

Abstract
We report an interesting case of a male toddler with global developmental delay, dysmorphic facies, seizures, and acyanotic heart disease. Detailed evaluation revealed absent corpus callosum with large doubly committed ventricular septal defect (VSD) and 8p23.3p23.1 deletion and 8p23.1p11.1 interstitial duplication syndrome. In comparison to similar reports of 8p deletion and inverted duplication syndrome, the uniqueness of this report lies in the fact that the congenital heart defect occurred without the GATA4 gene involvement, and the nervous system involvement was more extensive.
Keywords: 8p23deletion, 8p23 interstitial duplication, cardiac defect, GATA4 gene
Introduction
8p23 deletion syndrome, though rare, is well described in the literature. 1 2 This syndrome is associated with developmental delay, abnormal facies, behavior problem/autism, and cardiac defects. The latter defect is implicated mostly due to GATA4 gene deletion. 3 4 5 This report describes a unique case in a toddler with 8p23.3p23.1 deletion and 8p23.1p11.1 interstitial duplication syndrome with a significant cardiac defect and associated agenesis of corpus callosum with colpocephaly and bilateral T2 hyperintensities in the semiovale region. The clinical presentation is very akin to 8p deletion and inverted duplication syndrome; however, a significant cardiac defect occurred in our patient without the involvement of the GATA4 gene and the neurological involvement was more extensive.
Case Report
A 15-month-old male toddler was firstborn to a second-degree consanguineous marriage at term through normal vaginal delivery with a birth weight of 3 kg. He had a normal perinatal transition. He presented with global developmental delay (developmental age 3–4 months), abnormal facies, and fast breathing, noted since early infancy. There was associated history of generalized seizures presently controlled on sodium valproate. Clinically, his weight was 8.5 kg, pulse was 110/minute, all peripheral pulses were felt, respiratory rate was 40/minute, blood pressure was 74/45 mm Hg, oxygen saturation was 98%, and occipitofrontal circumference was 41 cm (< 3 centile, microcephaly). The patient had dysmorphic facies in the form of broad forehead, low set ears, thick lips, and prominent philtrum ( Fig. 1 ). Harrison sulcus was present. Systemic examination revealed hyperactive precordium, normal S1, wide split S2, and short systolic murmur at the left sternal border. Neurologically, he had generalized hypotonia with depressed tendon jerks. Babinski reflex was unequivocal, and other systems examination were normal.
Fig. 1.
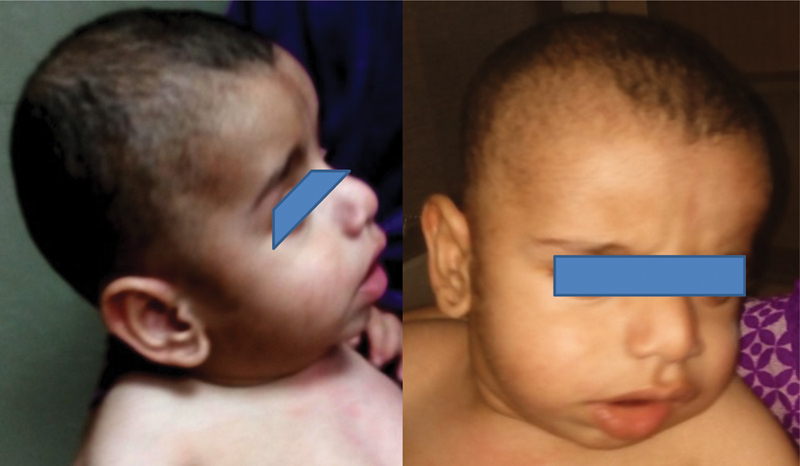
Frontal and side profile of the patient showing low-set ears, wide forehead, broad cheeks, depressed nasal bridge, thick and broad nose, thick lips, and prominent philtrum.
