

Abstract
Iron-sulfur cluster assembly 1 (ISCA1) is one of the essential proteins operating in the mitochondrial iron-sulfur (Fe-S) cluster biogenesis pathway. We reported the variant c.259G > A [p.(Glu87Lys)] in homozygous state in exon 4 of the ISCA1 gene as the likely cause of multiple mitochondrial dysfunction syndrome 5 in a previous publication. We now report the third patient with the same phenotype and variant, further supporting the possibility of a founder event. Our observation confirms the clinical presentation associated with a probable founder variant in this condition.
Keywords: multiple mitochondrial dysfunctions syndrome 5, ISCA1, founder effect
Introduction
Iron-sulfur cluster assembly 1 (ISCA1) is a mitochondrial protein involved in the biogenesis of iron-sulfur clusters (ISCs) essential for electron transport reaction. 1 2 3 Recently, we reported four patients from two unrelated families with a homozygous missense variant in ISCA1 leading to multiple mitochondrial dysfunctions syndrome 5 (MMDS5; MIM #617613). 4 Here, we report the same variant in another family with similar clinical and neuroimaging findings. Our findings further confirm this nosological entity.
Materials and Methods
Clinical Report
A 6-month-old male child was the first born to a nonconsanguineous couple by lower segment cesarean section ( Fig. 1A ). At birth, weight was 2.6 kg (–1 standard deviation [SD]), length was 47 cm (normal), and head circumference was 33 cm (normal). He had no feeding difficulty and the perinatal history was unremarkable. He presented with myoclonic jerks starting at the age of 3 months and developmental delay. On examination at 6 months of age, the head circumference was noted to be 40.5 cm (–2 SD), total length was 66 cm (normal), and weight was 6.5 kg (normal). He had mild spasticity in all four limbs. He had normal deep tendon reflexes. The rest of the systemic examination was unremarkable. Testing for serum amino acids, acylcarnitine profile, and thyroid function showed normal results. Magnetic resonance imaging of the brain at age 6 months showed pachygyria, dilated ventricles, and diffuse white matter abnormalities in the cerebrum, cerebellum, as well as brain stem ( Fig. 1B , C ). Magnetic resonance spectroscopy showed an elevated lactate peak. The patient was recruited for further investigations after obtaining informed consents. This work has been approved by the local Institutional Ethics Committee.
Fig. 1.
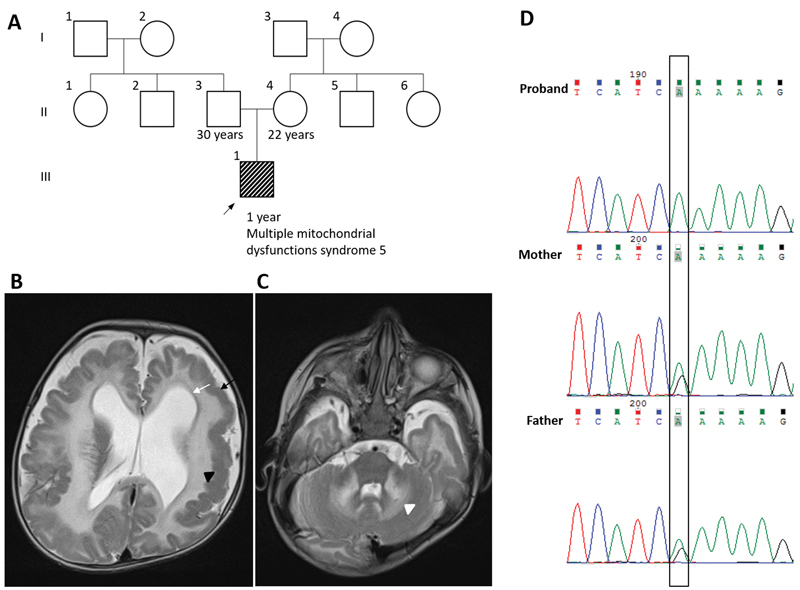
( A ) Pedigree of the family. ( B ) T2-weighted magnetic resonance imaging of brain shows pachygyria (black arrow), dilated ventricles (white arrow), and extensive hyperintensities in cerebral deep white matter (black arrowhead). ( C ) Extensive hyperintensities were also seen in deep cerebellar nuclei (white arrowhead) on T2-weighted magnetic resonance imaging of brain. ( D ) Homozygous variant c.259G > A [p. (Glu87Lys)] was observed on targeted Sanger sequencing in patient index, parents were heterozygous carriers.
