantar region. Ten minutes after injection, another three test readings were taken and the results are shown as the change in threshold from the baseline. The vehicle-1 (Vh1) was a saline injection and the test drugs were 50 μl of PAR2-AP (50 μm) or XE991 (200 μm).
Consistent with previous findings (Vergnolle et al., 2001; Shimada et al., 2006; Paszcuk et al., 2008), injection of PAR2-AP (50 μm) also induced spontaneous nocifensive behavior (Fig. 5B); in addition (also consistent with the above literature), PAR2-AP induced thermal (Fig. 5C) and mechanical (Fig. 5D) hyperalgesia [measured using Hargreaves test (Hargreaves et al., 1988) and Ugo Basile Dynamic Plantar Aesthesiometer, respectively]. Interestingly, XE991 not only failed to induce mechanical or thermal hyperalgesia but even somewhat increased thermal withdrawal latency. This difference in the hyperalgesic action of PAR2-AP and XE991 may reflect the fact that PAR-2 triggering has been reported to sensitize temperature-sensitive TRPV1 (Amadesi et al., 2004; Dai et al., 2004) and potentially mechanosensitive TRPV4 channels in DRG nociceptors (Grant et al., 2007; Sipe et al., 2008), whereas no such effect of XE991 is reported or expected. In addition, XE991-induced depolarization may produce some degree of desensitization. To test possible central side effects of PAR2-AP and XE991, we tested thermal withdrawal latencies on the paw contralateral to injected. No significant effects of the drugs were observed on the contralateral paw, indicating that ipsilateral effects were mediated peripherally (data not shown).
We then reasoned that if spontaneous nociception induced by XE991 and PAR2-AP are both mediated (at least in part) by neuronal depolarization caused by M current inhibition, then coapplication of XE991 and PAR2-AP should not produce as much nocifensive behavior as one would expect from simple summation of the effects of these drugs applied separately. We performed an experiment where 200 μm XE991 was injected 15 min before the PAR2-AP (50 μm) injection; there was no significant increase in nocifensive behavior compared with either PAR2-AP or XE991 injected separately (Fig. 5B). Nociceptive effects of XE991 and PAR2-AP were persistent over the period of observation (45 min). Similarly to spontaneous nociception, preinjection of XE991 produced a slight increase in mechanical hyperalgesia induced by PAR2-AP although it was not statistically significant (data not shown).
Discussion
It is well established that activation of PAR-2 in dorsal root and trigeminal sensory neurons induces (1) peripheral spontaneous pain and hyperalgesia (Vergnolle et al., 2001; Dai et al., 2004; Patwardhan et al., 2006; Shimada et al., 2006; Paszcuk et al., 2008); (2) depolarization and an increase in action potential firing of dissociated neurons and those in intact ganglia (Patwardhan et al., 2006; Kayssi et al., 2007; Alier et al., 2008); and (3) inhibition of a fraction of the outwardly rectifying K+ current (Kayssi et al., 2007). In this study we have identified the current inhibited by PAR-2 as being almost exclusively an M current. Inhibition occurs via PLC activation and is conferred by a coincident action of intracellular Ca2+ rises and PIP2 depletion. We also demonstrated that PAR-2 activation (similar to pharmacological block of M channels) induces depolarization of DRG neurons. Using behavioral tests we found that peripheral (intraplantar) administration of an M channel blocker induced acute nociception, which was not augmented when the PAR-2 agonist and M channel blocker were coadministered. Altogether our findings suggest that inflammatory pain induced by the activation of PAR-2 is mediated, at least in part, by the receptor-induced inhibition of M current.
