

Abstract
Interferons (IFNs) exhibit forceful inhibitory activities against numerous viruses by inducing synthesis of anti-viral proteins or promoting immune cell functions, which help eradicate the vicious microbes. Consequently, the degree to which viruses evade or counterattack IFN responses influences viral pathogenicity. Viruses have developed many strategies to interfere with the synthesis of IFNs or IFN receptor signaling pathway. Furthermore, multiple viruses decrease levels of IFN receptors via diverse tactics, which include decreasing type I IFN receptor mRNA expression, blocking post-translational modification of the receptor, and degrading IFN receptors. Recently, influenza virus was found to induce CK1α-induced phosphorylation and subsequent degradation of the receptor for type I and II IFNs. In this review, viral mechanisms that remove IFN receptors are summarized with an emphasis on the mechanisms for virus-induced degradation of IFN receptors.
Keywords: Type I IFN receptor, type II IFN receptor, Casein kinase 1α
1. Introduction
Type I IFN was first found to interfere with the replication of influenza virus by Isaacs and Lindenmann in 1957 (Isaacs and Lindenmann, 1957). Since then, the potent antiviral function of IFN has been confirmed in versatile systems (Hoffmann et al., 2015; Katze et al., 2002; Muller et al., 1994). Cellular sensing of viral genetic information triggers a signal cascade to induce and secrete type I IFNs, which can bind to the cell (self) or neighboring cells via the interaction of IFN with its cognate receptor. This autocrine and paracrine signaling activates the renowned JAK/STAT signaling pathway to induce expression of IFN-stimulated genes (/SGs), whose products exert antiviral functions and lead to the establishment of an antiviral state (Aaronson and Horvath, 2002; Darnell et al., 1994; Schoggins, 2014; Sharma et al., 2003). Type I IFNs that include multiple IFN-α subtypes and IFN-ß directly obstruct the replication of viruses, while type II IFN (IFN-γ) is produced from specific immune cells such as T cells and NK cells displaying immunoregulatory activities (Alspach et al., 2018; Schroder et al., 2004).
Viruses strive to escape host protective IFN responses to maximize viral propagation. One strategy, which has been elucidated for many viruses, is to interfere with the production of type I IFNs, thereby reducing anti-viral signaling. (Garcia-Sastre, 2017; Hoffmann et al., 2015; Horner and Gale, 2013; Nan et al., 2014). For example, influenza virus was shown to use a viral protein to block activation of RIG-I, a cellular sensor of viral RNAs involved in the interferon response (Gack et al., 2009). Nevertheless, completely blocking the production of type I IFNs seems difficult, given that type I IFNs were detected in diverse experimental systems as well as flu patients (de Jong et al., 2006; Garcia-Sastre, 2011; Kash et al., 2006; Killip et al., 2015). Furthermore, type I IFNs can be produced from uninfected cells through soluble secondary messengers produced from cGAS-STING pathway, indirect activation of plasmacytoid dendritic cells, or detection of viral genetic elements delivered from apoptotic cells by conventional dendritic cells and macrophages (Colonna et al., 2004; Ma and Damania, 2016; Seo and Hahm, 2010). Thus, for efficient replication, viruses may need to interfere with IFN receptor signaling as well. Indeed, viruses can impair the activation of receptor signaling by using diverse strategies, which include viral decoy to bind type I IFN to prevent IFN recognition by the receptor, inhibition of JAK/STAT signaling, and regulation of the antiviral function of ISG products (Garcia-Sastre, 2017; Symons et al., 1995; Xu et al., 2012; Xu et al., 2014).
Recently, influenza virus was demonstrated to induce degradation of receptors for type I and type II IFNs (Xia et al., 2015; Xia et al., 2018). This suggests an additional mechanism of anti-IFN defense, enabling influenza virus to interfere with the IFN response at both the level of IFN receptor signaling as well as IFN synthesis. Indeed, several other viruses were also shown to induce the downregulation of type I IFN receptor (IFNAR) (Chandra et al., 2014; Cho et al., 2012; Jarret et al., 2016; Liu et al., 2009b; Lu et al., 2012; Shah et al., 2009; Zhang et al., 2017b). Viral antagonism targeting IFNAR seems to be accomplished through a variety of mechanisms, which include the regulation of IFNAR1 mRNA expression, IFNAR1 degradation, and the inhibition of post-translational modification of IFNAR1. Additionally, a few reports indicate that viruses can cause downregulation of type II IFN receptor (IFNGR) (Chandra et al., 2014; Li et al., 2007; Shah et al., 2009). However, the degradation process of IFNGR remains largely unexplored. This review compiles reports on IFN receptor degradation and downregulation, particularly upon virus infection, and discusses the underlying mechanisms, which contain the findings from influenza virus-induced destruction of IFN receptors.
2. IFNAR degradation
2.1. Cellular conditions for IFNAR degradation.
IFNAR is composed of IFNAR1 and IFNAR2 (Uze et al., 2007). Detailed mechanisms for the regulation of IFN receptor endocytosis and degradation processes are found in an excellent review article written by Fuchs SY (Fuchs, 2013) and are briefly summarized in this introductory section. The IFNAR complex undergoes clathrin-dependent endocytosis when the Tyrosine (Y466 of human IFNAR1)-based endocytic motif (YVFF), which is present on the cytoplasmic domain of IFNAR1, is exposed to the adaptin protein-2 (AP-2) complex. Internalized IFNAR1 generally undergoes subsequent lysosomal degradation, while the IFNAR2 subunit tends to recycle back to the cell surface. Therefore, the level of IFNAR1 seems to be targeted for regulation and important for triggering the IFNAR downstream signaling pathway (Fuchs, 2012, 2013; Hwang et al., 1995; Marijanovic et al., 2007).
In unstimulated cells, the endocytic motif of IFNAR1 is engaged for association with TYK2 kinase, blocking the internalization and degradation of IFNAR1. This process is independent of the catalytic activity of TYK2 (Kumar et al., 2008). However, when stimulated by type I IFN, human IFNAR1 is ubiquitinated at K501, K525, and K526, which is critical for recruiting of AP2 complex to IFNAR1 to initiate its endocytosis (Kumar et al., 2007; Kumar et al., 2004). Importantly, Skp1-Cullin1-HOS-Roc1 (SCFHOS, also known as β-TrCP2) has been identified to mediate the ubiquitination of IFNAR1 (Kumar et al., 2003). Furthermore, serine phosphorylation of IFNAR1 is required for the interaction of IFNAR1 with β-TrCP2 and the consequent degradation of IFNAR1. The phosphorylation site (S535 in human IFNAR1) of IFNAR1 is located in the degradation motif DSGNYS (amino acid, aa 534–539), which is conserved in all known species (Kumar et al., 2004; Kumar et al., 2003).