Investigations showed normal hemogram and biochemical parameters (renal function, liver function tests, and electrolytes). Ultrasound for kidneys was normal. Thyroid function test was normal. Karyotyping was done using the G banding method (15 metaphases counted, analyzed, and karyotyped at band resolution of 500) and showed 46,XY ?additional (8p)(23) ( Fig. 2 ). Chromosomal microarray, Illumina HumanCytoSNP-12 (resolution: ∼30 KB), showed a male karyotype with hemizygous deletion at cytoband 8p23.3p23.1, boundaries—Chr8: 17,68,18–69,740,50 (start and end coordinates): 6.7 MB pathological variant. Interstitial duplication at cytoband 8p23.1p11.1, boundaries—Chr8: 12,551,156–43,646,413 (start and end coordinates): 31 MB pathological variant, genomic coordinates for the GATA4 gene is Chr 8: 11,534,468–11,617,511. GATA4 gene is lying between duplication and deletion event but is not falling inside the genomic coordinates of the observed deletion or duplication.
Fig. 2.
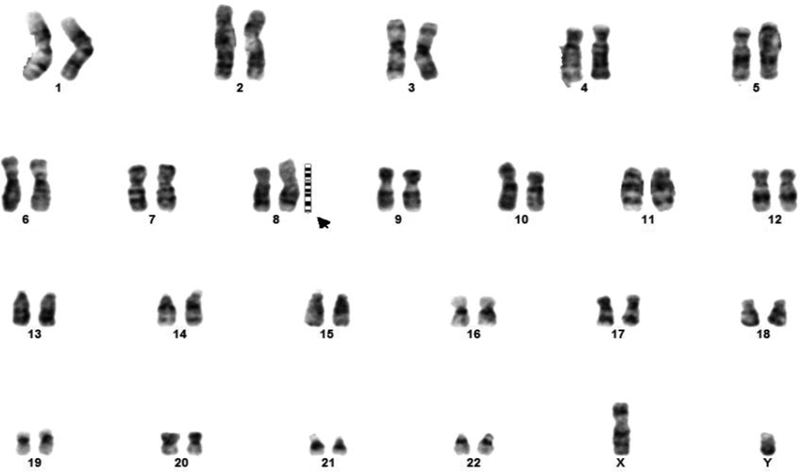
Karyotype showing X,Y ? additional (8P)(23). The symbol "?" was given in the report of karyotype due to unsurety of additional findings.
A further duplication at cytoband 11p11.2p11.12 is likely a benign variant and also a duplication found at cytoband 18p11.21. The sample also showed a copy-neutral loss of heterozygosity (cnLOH) on chromosomes 1, 2, 3, 4, 8, 9, 11, 12, 13, 18, and 19. In 8p23.1 deletion syndrome, the candidate genes including MCPH1 , SOX7 , TNKS , and GATA are deleted. However, in this patient only the MCPH1 gene was deleted ( Fig. 3 ). Echocardiography showed large doubly committed ventricular septal defect (VSD) with left to right shunt and severe hyperkinetic pulmonary artery hypertension ( Fig. 4 ). Magnetic resonance imaging (MRI) of the brain in T2-weighted image showed marked colpocephaly with complete absence of corpus callosum, prominent ventricles, and bilateral T2-weighted hyperintensities in the central semiovale region ( Fig. 5 ).
Fig. 3.

CMA showing deletion on the chromosome 8p23.3p23.1 cytoband region. CMA, chromosome microarray analysis.
Fig. 4.