Signaling cascades for M channel inhibition in DRG
PAR-2 belongs to the family of Gq/11-coupled GPCRs (Bushell et al., 2006), which act via PLC and its downstream signaling routes, including PIP2 depletion, Ca2+ transients, and DAG–PKC pathways (Berridge and Irvine, 1989). M channels are sensitive to Ca2+ (in concert with calmodulin) and to PIP2 depletion [for review, see Delmas and Brown (2005) and Gamper and Shapiro (2007b)]; however, in sympathetic neurons Gq/11-mediated inhibition of M current occurs either through PIP2 depletion or Ca2+/CaM but rarely through both (Gamper et al., 2004; Delmas et al., 2005; Brown et al., 2007; Gamper and Shapiro, 2007b; Zaika et al., 2007). Thus, reportedly, in SCG neurons Gq/11-coupled GPCRs that are spatially disconnected from the IP3-sensitive Ca2+ stores (such as muscarinic M1 and angiotensin AT1) do not produce Ca2+ transients but induce strong depletion of membrane PIP2, whereas those receptors that are able to induce Ca2+ transients (e.g., bradykinin B2 and purinergic P2Y6) produce only minor PIP2 depletion because of Ca2+-dependent activation of PIP2 resynthesis (Delmas et al., 2002; Gamper et al., 2004; Delmas et al., 2005; Zaika et al., 2006; Gamper and Shapiro, 2007a,b; Zaika et al., 2007). It was also suggested that disruption of Ca2+ signaling can increase the degree of PIP2 depletion induced by activation of bradykinin and P2Y receptors (Gamper et al., 2004; Delmas et al., 2005; Brown et al., 2007; Gamper and Shapiro, 2007a,b).
We investigated the signaling pathways linking PAR-2 and M channels in DRG neurons, and our data suggest that in these cells PAR-2 stimulation does induce robust Ca2+ transients but chelating of cytosolic Ca2+ or inhibition of IP3 receptors apparently does not significantly enhance membrane PIP2 depletion because under such conditions only modest inhibition of M channels by PAR-2 was observed. Thus we believe that global PIP2 levels in DRG neurons are well buffered so that PAR-2 activation produces only a modest (yet significant) drop in PIP2 and that the main inhibitory action of PAR-2 is therefore mediated by Ca2+. Our experiments with the PIP2/IP3-sensitive probe (PLCδ-PH-GFP) do show translocation of the probe in response to PAR2-AP but these data cannot be used to quantitate the degree of membrane PIP2 depletion because of the ∼20-fold higher affinity of the probe for IP3 (Várnai and Balla, 1998; Hirose et al., 1999). Buffering of PIP2 by dialysis of diC8-PIP2 through the whole-cell pipette does however reduce PAR-2 inhibition of M current, thus, we believe that although the Ca2+ pathway is predominant, both the Ca2+ rise and PIP2 depletion work in concert to ensure maximal M current inhibition. Interestingly, it has been suggested that PIP2 and CaM binding sites within the Kv7 channels overlap (Delmas and Brown, 2005; Haitin and Attali, 2008), accordingly, binding of PIP2 and PIP3 to Kv7.1 have been shown to compete against CaM (Kwon et al., 2007). We can therefore speculate that PIP2 depletion induced by PAR-2, although not robust enough to strongly inhibit M current by itself, may increase affinity of the CaM-Kv7 interaction and thus enhance the inhibitory action of Ca2+.
In a recent study it has been suggested that PAR-2 can induce depolarization of mouse DRG neurons via PKC and ERK1/2 cascades (Kayssi et al., 2007). Further research is required to find out whether there is a role for the PKC pathway in the PAR-2-mediated inhibition of M current in DRG; such a role has been recently proposed for the sensitization of M channels toward the PIP2-mediated modulation by muscarinic M1 receptors in sympathetic neurons (Hoshi et al., 2003, 2005).
Novel mechanism for inflammatory pain
It is now well established that some Gq/11-coupled inflammatory mediators such as proteases, ATP, and bradykinin are able to sensitize members of the TRP channel family: TRPV1 (Chuang et al., 2001; Amadesi et al., 2004; Dai et al., 2004; Tang et al., 2004; Amadesi et al., 2006), TRPV4 (Grant et al., 2007; Sipe et al., 2008), and TRPA1 (Bandell et al., 2004; Dai et al., 2007) probably by the mechanism(s) involving receptor-induced PIP2 depletion (Chuang et al., 2001; Dai et al., 2007) [although this remains controversial, see Gamper and Shapiro (2007b) for review] and/or protein kinase C-dependent phosphorylation (Amadesi et al., 2004, 2006; Dai et al., 2004; Grant et al., 2007). The sensitization of thermosensitive and possibly mechanosensitive TRP channels could be responsible for the thermal and mechanical hyperalgesia induced by inflammation (Amadesi et al., 2004; Dai et al., 2004; Dai et al., 2007; Grant et al., 2007; Sipe et al., 2008). However the role of these mechanisms in spontaneous inflammatory pain is less obvious because the sensitization of these TRP channels does not necessarily imply their activation at resting conditions in the absence of external sensory stimulation. Indeed, PAR-2 induced an increased response to both capsaicin and heat for TRPV1 (Amadesi et al., 2004; Dai et al., 2004) and to cinnamaldehyde for TRPA1 (Bandell et al., 2004; Dai et al., 2007), but no channel activity has been recorded in response to the PAR-2 agonist itself. Thus, receptor-induced sensitization of TRP channels is more likely to account for the inflammatory hyperalgesia than for spontaneous pain. However, lowering the temperature threshold for TRPV1 activation below the normal body temperature (Dai et al., 2004) may result in some spontaneous pain.