In a process called eliminative signaling, type I IFN bound to its receptor initiates internalization and degradation of IFNAR1 (Huangfu and Fuchs, 2010; Kumar et al., 2003). This seems to be a negative regulatory mechanism that could attenuate signaling when amounts of IFN reach a certain level (Fuchs, 2013). Perhaps, our cells intend to block the inflammatory response by downregulating IFNAR. In support of this, inflammatory experimental conditions and TLR stimulations could cause the downregulation of IFNAR (Bhattacharya et al., 2014; Katlinskaya et al., 2016; Qian et al., 2011). Cellular kinase, serine/threonine protein kinase D2 (PKD2) was reported to mediate the phosphorylation of IFNAR1 when type I IFN triggers the degradation process of IFNAR1 (Figure 1) (Zheng et al., 2011a; Zheng et al., 2011c). Although TYK2 is important for protecting IFNAR1 from endocytosis by masking the endocytic motif, the enzymatic activity of TYK2 is required for IFN-induced PKD2 activation for IFNAR1 degradation (Zheng et al., 2011a). PKD2 can be also activated by other stimuli, leading to IFNAR1 degradation. For example, vascular endothelial growth factor (VEGF), an angiogenic cytokine that is widely expressed in tumors, activates PKD2. This leads to the phosphorylation of IFNAR1 and consequent attenuation of type I IFN signaling during angiogenesis (Zheng et al., 2011b). BCR-ABL also activates PKD2 in chronic myeloid leukemia cells, resulting in the phosphorylation and subsequent degradation of IFNAR1 (Bhattacharya et al., 2011b; Mihailovic et al., 2004). This implies that IFNAR1 degradation is involved in BCR-ABL-mediated tumorigenesis.
Fig 1. Type I IFN-dependent pathway and ER stress response-induced pathway of IFNAR1 degradation.
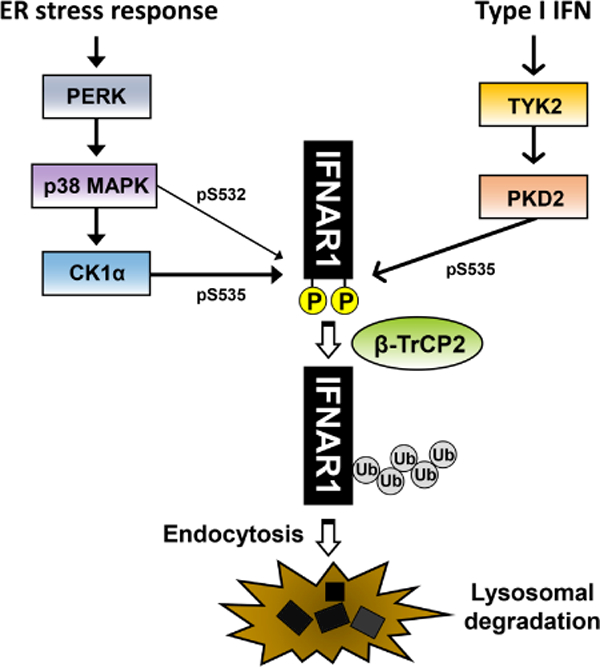
Two representative pathways of IFNAR1 degradation, i.e., ligand (IFN)-dependent pathway and ligand-independent but ER stress-induced pathway are depicted. PERK-mediated ER stress response activates p38 MAPK. IFNAR1 is phosphorylated by p38 MAPK at S532, and then subsequently phosphorylated again by CK1α at S535. Binding of type I IFN to the receptor also induces phosphorylation of IFNAR1 at S535, which is catalyzed by PKD2 and requires the activity of TYK2. The phosphorylation of IFNAR1 then recruits the E3 ubiquitin ligase β-TrCP2, which catalyzes poly-ubiquitination on IFNAR1, leading to the endocytosis and lysosomal degradation of IFNAR1.
IFNAR1 can be degraded in a manner independent of its ligand (type I IFN) such as the inflammatory response (Bhattacharya et al., 2014; Huangfu et al., 2012) and the endoplasmic reticulum (ER) stress response (Bhattacharya et al., 2013; Liu et al., 2009b). ER stress response mainly occurs through activation of one or more of the following regulatory proteins: activating transcription factor 6 (ATF6), PKR-like ER kinase (PERK), or inositol-requiring enzyme 1 (IRE1) (Hassan et al., 2012; Xu et al., 2005). Initially, forcibly overexpressed IFNAR1 at high levels was shown to undergo phosphorylation within its destruction motif. This prompted investigation into the relationship between the ER stress response and IFNAR1 degradation (Liu et al., 2009b). Pharmacologic induction of ER stress response such as the treatment with thapsigargin induced IFNAR1 degradation, which was demonstrated to be reliant on the PERK pathway (Liu et al., 2009b; Yu et al., 2015). While it is unclear how the PERK-dependent ER stress response activates p38 MAPK, p38 MAPK phosphorylates IFNAR1 at S532. This pre-phosphorylation subsequently induces the phosphorylation of IFNAR1 at S535 by casein kinase 1α (CK1α), which is independent of type I IFN (ligand) (Figure 1) (Bhattacharya et al., 2010; Bhattacharya et al., 2011a; Liu et al., 2009a; Liu et al., 2009b). Furthermore, inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and IL-1 β were also shown to induce the phosphorylation and degradation of IFNAR1 through activation of p38 MAPK and CK1α (Huangfu et al., 2012). Viral infections can cause inflammatory responses by inducing the synthesis of TNF-03b1 or IL-1 β, which may also lead to IFNAR1 degradation. Possibly, viruses could utilize this cellular mechanism of IFNAR1 degradation for their own benefit, which warrants future investigation. The phosphorylated IFNAR1 becomes a substrate for the ubiquitinase, β-TrCP2, leading to the degradation of poly-ubiquitinated IFNAR1 (Kumar et al., 2004; Kumar et al., 2003). After IFNAR1 is phosphorylated by either CK1α or PKD2, the following ubiquitination step between the ligand (type I IFN)-dependent and ligand-independent (ER stress response) degradation of IFNAR1 appears to be the same.
The phosphorylation-incompetent mutant of IFNAR1 (S535A) was shown to become resistant to degradation signaling (Kumar et al., 2004). Moreover, IFNAR1 knock-in mice that have a mutation at the phosphorylation site (S526A) of murine IFNAR1 was created. IFNAR1 (S526A) knock-in mice developed normally, but under the experimental inflammatory disease condition of pancreatitis, mice developed increased IFN responses and enhanced IFN-stimulated gene expression, leading to heightened inflammatory responses (Bhattacharya et al., 2014). The possible role of IFNAR degradation during tumorigenesis and tumor escape response is discussed in another review (Fuchs, 2013); this review will focus on viral regulation of IFNAR.
2.2. Viral elimination of IFNAR.
Many viruses have been reported to downregulate the level of IFNAR1. RNA viruses including the flaviviruses, hepatitis C virus (HCV), vesicular stomatitis virus (VSV), enterovirus 71 (EV71), and influenza A virus (IAV), as well as DNA viruses such as hepatitis B virus (HBV), herpes simplex virus (HSV), and Epstein-Barr virus (EBV) were shown to decrease the level of IFNAR1 (Table 1) (Chandra et al., 2014; Chee and Roizman, 2004; Cho et al., 2012; Jarret et al., 2016; Liu et al., 2009b; Lu et al., 2012; Lubick et al., 2015; Shah et al., 2009; Zhang et al., 2017b). It seems that the mechanisms to reduce IFNAR1 levels during virus infections are diverse.
Table 1.