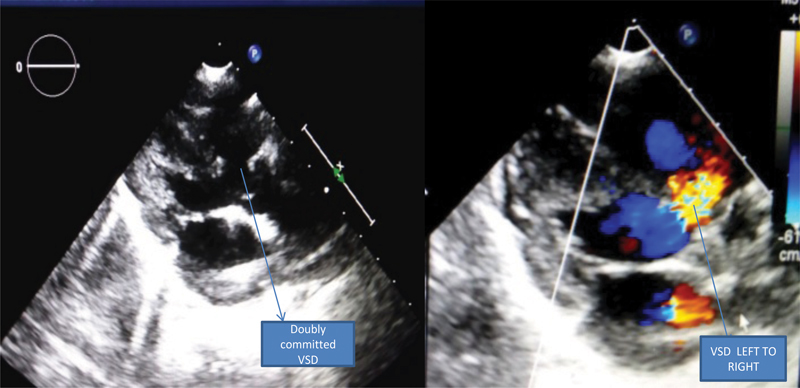
Parasternal long-axis view of 2D echo and color Doppler showing large doubly committed VSD shunting left to right. 2D, two-dimensional; VSD, ventricular septal defect.
Fig. 5.
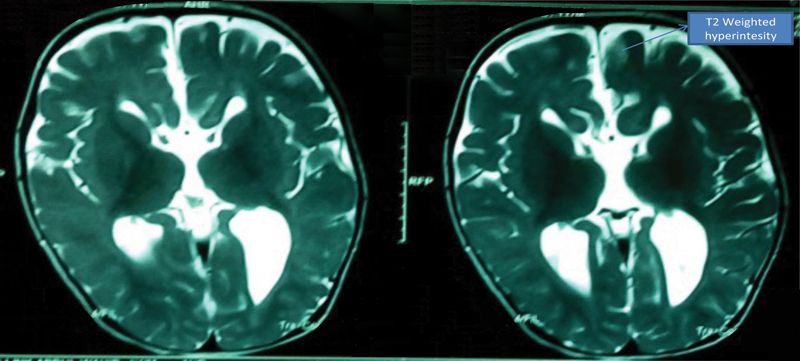
T2-weighted image on MRI showing marked colpocephaly with complete absence of the corpus callosum prominent ventricles and bilateral T2-weighted hyperintensities in the central semiovale region. MRI, magnetic resonance imaging.
Patient's antiepileptic drug (sodium valproate) was continued, and antifailure in the form of syrup furosemide at 2 mg/kg/day and tablet enalapril at 0.1 mg/kg/day was added. Case was discussed with the parents about the disease and long-term prognosis of the patient. Parents refused corrective surgery (VSD closure) for the patient. The decision was endorsed and the patient was advised to continue the drugs with physiotherapy. Patient on the first follow-up after 1 month was seizure-free and gaining weight albeit at a lower rate. Determining the origin of the chromosomal aberration was not possible, since the patient's parents refused to provide blood samples. Also, further validation of the chromosomal abnormalities identified here was not possible because additional samples from the patient could not be obtained.
Discussion
The 8p23 deletion syndrome is well described in the literature and it is associated with developmental delay, microcephaly, behavioral problems, and congenital heart defects. The latter are present in almost 75% of the patients. 1 2 Congenital heart defect is mainly related to GATA4 gene deletion on 8p23. 3 4 5 The present report describes a male toddler with global developmental delay, microcephaly, seizures, and large VSD. Evaluation showed 8p23.3p23.1 deletion and 8p23.1p11.1 interstitial duplication. Brain MRI revealed complete agenesis of corpus callosum with colpocephaly and bilateral T2-weighted hyperintensities in the central semiovale region. This case report is unique because the genetic aberration does not involve the GATA4 gene and the central nervous system (CNS) involvement is more extensive.