We suggest here a novel mechanism that may contribute to spontaneous inflammatory pain: our data suggest that such pain may be produced by PAR-2-mediated inhibition of M current. Indeed, as mentioned earlier, M current “clamps” the resting membrane potential of neurons at hyperpolarized values and prevents discharge of action potentials. The unique biophysical properties of Kv7 channels, such as a negative (−60 mV or less) threshold for activation, slow kinetics, and no inactivation, make them a powerful force counteracting any depolarizing input (Delmas and Brown, 2005). Numerous studies have demonstrated dramatic excitatory effect that inhibition (even partial) of M channels puts on the neurons of different types (Jones et al., 1995; Gu et al., 2005; Peters et al., 2005; Shen et al., 2005; Gamper et al., 2006; Zaika et al., 2006), including DRG sensory neurons (Passmore et al., 2003). Recent studies also suggest that augmentation of M current in peripheral sensory fibers (including C fibers) may have anti-nociceptive effect (Blackburn-Munro and Jensen, 2003; Hirano et al., 2007; Lang et al., 2008; Roza and Lopez-Garcia, 2008); accordingly, the role which M current regulation may play in different chronic pain states is now being increasingly recognized (for review, see Gribkoff, 2008).
Prototypic inflammatory pain pathway?
PAR-2 is not the only type of inflammation-related Gq/11-coupled receptors expressed in DRG neurons; among others are purinergic P2Y (Ruan and Burnstock, 2003), histamine H1 (Kashiba et al., 1999), bradykinin B2 (Seabrook et al., 1997), and Substance P NK1 (Szucs et al., 1999). Agonists of these receptors are released (or can be released) locally at the site of inflammation or injury (McMahon et al., 2006); all of them can trigger signaling cascades of PLC and, thus, potentially are able to inhibit M channels in peripheral nociceptive terminals and cause excitation in corresponding spinal nerve fibers. Simultaneous release of several such inflammatory agents often observed in inflammation may strengthen the M current inhibition and, thus, the excitatory input and pain. We therefore believe that the signaling pathway elucidated here may represent one of the general mechanisms of spontaneous and acute inflammatory pain, whereas pharmacological augmentation of M currents in peripheral sensory neurons may provide new ground for therapeutic targeting of such types of pain.
Footnotes
This work was supported by Wellcome Trust Grant 080593/Z06/Z to N.G., by Wellcome Trust Grants 070740 and 080833/Z/06/Z to B.R. and N.G., by Medical Research Council Grant 82507 to N.G. and B.R., and by National Institutes of Health R01 Grant DE017696 to A.N.A. We thank Hannah Kirton for expert technical assistance; we also thank Drs. Mark Shapiro, Malcolm Hunter, Carine Dalle, and Gayle Passmore for helpful discussions and advice.