Regulation of type I and type II IFN receptors during virus infections.
| Viruses | Impact on IFN receptors | Viral protein(s) | Molecular Mechanisms | References | ||
|---|---|---|---|---|---|---|
| Hepattis C virus (HCV) | 1) Reduces mRNA expression of IFNAR1 | 1) ND | 1) Infection induces aberrant expression of host microRNAs | 1) Jarret et al., Nat Med, 2016 | ||
| 2) Accelerates erdccytoss and degradation cf IFNAR1 | 2) ND | 2) ER-stress response mediates phosphorylation and ubiquitination of IFNAR1 | 2) Liu et al., Cell host Microbe, 2009. | |||
| Vesicular stomatitis virus (VSV) | Accelerates endocytosis and degradation of IFNAR1 | ND | ER-stress response mediates phosphorylation and ubiquitination of IFNAR1 | Liu et al., Cell host Microbe, 2009. | ||
| RNA viruses | Flaviviruses:West nile virus (WNV) and Tickborne encephalitis virus (TBEV) | 1)WNV downregulates IFNAR1 at protein level | 1) WNV Nonstructural proteins | 1) Non-canonical pathway: dependent on lysosomal and proteasome degradation pathway but independent of IFNAR1 phosphorylation | 1) Evans et al., Viral Immunol,2011 | |
| 2) WNV and TBEV inhibits maturation (glycosylation) of IFNAR1 | 2) NS5 of WNV and TBEV associates with cellular prolidase to inhibit IFNAR1 maturation | 2) NS5 of WNV and TBEV | 2) Lubck et al., Cell host Mcrcte. 2015 | |||
| Enterovirus 71 (EV71) | Downregulation of IFNAR1 | Protease 2A | ND | Lu et al., J Virol. 2012 | ||
| Influenza A virus (IAV) | Induces degradation of IFNAR1 | HA | HA induces phosphorylation and ubiquitination of IFNAR1 via CK1α |
Xia et al., J Virol. 2015; Xia et al., J Virol. 2018 |
||
| IFNAR1 | ||||||
| Human metapneumovirus (hMPV) |
Reduces surface expression but not total level cf IFNAR1 | ND | ND | Ren et al., PloS One, 2011 | ||
| Herpes simplex virus (HSV) | 1) Accelerates endocytosis and degradation cf IFNAR1 | ND | 1) PERK-indeperdent pathway; Possible involvement cf TLR signaling | 1) Qian et al., PloS Pathogen. 2011 | ||
| 2) Reduces the level of IFNAR1 | 2) ND | 2) Chee et al., J Virol, 2004 | ||||
| Murine gammaherpesvirus-68 (MHV-68) | Induces degradation of IFNAR1 | ORF54 (dUTPase) |
ND | Leang et al., PloS Pathogen. 2011 | ||
| DNA viruses |
Epstein-Barr virus (EBV) | Reduces the level of intracellular pool of IFNAR1 without affecting surface level | LMP2A and LMP2B | ND | Shah et al., Oncogene. 2009 | |
| Hepatitis B virus (HBV) | 1) Reduces RNA expression of IFNAR1 | Protein X | 1) ND | Cho et al., Int J Mol Med, 2012 | ||
| 2) Dmmshes IFNAR1 surface expression | (HBx) | 2) HBx reduces expression of TYK2 | ||||
| Pseudorabies virus (PRV) | Accelerates lyscscmal degradatici cf IFNAR1 | UL50 (dUTPase) |
ND | Zhang et al., J Virol. 2017 | ||
| RNA viruses |
Hepattis C virus (HCV) | Expression of IFNGR1 is mppaired | ND | ND | Chancra et al., PLoS Cne. 2014 | |
| Influenza A virus (IAV) | Induces degradation of IFNGR1 | HA | HA induces phosphorylation and ubiquitination of IFNGR1 via CK1α | Xia et al ., J Virol, 2018 | ||
| IFNGR1 | DNA viruses |
Kaposi's sarcoma-associated herpesvrus (KSHV) | Reduces surface expression of IFNGR1 | K3 and K5 | K3 and K5 induces ubiquitination and endocytosis of IFNGR1 | Li et al., J Virol, |
| Epstein-Barr virus (EBV) | Reduces the level of intracellular pool of IFNGR1 without | LMP2A and LMP2B | ND | Shah et al., Oncogene, 2009 | ||
ND: Not determined.
In some cases, both RNA and protein levels of IFNAR1 are modulated during virus infection. HCV infection was demonstrated to decrease the mRNA expression of IFNAR1, but not IFNAR2 (Jarret et al., 2016). Intriguingly, HCV induces aberrant expression of host microRNAs, miR-208b and miR-499a-5p encoded by myosin genes, which bind 3’UTR of IFNAR1 to destabilize the IFNAR1 mRNA leading to the inhibition of antiviral type I IFN signaling in HCV-infected hepatocytes (Jarret et al., 2016). Furthermore, ER stress markers were detected in HCV-induced chronic liver disease and liver cirrhosis patients, which was associated with the impaired expression of IFNAR1 (Chandra et al., 2014). Infection by HCV or VSV induces the PERK pathway of ER-stress response, which results in the phosphorylation and ubiquitination of IFNAR1, followed by endocytosis and degradation of the receptor (Chandra et al., 2014; Liu et al., 2009b). Therefore, during HCV infection, IFNAR1 seems to be controlled at both RNA and protein levels. HBV reduces the RNA expression of IFNAR1 by viral protein X (HBx) (Cho et al., 2012). HBx also diminished the expression of TYK2, which was proven to be crucial for the stabilization of IFNAR1 (Ragimbeau et al., 2003), and induced translocation of IFNAR1 into cytoplasm (Cho et al., 2012). Therefore, it is likely that HBx decreases the level of IFNAR1 protein by reducing the expression of TYK2. This means that HBx regulates IFNAR1 at the post-translational level as well as the transcriptional stage. However, the detailed molecular mechanism remains to be defined.
West Nile virus (WNV) was reported to induce depletion of IFNAR1 protein. WNV infection or expression of viral nonstructural (NS) proteins in subgenomic replicon-bearing cells caused downregulation of IFNAR1 without affecting the level of IFNAR1 mRNAs. Although this decrease of IFNAR1 was reliant on the lysosomal or proteasome degradation pathway, it was independent of the ER stress response and phosphorylation of S535 in IFNAR1, suggesting the presence of a non-canonical pathway (Evans et al., 2011). Another study reported that Flaviviruses including TickBorne Encephalitis virus (TBEV) and WNV inhibit IFNAR1 surface expression by suppressing post-translational modification such as glycosylation (maturation) of IFNAR1 (Lubick et al., 2015). IFNAR1 is known to be heavily glycosylated mostly through N-linked modifications (Ling et al., 1995). In this case, viral NS5 protein binds to the host factor prolidase, which prevents the glycosylation and surface expression of IFNAR1 (Lubick et al., 2015). This study elucidated the function of prolidase as being critical for post-translational modification of IFNAR1 and therefore a target for viral interference. Their results also suggest that the glycosylation of IFNAR1 is important for accumulation of functional IFNAR1 on the surface of cells. Human metapneumovirus (hMPV) infection reduced the surface expression of type I IFN receptor (IFNAR) but total cellular level of IFNAR1 appeared unchanged, suggesting that hMPV facilitated the internalization of IFNAR1 (Ren et al., 2011). The decreased level of IFNAR1 on cells could be due to the decreased level of TYK2 caused by hMPV infection. In contrast to hMPV, EBV has been shown to reduce the level of intracellular IFNAR1 without altering IFNAR1 levels on the surface of cells (Shah et al., 2009). In this research, LMP2A and LMP2B proteins of EBV were shown to accelerate the turnover of IFNAR1 through processes requiring endosomal and lysosomal function (Shah et al., 2009). However, it is unclear how the decrease of the intracellular IFNAR1 without altering surface level of IFNAR1 inhibits antiviral IFN pathway. It is possible that LMP2A/LMP2B have an as-yet unidentified mechanism to impair the functionality of IFN receptors on cell surfaces and decrease intracellular pool of IFNAR that can replace the non-functional receptors.