Molecular Analysis
Targeted Sanger sequencing of exon 4 and flanking intronic regions of ISCA1 was done for the proband and his parents as the clinical and radiographic features suggested possibility of MMDS5 as the likely diagnosis. Further, whole-exome sequencing was done to rule out other pathogenic variants and to analyze the founder effect as described earlier. 4 Variant prioritization and filtering strategy is outlined in Supplementary Table S1 (online only). Autozygosity mapping was done using FILTUS v.1.0.4 (Oslo University Hospital and University of Oslo, Oslo, Norway). 5
Results
Targeted Sanger sequencing confirmed homozygous missense variant, c.259G > A [p.(Glu87Lys)] in exon 4 of ISCA1 (NM_030940.3). Biallelic segregation of the variant was validated in the parents of the proband ( Fig. 1D ). The variant was observed on integrated genomic viewer with read frequency of 100% from exome data. 6 No other clinically relevant variants were observed in any other gene in the proband's exome. A total of 587 regions of homozygosity were observed from exome sequencing data spanning 311 Mb. ISCA1 was seen in one of the large regions of homozygosity of 8.25 Mb (Chr9:82006605–90258248). This block was then manually checked for the haplotype reported earlier in patients with the same variant in ISCA1 . 4 All three families were found to carry a common haplotype around the variant ( Supplementary Table S2 , online only).
Discussion
Biogenesis of ISCs operate in the mitochondria, cytosol, and nucleus of eukaryotic cells and is imperative for several cellular functions such as respiration, translation, DNA repair, and gene expression regulation. 7 8 In vivo functional studies in mice demonstrate critical roles of ISCA1 in maturation and maintenance of mitochondrial ISCs of neuronal cells. 9 Recently, we reported a homozygous missense variant c.259G > A [p.(Glu87Lys)] in ISCA1 associated with MMDS5. 4 We hereby report the third family with the same phenotype and the genotype confirming the association. Clinical features in the proband and the affected individuals reported earlier include early-onset seizures, developmental delay, spasticity, and early demise. Characteristic brain imaging findings of pachygyria, ventriculomegaly, and extensive bilateral white matter abnormalities in cerebral and cerebellar hemispheres and brain stem were noted to be identical in all affected individuals. The detailed clinical characteristics of the previously reported and the present proband are provided in Table 1 .
Table 1. Clinical findings observed in the proband and families reported earlier.
| Clinical findings | Present study | Shukla et al, 2017 | |||
|---|---|---|---|---|---|
| Family 1 | Family 2 | ||||
| III.1 | II.3 | II.4 | II.1 | II.2 | |
| Sex | Male | Female | Male | Female | Male |
| Age at assessment | 6 mo | 9 mo | 8 mo | 7 mo | 1 y 11 mo |
| Age at demise | NA | 1 y 7 mo | 5 y | 11 mo | 2 y 3 mo |
| Birth weight (kg) | 2.6 | 3 | 3.6 | 2.5 | 2.7 |
| Weight (kg/SD) | 6.5 (normal) | 7/–1 | NA | 6.4/–2 | 5.8/–6 |
| Length (cm/SD) | 66 (normal) | 63/–1 | NA | NA | 76/–5 |
| OFC (cm/SD) | 40.5/–2 | 41/–2 | 42/–2 | 42.5/–1 | 43.5/–6 |
| Seizures | + | + | + | + | + |
| Onset of seizures | 3 mo | NA | 4 mo | 3 mo | 2 mo |
| Developmental delay | + | + | + | + | + |
| Milestones achieved | Social smile sometimes | Partial head control | No milestones achieved | ||
| Feeding difficulty | – | + | + | + | + |
| Tone | Spasticity | Spasticity | Spasticity | Spasticity | Spasticity |
| Deep tendon reflexes | Normal | Exaggerated | Exaggerated | Exaggerated | Exaggerated |
| Strabismus | – | + | – | – | + |
| Other clinical findings | – | History of incessant cry, nystagmus | History of incessant cry, tremors in hands | – | – |
| Pachygyria | + | + | + | NA | – |
| Cerebral white matter abnormalities | + | + | + | + | + |
| Cerebellar white matter abnormalities | + | + | + | + | + |
| Cerebral ventriculomegaly | + | + | + | + a | + |
| MRS findings | Elevated lactate peak | Elevated lactate peak | Elevated lipid-lactate peak | NA | Elevated lipid-lactate peak |
| EEG findings | NA | Normal | NA | Abnormal | Normal |
| Ophthalmological findings | NA | Normal VEP and fundus | Stippled pigmentation of fundus | NA | NA |
| Hearing evaluation | Normal | NA | NA | Impaired | Normal |
| Blood lactate (mg/dL) | NA | 20.7 | 36 | 65.8 | 40.5 |
| CPK (normal: 22–198 U/L) | NA | 132 | 568 | NA | 42 |
Abbreviations: +, present; −, absent; CPK, creatine phosphokinase; EEG, electroencephalogram; MRI, magnetic resonance imaging; MRS, magnetic resonance spectroscopy; NA, not available; OFC, occipitofrontal circumference; SD, standard deviation; VEP, visual evoked potential.