References
- Alier KA, Endicott JA, Stemkowski PL, Cenac N, Cellars L, Chapman K, Andrade-Gordon P, Vergnolle N, Smith PA. Intrathecal administration of proteinase-activated receptor-2 agonists produces hyperalgesia by exciting the cell bodies of primary sensory neurons. J Pharmacol Exp Ther. 2008;324:224–233. doi: 10.1124/jpet.107.129171. [DOI] [PubMed] [Google Scholar]
- Amadesi S, Nie J, Vergnolle N, Cottrell GS, Grady EF, Trevisani M, Manni C, Geppetti P, McRoberts JA, Ennes H, Davis JB, Mayer EA, Bunnett NW. Protease-activated receptor 2 sensitizes the capsaicin receptor transient receptor potential vanilloid receptor 1 to induce hyperalgesia. J Neurosci. 2004;24:4300–4312. doi: 10.1523/JNEUROSCI.5679-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadesi S, Cottrell GS, Divino L, Chapman K, Grady EF, Bautista F, Karanjia R, Barajas-Lopez C, Vanner S, Vergnolle N, Bunnett NW. Protease-activated receptor 2 sensitizes TRPV1 by protein kinase Cepsilon- and A-dependent mechanisms in rats and mice. J Physiol. 2006;575:555–571. doi: 10.1113/jphysiol.2006.111534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, Petrus MJ, Earley TJ, Patapoutian A. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron. 2004;41:849–857. doi: 10.1016/s0896-6273(04)00150-3. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Irvine RF. Inositol phosphates and cell signalling. Nature. 1989;341:197–205. doi: 10.1038/341197a0. [DOI] [PubMed] [Google Scholar]
- Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, Steinlein OK. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- Binshtok AM, Bean BP, Woolf CJ. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature. 2007;449:607–610. doi: 10.1038/nature06191. [DOI] [PubMed] [Google Scholar]
- Blackburn-Munro G, Jensen BS. The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. Eur J Pharmacol. 2003;460:109–116. doi: 10.1016/s0014-2999(02)02924-2. [DOI] [PubMed] [Google Scholar]
- Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- Brown DA, Hughes SA, Marsh SJ, Tinker A. Regulation of M(Kv7.2/7.3) channels in neurons by PIP2 and products of PIP2 hydrolysis: significance for receptor-mediated inhibition. J Physiol. 2007;582:917–925. doi: 10.1113/jphysiol.2007.132498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushell TJ, Plevin R, Cobb S, Irving AJ. Characterization of proteinase-activated receptor 2 signalling and expression in rat hippocampal neurons and astrocytes. Neuropharmacology. 2006;50:714–725. doi: 10.1016/j.neuropharm.2005.11.024. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Cenac N, Andrews CN, Holzhausen M, Chapman K, Cottrell G, Andrade-Gordon P, Steinhoff M, Barbara G, Beck P, Bunnett NW, Sharkey KA, Ferraz JG, Shaffer E, Vergnolle N. Role for protease activity in visceral pain in irritable bowel syndrome. J Clin Invest. 2007;117:636–647. doi: 10.1172/JCI29255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJ, Leppert M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53–55. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957–962. doi: 10.1038/35082088. [DOI] [PubMed] [Google Scholar]
- Dai Y, Moriyama T, Higashi T, Togashi K, Kobayashi K, Yamanaka H, Tominaga M, Noguchi K. Proteinase-activated receptor 2-mediated potentiation of transient receptor potential vanilloid subfamily 1 activity reveals a mechanism for proteinase-induced inflammatory pain. J Neurosci. 2004;24:4293–4299. doi: 10.1523/JNEUROSCI.0454-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Wang S, Tominaga M, Yamamoto S, Fukuoka T, Higashi T, Kobayashi K, Obata K, Yamanaka H, Noguchi K. Sensitization of TRPA1 by PAR2 contributes to the sensation of inflammatory pain. J Clin Invest. 2007;117:1979–1987. doi: 10.1172/JCI30951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Delmas P, Wanaverbecq N, Abogadie FC, Mistry M, Brown DA. Signaling microdomains define the specificity of receptor-mediated InsP3 pathways in neurons. Neuron. 2002;34:209–220. doi: 10.1016/s0896-6273(02)00641-4. [DOI] [PubMed] [Google Scholar]