Protease 2A of EV71 was reported to reduce IFNAR1 levels, attenuating IFN receptor signaling. The protease activity of EV71 2A is required for this inhibitory effect, but the target of 2A protease is unknown (Lu et al., 2012). dUTPase UL50 of pseudorabies virus (PRV) was shown to induce the lysosomal degradation of IFNAR1; however, the degradation of IFNAR1 was independent of the dUTPase activity of UL50 of PRV (Zhang et al., 2017b). Similarly, dUTPase enzymatic activity of ORF54 was not required for downregulation of IFNAR1 protein during murine gammaherpesvirus-68 (MHV-68) infection (Leang et al., 2011). In this study, ORF54’s anti-IFN function was shown to be critical for persistent infection of MHV-68. Therefore, viral proteins may or may not utilize their well-known enzymatic function in the induction of IFNAR1 downregulation.
The level of IFNAR1 was shown to decrease following HSV infection in vitro (Chee and Roizman, 2004). IFNAR1 degradation caused by HSV infection was reported to be independent of PERK, but possibly mediated by p38 MAP kinase upon stimulation of toll-like receptors (TLRs) (Ma and He, 2014; Qian et al., 2011 ).
Recently, we have shown that IAV induces the reduction of IFNAR1 at a post-transcriptional stage (Xia et al., 2015). In fact, the viral hemagglutinin (HA) was proven to trigger phosphorylation and ubiquitination of IFNAR1, leading to the degradation of this receptor (Figure 2). IAV HA-induced IFNAR1 degradation appeared to be dependent on both the proteasome and lysosome degradation pathways (Xia et al., 2015). Traditionally, degradation of the cell-surface receptor IFNAR1 is thought to depend primarily on the lysosomal degradation pathway (Katzmann et al., 2001; Kumar et al., 2003). However, HA-induced degradation of IFNAR1 is reliant on the proteasome-dependent pathway as well as lysosome pathway (Xia et al., 2015). Possibly, influenza viral HA also regulates the stability of IFNAR1 protein from the intracellular pool existing within the cells before the receptor is localized on the plasma membrane of cell surface.
Fig 2. Influenza HA-induced degradation of IFNAR1 blocks the IFNAR signaling pathway.
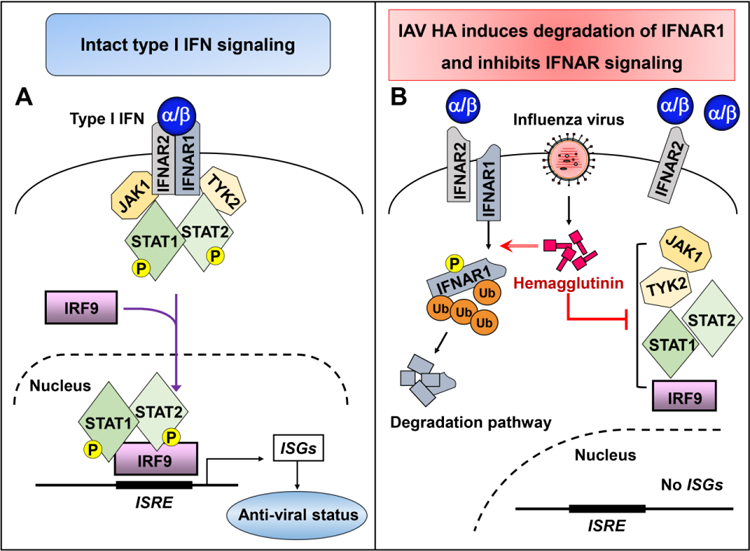
(A)Type I IFNs (IFN-α/β) bind to their receptor IFNAR, which is composed of IFNAR1 and IFNAR2. This interaction elicits the JAK/STAT signaling pathway. JAK1 and TYK2 phosphorylate STAT1 and STAT2, which form a trimeric complex with IRF9, translocate to the nucleus, and bind to interferon stimulated response elements (ISRE), leading to the expression of interferon-stimulated genes (ISGs). ISGs encode antiviral proteins, which establish an anti-viral status. (B) During IAV infection, hemagglutinin protein induces phosphorylation and ubiquitination of IFNAR1, leading to the degradation of this receptor. This process will deplete cellular IFNAR1, which in turn suppresses the JAK/STAT activation and inhibits the expression of ISGs.
2.3. Phosphorylation of IFNAR by viral infection.
Although several viruses were shown to induce degradation of IFNAR1 during infection, phosphorylation of IFNAR1 was demonstrated to take place during infection by only a few viruses such as VSV and HCV via the ER-stress response, or by IAV HA (Liu et al., 2009b; Xia et al., 2015). In the context of VSV or HCV infection, PERK-mediated ER stress responses cause p38 MAPK-induced phosphorylation of IFNAR1, leading to degradation of IFNAR1. Recently, CK1α was proven to be critical in IAV HA-mediated IFNAR1 degradation, as pharmacologic inhibition of CK1α as well as siRNA-based knockdown of CK1α attenuated the degradation process of IFNAR1 triggered by IAV infection or IAV HA expression (Figure 3) (Xia et al., 2018).
Fig 3. CK1α controls HA-induced IFN receptor degradation, which regulates influenza viral replication and the host immunity to infection.
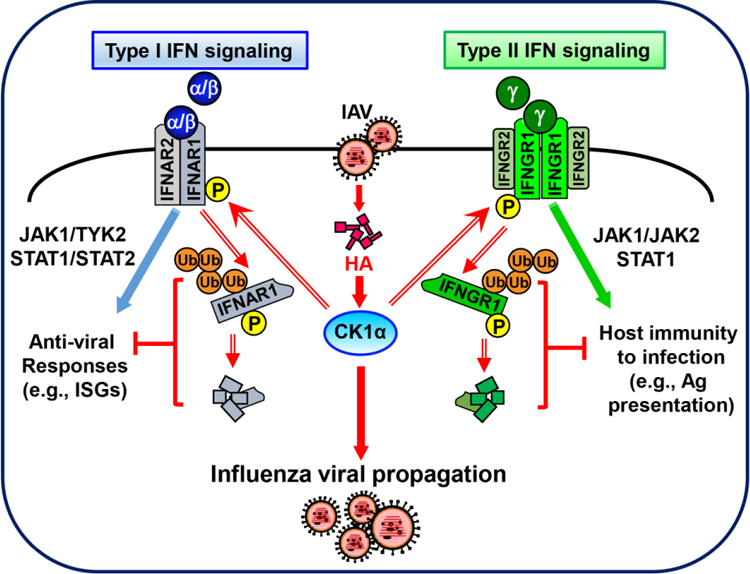
During IAV infection, hemagglutinin induces activation of casein kinase 1α (CK1α), which mediates the phosphorylation and ubiquitination of IFNAR1 and IFNGR1, leading to the degradation of both receptors. The elimination of IFN receptors suppresses both the IFNAR-mediated anti-viral responses and the IFNGR-triggered host immunity to virus infection.