Observed on computed tomography.
The marked similarity between the clinical presentation of the proband described here when compared with our previously reported subjects with MMDS5 led us to perform targeted testing for the same genetic variant, c.259G > A [p.(Glu87Lys)] in ISCA1 . The in silico prediction tools, Poly-Phen-2, Sorting Intolerant from Tolerant (SIFT), and MutationTaster, are consistent in predicting the pathogenicity of the said genetic variant. 10 11 12 The variant is not present in homozygous state in the 1000 Genomes Project, the Exome Aggregation Consortium (ExAC), gnomAD, and in our in-house exome data of 377 unrelated individuals.
All the patients and families with the reported pathogenic variant are from the same geographical region. Exome sequencing was done to validate the founder region haplotype as observed in the previously reported patients who demonstrated an identical haplotype (G-A-T-G-C-G-A-A-T-T-G-T-T-T-C-G) in all the three families, thus confirming the founder effect.
This report further validates ISCA1 as a causative gene for MMDS5. It also illustrates that MMDS5 has very characteristic and recognizable clinical and radiological findings. Also, the confirmation of a founder effect is likely to aid in diagnosis of MMDS5 by targeted variant testing for individuals from this geographic region.
Acknowledgments
We thank the patient and his family for participating in the study. We thank the Department of Health Research, Ministry of Health and Family Welfare, Government of India, for funding the project entitled “Clinical and molecular characterization of leukodystrophies in Indian children” (V.25011/379/2015-GIA/HR).
Footnotes
Conflict of Interest None.
Supplementary Material
References
- 1.Cózar-Castellano I, del Valle Machargo M, Trujillo E et al. hIscA: a protein implicated in the biogenesis of iron-sulfur clusters. Biochim Biophys Acta. 2004;1700(02):179–188. doi: 10.1016/j.bbapap.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 2.Song D, Tu Z, Lee F S. Human ISCA1 interacts with IOP1/NARFL and functions in both cytosolic and mitochondrial iron-sulfur protein biogenesis. J Biol Chem. 2009;284(51):35297–35307. doi: 10.1074/jbc.M109.040014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheftel A D, Wilbrecht C, Stehling O et al. The human mitochondrial ISCA1, ISCA2, and IBA57 proteins are required for [4Fe-4S] protein maturation. Mol Biol Cell. 2012;23(07):1157–1166. doi: 10.1091/mbc.E11-09-0772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shukla A, Hebbar M, Srivastava A et al. Homozygous p.(Glu87Lys) variant in ISCA1 is associated with a multiple mitochondrial dysfunctions syndrome. J Hum Genet. 2017;62(07):723–727. doi: 10.1038/jhg.2017.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vigeland M D, Gjøtterud K S, Selmer K K. FILTUS: a desktop GUI for fast and efficient detection of disease-causing variants, including a novel autozygosity detector. Bioinformatics. 2016;32(10):1592–1594. doi: 10.1093/bioinformatics/btw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thorvaldsdóttir H, Robinson J T, Mesirov J P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(02):178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brancaccio D, Gallo A, Mikolajczyk M et al. Formation of [4Fe-4S] clusters in the mitochondrial iron-sulfur cluster assembly machinery. J Am Chem Soc. 2014;136(46):16240–16250. doi: 10.1021/ja507822j. [DOI] [PubMed] [Google Scholar]
- 8.Netz D J, Mascarenhas J, Stehling O, Pierik A J, Lill R. Maturation of cytosolic and nuclear iron-sulfur proteins. Trends Cell Biol. 2014;24(05):303–312. doi: 10.1016/j.tcb.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 9.Beilschmidt L K, Ollagnier de Choudens S, Fournier M et al. ISCA1 is essential for mitochondrial Fe 4 S 4 biogenesis in vivo . Nat Commun. 2017;8:15124. doi: 10.1038/ncomms15124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwarz J M, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7(08):575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 11.Adzhubei I A, Schmidt S, Peshkin L et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(04):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar P, Henikoff S, Ng P C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(07):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.