- Delmas P, Coste B, Gamper N, Shapiro MS. Phosphoinositide lipid second messengers: new paradigms for calcium channel modulation. Neuron. 2005;47:179–182. doi: 10.1016/j.neuron.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Elmedyb P, Calloe K, Schmitt N, Hansen RS, Grunnet M, Olesen SP. Modulation of ERG channels by XE991. Basic Clin Pharmacol Toxicol. 2007;100:316–322. doi: 10.1111/j.1742-7843.2007.00048.x. [DOI] [PubMed] [Google Scholar]
- Ford CP, Stemkowski PL, Light PE, Smith PA. Experiments to test the role of phosphatidylinositol 4,5-bisphosphate in neurotransmitter-induced M-channel closure in bullfrog sympathetic neurons. J Neurosci. 2003;23:4931–4941. doi: 10.1523/JNEUROSCI.23-12-04931.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Shapiro MS. Calmodulin mediates Ca2+-dependent modulation of M-type K+ channels. J Gen Physiol. 2003;122:17–31. doi: 10.1085/jgp.200208783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Shapiro MS. Target-specific PIP2 signalling: how might it work? J Physiol. 2007a;582:967–975. doi: 10.1113/jphysiol.2007.132787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Shapiro MS. Regulation of ion transport proteins by membrane phosphoinositides. Nat Rev Neurosci. 2007b;8:921–934. doi: 10.1038/nrn2257. [DOI] [PubMed] [Google Scholar]
- Gamper N, Reznikov V, Yamada Y, Yang J, Shapiro MS. Phosphatidylinositol 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. J Neurosci. 2004;24:10980–10992. doi: 10.1523/JNEUROSCI.3869-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Stockand JD, Shapiro MS. The use of Chinese hamster ovary (CHO) cells in the study of ion channels. J Pharmacol Toxicol Methods. 2005a;51:177–185. doi: 10.1016/j.vascn.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Gamper N, Li Y, Shapiro MS. Structural requirements for differential sensitivity of KCNQ K+ channels to modulation by Ca2+/calmodulin. Mol Biol Cell. 2005b;16:3538–3551. doi: 10.1091/mbc.E04-09-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Zaika O, Li Y, Martin P, Hernandez CC, Perez MR, Wang AY, Jaffe DB, Shapiro MS. Oxidative modification of M-type K+ channels as a mechanism of cytoprotective neuronal silencing. EMBO J. 2006;25:4996–5004. doi: 10.1038/sj.emboj.7601374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant AD, Cottrell GS, Amadesi S, Trevisani M, Nicoletti P, Materazzi S, Altier C, Cenac N, Zamponi GW, Bautista-Cruz F, Lopez CB, Joseph EK, Levine JD, Liedtke W, Vanner S, Vergnolle N, Geppetti P, Bunnett NW. Protease-activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. J Physiol. 2007;578:715–733. doi: 10.1113/jphysiol.2006.121111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribkoff VK. The therapeutic potential of neuronal K V 7 (KCNQ) channel modulators: an update. Expert Opin Ther Targets. 2008;12:565–581. doi: 10.1517/14728222.12.5.565. [DOI] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol. 2005;566:689–715. doi: 10.1113/jphysiol.2005.086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haitin Y, Attali B. The C-terminus of Kv7 channels: a multifunctional module. J Physiol. 2008;586:1803–1810. doi: 10.1113/jphysiol.2007.149187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hirano K, Kuratani K, Fujiyoshi M, Tashiro N, Hayashi E, Kinoshita M. Kv7.2–7.5 voltage-gated potassium channel (KCNQ2–5) opener, retigabine, reduces capsaicin-induced visceral pain in mice. Neurosci Lett. 2007;413:159–162. doi: 10.1016/j.neulet.2006.11.043. [DOI] [PubMed] [Google Scholar]
- Hirose K, Kadowaki S, Tanabe M, Takeshima H, Iino M. Spatiotemporal dynamics of inositol 1,4,5-trisphosphate that underlies complex Ca2+ mobilization patterns. Science. 1999;284:1527–1530. doi: 10.1126/science.284.5419.1527. [DOI] [PubMed] [Google Scholar]