Unlike during the ER stress response, influenza viral HA-induced IFNAR1 degradation does not require the activation of p38 MAPK (Xia et al., 2018). Thus, HA seems to have a unique strategy to activate CK1α, bypassing the pre-phosphorylation step of IFNAR1 by p38 MAPK. IAV-induced CK1α activation appeared to be independent of the ligand, type I IFN. This is because viral HA did not induce IFN response but caused IFNAR1 to degrade. Furthermore, IAV infection could effectively induce IFNAR1 degradation in IFN production-incompetent Vero cells, which was also blocked by treatment with CK1α -specific inhibitor (Xia et al., 2018).
It is unknown if FIA directly associates with CK1α or if HA utilizes another cellular factor to recruit CK1α to IFNAR1. It is also uncertain whether IAV HA displays any function inside cells to activate CK1α or if HA on virus particle interacts with a cellular surface protein triggering a signal cascade to regulate host IFNAR1 stability. Interestingly, when cells were infected with an HA gene-deficient influenza virus, IFN receptor was not downregulated (Xia et al., 2018). HA gene-deficient influenza virus was made using HA protein-expressing cells and thus retains HA proteins on the virus, allowing one cycle of viral infection (Marsh et al., 2007; Martinez-Sobrido et al., 2010). Thus, the engagement of HA protein with the cellular receptor, sialic acid, does not appear to cause downregulation of IFNAR1. Rather, HA proteins newly expressed in the infected cells seem to trigger a signal path for the receptor degradation. Indeed, the degradation of IFNAR1 is correlated with the increased expression level of viral proteins in the cells (Xia et al., 2015; Xia et al., 2018). The possibility of specific localization of HA inside cellular organelles such as the ER or Golgi being important for initializing the degradation pathway remains to be further investigated.
While viruses such as TBEV were shown to downregulate functional IFNAR to promote viral spread (Lubick et al., 2015), the importance of IFNAR1 degradation during IAV infection has not been investigated. It is conceivable that the downregulation of IFNAR1 is a cellular process that takes place at the late stage of virus replication to prevent extreme activation of IFN signal. Fortunately, the identification of CK1α as a cellular protein to control IFNAR1 degradation allowed us to directly address this question during IAV infection. Indeed, CK1α was demonstrated to display a pro-viral function by inducing degradation of IFNAR1. Treatment of A549 cells with a CK1α-specific inhibitor, (D4476), massively suppressed the production of infectious influenza virus by over 100 fold. This effect was observed throughout the course of infection with time points taken at 24, 48, and 72 hours post-infection. This was also associated with an increase in IFN-induced gene expressions (Xia et al., 2018). These results demonstrate the importance of IFNAR1 degradation caused by influenza and show how strongly influenza virus suppresses host IFN system by eliminating IFNAR1. Nullifying the viral immune-evasive strategy of IFNAR1 degradation presents a possible target to potently repress influenza virus replication in this culture system.
Recently, CK1α was reported to interact with the VP2 protein of the infectious bursal disease virus (IBDV) resulting in regulation of viral growth (Zhang et al., 2017a). It was also shown that CK1α was downregulated during IBDV replication. The siRNA-mediated knockdown of CK1α inhibited IBDV replication, whereas overexpression of CK1α enhanced IBDV propagation. Although it is uncertain whether IBDV induces CK1α-mediated degradation of IFNAR, the results suggest that CK1α exhibits a pro-viral function during IBDV infection. CK1α may represent an attractive cellular target to impair replication processes of multiple viruses in our effort to cure viral diseases.
3. Viral degradation of IFNGR
The IFNGR complex consists of IFNGR1 and IFNGR2 subunits, and it undergoes clathrin-dependent endocytosis upon binding to IFN-γ (Bach et al., 1997; Blouin and Lamaze, 2013; Marchetti et al., 2006). It was reported that both of the two subunits contain putative AP-2 binding motifs: A leucine-isoleucine (LI) doublet (aa 270–271) and a typical tyrosine-based endocytic motif (YVSL, aa 287–290) are found in IFNGR1; a LI doublet (aa 255–256) and an YRGL motif (aa 273–276) are present in IFNGR2. Deletion of these motifs impaired cellular response to IFN-γ as well as the receptor internalization (Blouin and Lamaze, 2013; Farrar et al., 1991). Knockdown of calthrin resulted in accumulation of IFNGR1 at the cell surface and suppressed the IFN-y uptake (Marchetti et al., 2006). However, it is unknown whether IFN-γ induces the phosphorylation or ubiquitination of endogenous IFNGR.
A few reports have documented downregulation of IFNGR1 during viral infection. Kaposi’s sarcoma-associated herpesvirus (KSHV) was shown to downregulate IFNGR1 by using viral proteins. In this elegant study, K3 and K5 proteins of KSHV were proven to increase the endocytosis rate of IFNGR1 expressed on the surface of lymphocytes. K5 induced internalization and ubiquitination of IFNGR1 more strongly than K3, and domains including the amino terminal ring finger motif in K5 were identified to be crucial for downregulation of IFNGR1 (Li et al., 2007). Further, EBV viral proteins LMP2A and LMP2B were reported to accelerate the turnover of IFNGR1 without affecting the level of cell surface IFNGR1 (Shah et al., 2009). HCV infection of primary human hepatocytes shows impaired expression of IFNGR1 as well as IFNAR1 (Chandra et al., 2014). Unlike IFNAR, the degradation mechanism for IFNGR has not been extensively investigated.
Our findings indicate that CK1α is required for efficient degradation of both IFNAR1 and IFNGR1 during influenza virus infection (Figure 3) (Xia et al., 2018). This suggests that the initial degradation pathways for these two receptors may have commonality in employing the same host factors during infection. This could be beneficial for the virus that pulls one trigger to twist both arms of IFN system (type I and type II IFNs). However, IFNAR1 degradation is dependent on both proteasome and lysosomal degradation pathways, whereas IFNGR1 degradation is solely dependent on lysosomal pathway and independent of proteasome pathway during IAV infection (Xia et al., 2015; Xia et al., 2018). The finding implies that these two pathways are different after the step of phosphorylation by CK1α during influenza viral infection. It is possible that the type of poly-ubiquitination or cytosolic and lysosome transport system are different between these two pathways, which could be the cellular intrinsic process designated for these receptors’ degradation.
It was reported that HCV-infected primary human hepatocytes showed a reduction of IFNGR1 and IFNAR1 expression. It was also shown that ER stress markers and autophagy responses were induced in HCV infected chronic liver disease and liver cirrhosis patients (Chandra et al., 2014). We also noted that the ER stress response could induce IFNGR1 degradation, as the treatment of cells with thapsigargin, an inhibitor of the autophagic process which in turn induces stress on the ER, caused downregulation of this receptor (Xia et al., 2018). However, unlike IFNAR1 degradation caused by ER stress response, IFNGR1 degradation in response to thapsigargin treatment is not dependent on p38 MAPK (Xia et al., 2018). Therefore, during PERK-mediated ER stress response, the degradation processes of IFNAR1 and IFNGR1 could be more divergent than the elimination of these receptors by influenza. It is possible that Influenza virus is using a mechanism that simplifies the degradation processes and rapidly destroys both receptors simultaneously.