The 8p deletion and inverted duplication happen because of nonallelic homologous recombination during maternal meiosis. 6 It is clinically characterized by developmental delay, hypotonia/hypertonia, absent corpus callosum, and cardiac defects. Chromosomal microarray abnormality commonly shows inverted duplication of approximately 31.3 Mb, from 8p23.1 to 8p11.1, and deletion of approximately 6.8 Mb, from 8p23.3 to 8p23.1. 1 6 Our patient, who shared similar clinical features, showed a 6.7-Mb deletion on chromosome 8p23.3p23.1 and a 31-Mb interstitial duplication on chromosome 8p23.1p11.1. Cardiac defects in 8p deletion syndrome are attributed to GATA 4 gene deletion. 3 4 5 7 Their occurrence in 8p deletion and inverted duplication could be ascribed to the GATA4 gene duplication or copy number variation at introns 4 to 5; 8 9 10 however, in our case, the GATA4 gene was not involved. The associated 11p and 18 p duplications were cited as benign variants, probably because of minimal gene involvement. However, reference to this effect was not found in the available literature. Interestingly, mosaic trisomy 8 bears partial similarity with these two syndromes showing the presence of cardiac defects and agenesis of the corpus callosum. 11
Conflict of Interest None.
Patient's Consent
The patient's consent was obtained for the study.
References
- 1.Páez M T, Yamamoto T, Hayashi K et al. Two patients with atypical interstitial deletions of 8p23.1: mapping of phenotypical traits. Am J Med Genet A. 2008;146A(09):1158–1165. doi: 10.1002/ajmg.a.32205. [DOI] [PubMed] [Google Scholar]
- 2.Reddy K S. A paternally inherited terminal deletion, del(8)(p23.1)pat, detected prenatally in an amniotic fluid sample: a review of deletion 8p23.1 cases. Prenat Diagn. 1999;19(09):868–872. doi: 10.1002/(sici)1097-0223(199909)19:9<868::aid-pd641>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 3.Molkentin J D, Lin Q, Duncan S A, Olson E N. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997;11(08):1061–1072. doi: 10.1101/gad.11.8.1061. [DOI] [PubMed] [Google Scholar]
- 4.Kuo C T, Morrisey E E, Anandappa R et al. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 1997;11(08):1048–1060. doi: 10.1101/gad.11.8.1048. [DOI] [PubMed] [Google Scholar]
- 5.Garg V, Kathiriya I S, Barnes Ret al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5 Nature 2003424(6947):443–447. [DOI] [PubMed] [Google Scholar]
- 6.Sireteanu A, Braha E, Popescu R, Gramescu M, Gorduza E V, Rusu C. Inverted duplication deletion of 8P: characterization by standard cytogenetic and SNP array analyses. Rev Med Chir Soc Med Nat Iasi. 2013;117(03):731–734. [PubMed] [Google Scholar]
- 7.Jordan M A, Marques I, Rosendorff J, de Ravel T J. Trisomy 8 mosaicism: a further five cases illustrating marked clinical and cytogenetic variability. Genet Couns. 1998;9(02):139–146. [PubMed] [Google Scholar]
- 8.Yu S, Zhou X G, Fiedler S D, Brawner S J, Joyce J M, Liu H Y. Cardiac defects are infrequent findings in individuals with 8p23.1 genomic duplications containing GATA4. Circ Cardiovasc Genet. 2011;4(06):620–625. doi: 10.1161/CIRCGENETICS.111.960302. [DOI] [PubMed] [Google Scholar]
- 9.Barber J C, Maloney V K, Huang S et al. 8p23.1 duplication syndrome; a novel genomic condition with unexpected complexity revealed by array CGH. Eur J Hum Genet. 2008;16(01):18–27. doi: 10.1038/sj.ejhg.5201932. [DOI] [PubMed] [Google Scholar]
- 10.Barber J C, James R S, Patch C, Temple I K. Protelomeric sequences are deleted in cases of short arm inverted duplication of chromosome 8. Am J Med Genet. 1994;50(03):296–299. doi: 10.1002/ajmg.1320500315. [DOI] [PubMed] [Google Scholar]
- 11.Giraldo G, Gómez A M, Mora L, Suarez-Obando F, Moreno O. Mosaic trisomy 8 detected by fibroblasts cultured of skin. Colomb Med (Cali) 2016;47(02):100–104. [PMC free article] [PubMed] [Google Scholar]