- Hoshi N, Zhang JS, Omaki M, Takeuchi T, Yokoyama S, Wanaverbecq N, Langeberg LK, Yoneda Y, Scott JD, Brown DA, Higashida H. AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat Neurosci. 2003;6:564–571. doi: 10.1038/nn1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Langeberg LK, Scott JD. Distinct enzyme combinations in AKAP signalling complexes permit functional diversity. Nat Cell Biol. 2005;7:1066–1073. doi: 10.1038/ncb1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Brown DA, Milligan G, Willer E, Buckley NJ, Caulfield MP. Bradykinin excites rat sympathetic neurons by inhibition of M current through a mechanism involving B2 receptors and G alpha q/11. Neuron. 1995;14:399–405. doi: 10.1016/0896-6273(95)90295-3. [DOI] [PubMed] [Google Scholar]
- Kashiba H, Fukui H, Morikawa Y, Senba E. Gene expression of histamine H1 receptor in guinea pig primary sensory neurons: a relationship between H1 receptor mRNA-expressing neurons and peptidergic neurons. Brain Res Mol Brain Res. 1999;66:24–34. doi: 10.1016/s0169-328x(98)00346-5. [DOI] [PubMed] [Google Scholar]
- Kayssi A, Amadesi S, Bautista F, Bunnett NW, Vanner S. Mechanisms of protease-activated receptor 2-evoked hyperexcitability of nociceptive neurons innervating the mouse colon. J Physiol. 2007;580:977–991. doi: 10.1113/jphysiol.2006.126599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y, Hofmann T, Montell C. Integration of phosphoinositide- and calmodulin-mediated regulation of TRPC6. Mol Cell. 2007;25:491–503. doi: 10.1016/j.molcel.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang PM, Fleckenstein J, Passmore GM, Brown DA, Grafe P. Retigabine reduces the excitability of unmyelinated peripheral human axons. Neuropharmacology. 2008;54:1271–1278. doi: 10.1016/j.neuropharm.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Li Y, Gamper N, Hilgemann DW, Shapiro MS. Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol (4,5)-bisphosphate. J Neurosci. 2005;25:9825–9835. doi: 10.1523/JNEUROSCI.2597-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Wang Y, Reiser G. Protease-activated receptors in the brain: receptor expression, activation, and functions in neurodegeneration and neuroprotection. Brain Res Rev. 2007;56:331–345. doi: 10.1016/j.brainresrev.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245–282. [PubMed] [Google Scholar]
- McMahon SB, Bennet DLH, Bevan S. Inflammatory mediators and modulators of pain. In: McMahon SB, Koltzenburg M, Wall PD, editors. Wall and Melzack's textbook of pain. Philadelphia: Elsevier Churchill Livingstone; 2006. pp. 49–72. [Google Scholar]
- Passmore GM, Selyanko AA, Mistry M, Al-Qatari M, Marsh SJ, Matthews EA, Dickenson AH, Brown TA, Burbidge SA, Main M, Brown DA. KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci. 2003;23:7227–7236. doi: 10.1523/JNEUROSCI.23-18-07227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszcuk AF, Quintão NL, Fernandes ES, Juliano L, Chapman K, Andrade-Gordon P, Campos MM, Vergnolle N, Calixto JB. Mechanisms underlying the nociceptive and inflammatory responses induced by trypsin in the mouse paw. Eur J Pharmacol. 2008;581:204–215. doi: 10.1016/j.ejphar.2007.11.025. [DOI] [PubMed] [Google Scholar]
- Patwardhan AM, Diogenes A, Berg KA, Fehrenbacher JC, Clarke WP, Akopian AN, Hargreaves KM. PAR-2 agonists activate trigeminal nociceptors and induce functional competence in the delta opioid receptor. Pain. 2006;125:114–124. doi: 10.1016/j.pain.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Peretz A, Sheinin A, Yue C, Degani-Katzav N, Gibor G, Nachman R, Gopin A, Tam E, Shabat D, Yaari Y, Attali B. Pre- and postsynaptic activation of M-channels by a novel opener dampens neuronal firing and transmitter release. J Neurophysiol. 2007;97:283–295. doi: 10.1152/jn.00634.2006. [DOI] [PubMed] [Google Scholar]
- Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci. 2005;8:51–60. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- Roza C, Lopez-Garcia JA. Retigabine, the specific KCNQ channel opener, blocks ectopic discharges in axotomized sensory fibres. Pain. 2008;138:537–545. doi: 10.1016/j.pain.2008.01.031. [DOI] [PubMed] [Google Scholar]
- Ruan HZ, Burnstock G. Localisation of P2Y1 and P2Y4 receptors in dorsal root, nodose and trigeminal ganglia of the rat. Histochem Cell Biol. 2003;120:415–426. doi: 10.1007/s00418-003-0579-3. [DOI] [PubMed] [Google Scholar]