Degradation of IFNGR1 induced by influenza virus infection rendered cells much less sensitive to recombinant IFN-γ as evidenced by the decreased expression of type II IFN-induced genes such as interferon regulatory factor-1 (IRF-1), transporter associated with antigen processing 1 (TAP-1), and low molecular weight polypeptide 2 (LMP-2) (Xia et al., 2018). Similar results were observed following FIA expression. The antiviral activity of CK1α shown in A549 cells was dependent on the level of IFNAR1, but independent of IFNGR1 expression (Xia et al., 2018). Obviously, type II IFN is produced from some immune cells such as T cells and NK cells and thus the level of IFNGR1 in A549 cells is not important for virus replication. Type II IFN is known to increase the level of proteins that mediate the antigen processing and presentation such as MHC molecules and the cell death pathway (Ferm et al., 1996; Novelli et al., 1997; Seliger et al., 1997). Also, type II IFN could heighten the capability of dendritic cells in producing IL-12 to promote the host protective immunity (Liu et al., 2005; Wang et al., 2000). Therefore, IAV infection is expected to attenuate IFN-γ-induced host immunity during infection (Figure 3). The biological role of IFNGR1 degradation during influenza requires further investigation. Developing new experimental systems, such as creation of the degradation-resistant IFNGR mutant knock-in mice could help address this issue. If the phosphorylation site in the IFNGR1 is defined, the degradation-resistant IFNGR1 mutant can be generated. Additionally, identifying amino acid position in HA protein that is responsible for IFNGR1 degradation may allow us to generate recombinant influenza virus where HA gene is mutated not to cause degradation of IFNGR1. Evaluating the host immunity to this recombinant virus could help us understand the importance of IFNGR1 degradation during virus infection.
A previous study found that high levels of IFN-γ were detected in mouse lungs during IAV infection. It was shown that IFN-γ regulates contraction of the influenza virus-specific CD8 T cells response (Prabhu et al., 2013). However, by using IFNGR−/−IFN-γ−/− mice, the authors demonstrated that the absence of IFN-γ or IFNGR did not compromise the survival of infected mice, virus titers, or rates of viral clearance in their experimental conditions (Prabhu et al., 2013). This implies that IAV has a good strategy to evade the type II IFN signaling, creating a condition favorable for IAV propagation. Our findings that IAV infection can cause rigorous degradation of IFNGR1 may partially explain the reason why deficiency of IFNGR or its binding cytokine ligand does not affect IAV eradication. However, we could not exclude the possibility that the removal of IFNGR1 is simply a byproduct of the viral antagonism of IFNAR pathway, as the IFNGR1 is linked to the mechanism for the elimination of IFNAR1.
4. Perspectives
In sum, a growing body of evidence demonstrates that viruses induce elimination of IFN receptors, presumably in order to maximize viral propagation and survival in the host. The molecular and cellular mechanisms for IFN destruction during virus infection warrants further exploration, which will greatly increase our knowledge about virus-host defense interaction. Since excessive IFN-signaling could cause detrimental inflammatory responses, direct targeting of IFN receptors to increase their expression may not be an ideal approach to designing therapeutic interventions for the treatment of viral infections. However, uncovering viral mechanisms that result in IFN receptor degradation could identify a novel cellular protein that can be targeted to treat viral diseases or autoimmune conditions associated with aberrant production of type I IFNs.
Acknowledgements
We thank the editor Dr. Knipe (Harvard University) for invitation of this review. This work was supported by NIH/NIAID grant R21AI127404 (B.H.) and Laboratory for Infectious Disease Research (LIDR) Jump Start Grant from University of Missouri (P.A.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aaronson DS, Horvath CM, 2002. A road map for those who don’t know JAK-STAT. Science 296, 1653–1655. [DOI] [PubMed] [Google Scholar]
- Alspach E, Lussier DM, Schreiber RD, 2018. Interferon gamma and Its Important Roles in Promoting and Inhibiting Spontaneous and Therapeutic Cancer Immunity. Cold Spring Harb Perspect Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach EA, Aguet M, Schreiber RD, 1997. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol 15, 563–591. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, HuangFu WC, Dong G, Qian J, Baker DP, Karar J, Koumenis C, Diehl JA, Fuchs SY, 2013. Anti-tumorigenic effects of Type 1 interferon are subdued by integrated stress responses. Oncogene 32, 4214–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, HuangFu WC, Liu J, Veeranki S, Baker DP, Koumenis C, Diehl JA, Fuchs SY, 2010. Inducible priming phosphorylation promotes ligandindependent degradation of the IFNAR1 chain of type I interferon receptor. J Biol Chem 285, 2318–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Katlinski KV, Reichert M, Takano S, Brice A, Zhao B, Yu Q, Zheng H, Carbone CJ, Katlinskaya YV, Leu NA, McCorkell KA, Srinivasan S, Girondo M, Rui H, May MJ, Avadhani NG, Rustgi AK, Fuchs SY, 2014. Triggering ubiquitination of IFNAR1 protects tissues from inflammatory injury. EMBO Mol Med 6, 384–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Qian J, Tzimas C, Baker DP, Koumenis C, Diehl JA, Fuchs SY, 2011a. Role of p38 protein kinase in the ligand-independent ubiquitination and down-regulation of the IFNAR1 chain of type I interferon receptor. J Biol Chem 286, 22069–22076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Zheng H, Tzimas C, Carroll M, Baker DP, Fuchs SY, 2011b. Bcr-abl signals to desensitize chronic myeloid leukemia cells to IFNalpha via accelerating the degradation of its receptor. Blood 118, 4179–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouin CM, Lamaze C, 2013. Interferon gamma receptor: the beginning of the journey. Front Immunol 4, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra PK, Gunduz F, Hazari S, Kurt R, Panigrahi R, Poat B, Bruce D, Cohen AJ, Bohorquez HE, Carmody I, Loss G, Balart LA, Wu T, Dash S, 2014. Impaired expression of type I and type II interferon receptors in HCV-associated chronic liver disease and liver cirrhosis. PLoS One 9, e108616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee AV, Roizman B, 2004. Herpes simplex virus 1 gene products occlude the interferon signaling pathway at multiple sites. J Virol 78, 4185–4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho IR, Oh M, Koh SS, Malilas W, Srisuttee R, Jhun BH, Pellegrini S, Fuchs SY, Chung YH, 2012. Hepatitis B virus X protein inhibits extracellular IFN-alpha-mediated signal transduction by downregulation of type I IFN receptor. Int J Mol Med 29, 581–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M, Trinchieri G, Liu YJ, 2004. Plasmacytoid dendritic cells in immunity. Nat Immunol 5, 1219–1226. [DOI] [PubMed] [Google Scholar]
- Darnell JE Jr., Kerr IM, Stark GR, 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264, 1415–1421. [DOI] [PubMed] [Google Scholar]
- de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC, Qui PT, Cam BV, Ha do Q, Guan Y, Peiris JS, Chinh NT, Hien TT, Farrar J, 2006. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 12, 1203–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JD, Crown RA, Sohn JA, Seeger C, 2011. West Nile virus infection induces depletion of IFNAR1 protein levels. Viral Immunol 24, 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar MA, Fernandez-Luna J, Schreiber RD, 1991. Identification of two regions within the cytoplasmic domain of the human interferon-gamma receptor required for function. J Biol Chem 266, 19626–19635. [PubMed] [Google Scholar]
- Ferm M, Gronberg A, Tally M, 1996. IFN-gamma treatment increases insulin binding and MHC class I expression in erythroleukemia cells. Immunol Invest 25, 37–47. [DOI] [PubMed] [Google Scholar]
- Fuchs SY, 2012. Ubiquitination-mediated regulation of interferon responses. Growth Factors 30, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs SY, 2013. Hope and fear for interferon: the receptor-centric outlook on the future of interferon therapy. J Interferon Cytokine Res 33, 211–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, Farzan M, Inoue S, Jung JU, Garcia-Sastre A, 2009. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5, 439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sastre A, 2011. Induction and evasion of type I interferon responses by influenza viruses. Virus Res 162, 12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sastre A, 2017. Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe 22, 176–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan IH, Zhang MS, Powers LS, Shao JQ, Baltrusaitis J, Rutkowski DT, Legge K, Monick MM, 2012. Influenza A viral replication is blocked by inhibition of the inositol-requiring enzyme 1 (IRE1) stress pathway. J Biol Chem 287, 4679–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann HH, Schneider WM, Rice CM, 2015. Interferons and viruses: an evolutionary arms race of molecular interactions. Trends Immunol 36, 124–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner SM, Gale M Jr., 2013. Regulation of hepatic innate immunity by hepatitis C virus. Nat Med 19, 879–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu WC, Fuchs SY, 2010. Ubiquitination-dependent regulation of signaling receptors in cancer. Genes Cancer 1, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu WC, Qian J, Liu C, Liu J, Lokshin AE, Baker DP, Rui H, Fuchs SY, 2012. Inflammatory signaling compromises cell responses to interferon alpha. Oncogene 31, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SY, Hertzog PJ, Holland KA, Sumarsono SH, Tymms MJ, Hamilton JA, Whitty G, Bertoncello I, Kola I, 1995. A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc Natl Acad Sci U S A 92, 11284–11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs A, Lindenmann J, 1957. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 147, 258–267. [PubMed] [Google Scholar]
- Jarret A, McFarland AP, Horner SM, Kell A, Schwerk J, Hong M, Badil S, Joslyn RC, Baker DP, Carrington M, Hagedorn CH, Gale M Jr., Savan R, 2016. Hepatitis-C-virus-induced microRNAs dampen interferon-mediated antiviral signaling. Nat Med 22, 1475–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash JC, Tumpey TM, Proll SC, Carter V, Perwitasari O, Thomas MJ, Basler CF, Palese P, Taubenberger JK, Garcia-Sastre A, Swayne DE, Katze MG, 2006. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 443, 578–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katlinskaya YV, Katlinski KV, Lasri A, Li N, Beiting DP, Durham AC, Yang T, Pikarsky E, Lengner CJ, Johnson FB, Ben-Neriah Y, Fuchs SY, 2016. Type I Interferons Control Proliferation and Function of the Intestinal Epithelium. Mol Cell Biol 36, 1124–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katze MG, He Y, Gale M Jr., 2002. Viruses and interferon: a fight for supremacy. Nat Rev Immunol 2, 675–687. [DOI] [PubMed] [Google Scholar]
- Katzmann DJ, Babst M, Emr SD, 2001. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell 106, 145–155. [DOI] [PubMed] [Google Scholar]
- Killip MJ, Fodor E, Randall RE, 2015. Influenza virus activation of the interferon system. Virus Res 209, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar KG, Barriere H, Carbone CJ, Liu J, Swaminathan G, Xu P, Li Y, Baker DP, Peng J, Lukacs GL, Fuchs SY, 2007. Site-specific ubiquitination exposes a linear motif to promote interferon-alpha receptor endocytosis. J Cell Biol 179, 935–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar KG, Krolewski JJ, Fuchs SY, 2004. Phosphorylation and specific ubiquitin acceptor sites are required for ubiquitination and degradation of the IFNAR1 subunit of type I interferon receptor. J Biol Chem 279, 46614–46620. [DOI] [PubMed] [Google Scholar]
- Kumar KG, Tang W, Ravindranath AK, Clark WA, Croze E, Fuchs SY, 2003. SCF(HOS) ubiquitin ligase mediates the ligand-induced down-regulation of the interferon-alpha receptor. EMBO J 22, 5480–5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar KG, Varghese B, Banerjee A, Baker DP, Constantinescu SN, Pellegrini S, Fuchs SY, 2008. Basal ubiquitin-independent internalization of interferon alpha receptor is prevented by Tyk2-mediated masking of a linear endocytic motif. J Biol Chem 283, 18566–18572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leang RS, Wu TT, Hwang S, Liang LT, Tong L, Truong JT, Sun R, 2011. The anti-interferon activity of conserved viral dUTPase ORF54 is essential for an effective MHV-68 infection. PLoS Pathog 7, e1002292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Means R, Lang S, Jung JU, 2007. Downregulation of gamma interferon receptor 1 by Kaposi’s sarcoma-associated herpesvirus K3 and K5. J Virol 81, 2117–2127.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling LE, Zafari M, Reardon D, Brickelmeier M, Goelz SE, Benjamin CD, 1995. Human type I interferon receptor, IFNAR, is a heavily glycosylated 120–130 kD membrane protein. J Interferon Cytokine Res 15, 55–61. [DOI] [PubMed] [Google Scholar]
- Liu J, Cao S, Kim S, Chung EY, Homma Y, Guan X, Jimenez V, Ma X, 2005. Interleukin-12: an update on its immunological activities, signaling and regulation of gene expression. Curr Immunol Rev 1, 119–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Carvalho LP, Bhattacharya S, Carbone CJ, Kumar KG, Leu NA, Yau PM, Donald RG, Weiss MJ, Baker DP, McLaughlin KJ, Scott P, Fuchs SY, 2009a. Mammalian casein kinase 1alpha and its leishmanial ortholog regulate stability of IFNAR1 and type I interferon signaling. Mol Cell Biol 29, 6401–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, HuangFu WC, Kumar KG, Qian J, Casey JP, Hamanaka RB, Grigoriadou C, Aldabe R, Diehl JA, Fuchs SY, 2009b. Virus-induced unfolded protein response attenuates antiviral defenses via phosphorylation-dependent degradation of the type I interferon receptor. Cell Host Microbe 5, 72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Yi L, Zhao J, Yu J, Chen Y, Lin MC, Kung HF, He ML, 2012. Enterovirus 71 disrupts interferon signaling by reducing the level of interferon receptor 1. J Virol 86, 3767–3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubick KJ, Robertson SJ, McNally KL, Freedman BA, Rasmussen AL, Taylor RT, Walts AD, Tsuruda S, Sakai M, Ishizuka M, Boer EF, Foster EC, Chiramel AI, Addison CB, Green R, Kastner DL, Katze MG, Holland SM, Forlino A, Freeman AF, Boehm M, Yoshii K, Best SM, 2015. Flavivirus Antagonism of Type I Interferon Signaling Reveals Prolidase as a Regulator of IFNAR1 Surface Expression. Cell Host Microbe 18, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, He B, 2014. Recognition of herpes simplex viruses: toll-like receptors and beyond. J Mol Biol 426, 1133–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Damania B, 2016. The cGAS-STING Defense Pathway and Its Counteraction by Viruses. Cell Host Microbe 19, 