- Seabrook GR, Bowery BJ, Heavens R, Brown N, Ford H, Sirinathsinghi DJ, Borkowski JA, Hess JF, Strader CD, Hill RG. Expression of B1 and B2 bradykinin receptor mRNA and their functional roles in sympathetic ganglia and sensory dorsal root ganglia neurones from wild-type and B2 receptor knockout mice. Neuropharmacology. 1997;36:1009–1017. doi: 10.1016/s0028-3908(97)00065-8. [DOI] [PubMed] [Google Scholar]
- Selyanko AA, Brown DA. Intracellular calcium directly inhibits potassium M channels in excised membrane patches from rat sympathetic neurons. Neuron. 1996;16:151–162. doi: 10.1016/s0896-6273(00)80032-x. [DOI] [PubMed] [Google Scholar]
- Shen W, Hamilton SE, Nathanson NM, Surmeier DJ. Cholinergic suppression of KCNQ channel currents enhances excitability of striatal medium spiny neurons. J Neurosci. 2005;25:7449–7458. doi: 10.1523/JNEUROSCI.1381-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada SG, Shimada KA, Collins JG. Scratching behavior in mice induced by the proteinase-activated receptor-2 agonist, SLIGRL-NH2. Eur J Pharmacol. 2006;530:281–283. doi: 10.1016/j.ejphar.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Sipe WE, Brierley SM, Martin CM, Phillis BD, Cruz FB, Grady EF, Liedtke W, Cohen DM, Vanner S, Blackshaw LA, Bunnett NW. Transient receptor potential vanilloid 4 mediates protease activated receptor 2-induced sensitization of colonic afferent nerves and visceral hyperalgesia. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1288–G1298. doi: 10.1152/ajpgi.00002.2008. [DOI] [PubMed] [Google Scholar]
- Stauffer TP, Ahn S, Meyer T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr Biol. 1998;8:343–346. doi: 10.1016/s0960-9822(98)70135-6. [DOI] [PubMed] [Google Scholar]
- Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS, Trevisani M, Hollenberg MD, Wallace JL, Caughey GH, Mitchell SE, Williams LM, Geppetti P, Mayer EA, Bunnett NW. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med. 2000;6:151–158. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron. 2002;35:507–520. doi: 10.1016/s0896-6273(02)00790-0. [DOI] [PubMed] [Google Scholar]
- Szucs P, Polgar E, Spigelman I, Porszasz R, Nagy I. Neurokinin-1 receptor expression in dorsal root ganglion neurons of young rats. J Peripher Nerv Syst. 1999;4:270–278. [PubMed] [Google Scholar]
- Tang HB, Inoue A, Oshita K, Nakata Y. Sensitization of vanilloid receptor 1 induced by bradykinin via the activation of second messenger signaling cascades in rat primary afferent neurons. Eur J Pharmacol. 2004;498:37–43. doi: 10.1016/j.ejphar.2004.07.076. [DOI] [PubMed] [Google Scholar]
- Várnai P, Balla T. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol. 1998;143:501–510. doi: 10.1083/jcb.143.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergnolle N, Bunnett NW, Sharkey KA, Brussee V, Compton SJ, Grady EF, Cirino G, Gerard N, Basbaum AI, Andrade-Gordon P, Hollenberg MD, Wallace JL. Proteinase-activated receptor-2 and hyperalgesia: A novel pain pathway. Nat Med. 2001;7:821–826. doi: 10.1038/89945. [DOI] [PubMed] [Google Scholar]
- Zaika O, Lara LS, Gamper N, Hilgemann DW, Jaffe DB, Shapiro MS. Angiotensin II regulates neuronal excitability via phosphatidylinositol 4,5-bisphosphate-dependent modulation of Kv7 (M-type) K+ channels. J Physiol. 2006;575:49–67. doi: 10.1113/jphysiol.2006.114074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaika O, Tolstykh GP, Jaffe DB, Shapiro MS. Inositol triphosphate-mediated Ca2+ signals direct purinergic P2Y receptor regulation of neuronal ion channels. J Neurosci. 2007;27:8914–8926. doi: 10.1523/JNEUROSCI.1739-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Craciun LC, Mirshahi T, Rohács T, Lopes CM, Jin T, Logothetis DE. PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron. 2003;37:963–975. doi: 10.1016/s0896-6273(03)00125-9. [DOI] [PubMed] [Google Scholar]
- Zimmermann K, Leffler A, Babes A, Cendan CM, Carr RW, Kobayashi J, Nau C, Wood JN, Reeh PW. Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature. 2007;447:855–858. doi: 10.1038/nature05880. [DOI] [PubMed] [Google Scholar]