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti M, Monier MN, Fradagrada A, Mitchell K, Baychelier F, Eid P, Johannes L, Lamaze C, 2006. Stat-mediated signaling induced by type I and type II interferons (IFNs) is differentially controlled through lipid microdomain association and clathrin-dependent endocytosis of IFN receptors. Mol Biol Cell 17, 2896–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marijanovic Z, Ragimbeau J, van der Heyden J, Uze G, Pellegrini S, 2007. Comparable potency of IFNalpha2 and IFNbeta on immediate JAK/STAT activation but differential down-regulation of IFNAR2. Biochem J 407, 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh GA, Hatami R, Palese P, 2007. Specific residues of the influenza A virus hemagglutinin viral RNA are important for efficient packaging into budding virions. J Virol 81,9727–9736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Sobrido L, Cadagan R, Steel J, Basler CF, Palese P, Moran TM, Garcia-Sastre A, 2010. Hemagglutinin-pseudotyped green fluorescent proteinexpressing influenza viruses for the detection of influenza virus neutralizing antibodies. J Virol 84, 2157–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihailovic T, Marx M, Auer A, Van Lint J, Schmid M, Weber C, Seufferlein T, 2004. Protein kinase D2 mediates activation of nuclear factor kappaB by Bcr-Abl in Bcr-Abl+ human myeloid leukemia cells. Cancer Res 64, 8939–8944. [DOI] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M, 1994. Functional role of type I and type II interferons in antiviral defense. Science 264, 1918–1921. [DOI] [PubMed] [Google Scholar]
- Nan Y, Nan G, Zhang YJ, 2014. Interferon induction by RNA viruses and antagonism by viral pathogens. Viruses 6, 4999–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novelli F, D’Elios MM, Bernabei P, Ozmen L, Rigamonti L, Almerigogna F, Forni G, Del Prete G, 1997. Expression and role in apoptosis of the alpha- and beta-chains of the IFN-gamma receptor on human Th1 and Th2 clones. J Immunol 159, 206–213. [PubMed] [Google Scholar]
- Prabhu N, Ho AW, Wong KH, Hutchinson PE, Chua YL, Kandasamy M, Lee DC, Sivasankar B, Kemeny DM, 2013. Gamma interferon regulates contraction of the influenza virus-specific CD8 T cell response and limits the size of the memory population. J Virol 87, 12510–12522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Zheng H, Huangfu WC, Liu J, Carbone CJ, Leu NA, Baker DP, Fuchs SY, 2011. Pathogen recognition receptor signaling accelerates phosphorylation-dependent degradation of IFNAR1. PLoS Pathog 7, e1002065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragimbeau J, Dondi E, Alcover A, Eid P, Uze G, Pellegrini S, 2003. The tyrosine kinase Tyk2 controls IFNAR1 cell surface expression. EMBO J 22, 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Kolli D, Liu T, Xu R, Garofalo RP, Casola A, Bao X, 2011. Human metapneumovirus inhibits IFN-beta signaling by downregulating Jak1 and Tyk2 cellular levels. PLoS One 6, e24496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, 2014. Interferon-stimulated genes: roles in viral pathogenesis. Curr Opin Virol 6, 40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Hertzog PJ, Ravasi T, Hume DA, 2004. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75, 163–189. [DOI] [PubMed] [Google Scholar]
- Seliger B, Hammers S, Hohne A, Zeidler R, Knuth A, Gerharz CD, Huber C, 1997. IFN-gamma-mediated coordinated transcriptional regulation of the human TAP-1 and LMP-2 genes in human renal cell carcinoma. Clin Cancer Res 3, 573–578. [PubMed] [Google Scholar]
- Seo YJ, Hahm B, 2010. Type I interferon modulates the battle of host immune system against viruses. Adv Appl Microbiol 73, 83–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah KM, Stewart SE, Wei W, Woodman CB, O’Neil JD, Dawson CW, Young LS, 2009. The EBV-encoded latent membrane proteins, LMP2A and LMP2B, limit the actions of interferon by targeting interferon receptors for degradation. Oncogene 28, 3903–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J, 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 300, 1148–1151. [DOI] [PubMed] [Google Scholar]
- Symons JA, Alcami A, Smith GL, 1995. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell 81, 551–560. [DOI] [PubMed] [Google Scholar]
- Uze G, Schreiber G, Piehler J, Pellegrini S, 2007. The receptor of the type I interferon family. Curr Top Microbiol Immunol 316, 71–95. [DOI] [PubMed] [Google Scholar]
- Wang IM, Contursi C, Masumi A, Ma X, Trinchieri G, Ozato K, 2000. An IFN-gamma-inducible transcription factor, IFN consensus sequence binding protein (ICSBP), stimulates IL-12 p40 expression in macrophages. J Immunol 165, 271–279. [DOI] [PubMed] [Google Scholar]
- Xia C, Vijayan M, Pritzl CJ, Fuchs SY, McDermott AB, Hahm B, 2015. Hemagglutinin of Influenza A Virus Antagonizes Type I Interferon (IFN) Responses by Inducing Degradation of Type I IFN Receptor 1. J Virol 90, 2403–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia C, Wolf JJ, Vijayan M, Studstill CJ, Ma W, Hahm B, 2018. Casein kinase 1alpha mediates degradation of receptors for type I and type II interferons caused by hemagglutinin of influenza A virus. J Virol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Bailly-Maitre B, Reed JC, 2005. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 115, 2656–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu RH, Rubio D, Roscoe F, Krouse TE, Truckenmiller ME, Norbury CC, Hudson PN, Damon IK, Alcami A, Sigal LJ, 2012. Antibody inhibition of a viral type 1 interferon decoy receptor cures a viral disease by restoring interferon signaling in the liver. PLoS Pathog 8, e1002475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Edwards MR, Borek DM, Feagins AR, Mittal A, Alinger JB, Berry KN, Yen B, Hamilton J, Brett TJ, Pappu RV, Leung DW, Basler CF, Amarasinghe GK, 2014. Ebola virus VP24 targets a unique NLS binding site on karyopherin alpha 5 to selectively compete with nuclear import of phosphorylated STAT1. Cell Host Microbe 16, 187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Zhao B, Gui J, Katlinski KV, Brice A, Gao Y, Li C, Kushner JA, Koumenis C, Diehl JA, Fuchs SY, 2015. Type I interferons mediate pancreatic toxicities of PERK inhibition. Proc Natl Acad Sci U S A 112, 15420–15425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Li H, Chen Y, Gao X, Lu Z, Gao L, Wang Y, Gao Y, Gao H, Liu C, Cui H, Zhang Y, Pan Q, Qi X, Wang X, 2017a. The down-regulation of casein kinase 1 alpha as a host defense response against infectious bursal disease virus infection. Virology 512, 211–221. [DOI] [PubMed] [Google Scholar]
- Zhang R, Xu A, Qin C, Zhang Q, Chen S, Lang Y, Wang M, Li C, Feng W, Zhang R, Jiang Z, Tang J, 2017b. Pseudorabies Virus dUTPase UL50 Induces Lysosomal Degradation of Type I Interferon Receptor 1 and Antagonizes the Alpha Interferon Response. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Qian J, Baker DP, Fuchs SY, 2011a. Tyrosine phosphorylation of protein kinase D2 mediates ligand-inducible elimination of the Type 1 interferon receptor. J Biol Chem 286, 35733–35741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Qian J, Carbone CJ, Leu NA, Baker DP, Fuchs SY, 2011b. Vascular endothelial growth factor-induced elimination of the type 1 interferon receptor is required for efficient angiogenesis. Blood 118, 4003–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Qian J, Varghese B, Baker DP, Fuchs S, 2011c. Ligand-stimulated downregulation of the alpha interferon receptor: role of protein kinase D2. Mol Cell Biol 31,710–720. [DOI] [PMC free article] [PubMed] [Google Scholar]