

Abstract
The biological interactions between glycosaminoglycans (GAGs) and other biomolecules are heavily influenced by structural features of the glycan. The structure of GAGs can be assigned using tandem mass spectrometry (MS2), but analysis of these data, to date, requires manually interpretation, a slow process that presents a bottleneck to the broader deployment of this approach to solving biologically relevant problems. Automated interpretation remains a challenge, as GAG biosynthesis is not template-driven, and therefore one cannot predict structures from genomic data, as is done with proteins. The lack of a structure database, a consequence of the non-template biosynthesis, requires a de novo approach to interpretation of the mass spectral data. We propose a model for rapid, high-throughput GAG analysis by using an approach in which candidate structures are scored for the likelihood that they would produce the features observed in the mass spectrum. To make this approach tractable, a genetic algorithm is used to greatly reduce the search-space of isomeric structures that are considered. The time required for analysis is significantly reduced compared to an approach in which every possible isomer is considered and scored. The model is coded in a software package using the MATLAB environment. This approach was tested on tandem mass spectrometry data for long chain, moderately sulfated chondroitin sulfate oligomers that were derived from the proteoglycan bikunin. The bikunin data was previously interpreted manually. Our approach examines glycosidic fragments to localize SO3 modifications to specific residues and yields the same structures reported in literature, only much more quickly.
Graphical Abstract
INTRODUCTION
Glycosaminoglycans (GAGs) are linear, polydisperse carbohydrates consisting of a repeating uronic sugar and amino sugar copolymer. GAGs serve a multitude of roles in biology including cell-cell and cell-matrix interactions, generation of energy, changes in proteins binding conformation, and molecular recognition[1–3]. Certain GAGs have also been observed as potential biomarkers for disease states[4]. The degree of GAG-protein binding has been shown to be highly dependent on their structure and, more specifically, the position of modifications within their generic repeating copolymer chain [5, 6].
Despite the simple polymeric backbone in GAGs, a single sugar residue can exhibit varying levels of three key modifications, namely O-sulfation, N-deacetylation/sulfation, and uronic sugar stereochemistry[2]. Moreover, the biosynthesis of GAGs is not template driven, resulting in nonuniform dispersion of these modifications across the chain[7, 8]. Database-derived approaches are widely used for protein mass spectra assignment (either top-down or bottom-up) due to the predictability of amino acid sequences from genome sequences, but fail when applied to biomolecules whose production is not template-derived [9, 10]. In contrast to the approaches that are successful for protein/peptide analysis, a de novo approach is required for the computer-based analysis of the tandem mass spectra of GAGs.
Considerable progress has been made in GAG analysis using mass spectrometry [1, 11]. At the MS1 level, a parts-per-million accurate mass measurement, using high resolution instruments such as Fourier transform ion cyclotron resonance mass spectrometry (FTICR-MS), allows assignment of composition, from which GAG chain length, number of modifications and types of modification can be assigned [12]. Tandem MS (MS2) of GAGs using various ion activation methods, such as collision-induced dissociation (CID)[13–15], infrared multiphoton dissociation [16–19], electron-detachment dissociation (EDD)[16, 18–24], and negative-electron transfer dissociation (NETD) [25–27] yields structurally informative fragment ions [28]. Glycosidic bond fragmentation provides monosaccharide composition, while cross-ring fragmentation is used to assign the location of modifications within each residue [29]. Because this is a de novo analytical approach, complete structure analysis requires an information-rich mass spectrum that contains sufficient fragment peaks to fully assign all the variable features. Recent developments in ion activation for GAGs has led to a variety of approaches to produce informative MS2 spectra [21, 23, 28, 30]. However, the interpretation of the such complex mass spectra is generally a tedious manual process that relies upon the expertise of the data analyst. A better understanding of the structural features that promote GAG activity would benefit from an automated, accurate and high-throughput analytical process.
The complexity of the data sets and the time required for analysis increases dramatically as the chain length and the number of modifications increase. Two families of GAGs, heparin/heparan sulfate (Hp/HS) and chondroitin/dermatan sulfate (CS/DS), often contain large numbers of labile sulfate modifications. For these compounds, conventional MS2 methods are often inadequate for complete structural determination, either because they do not produce a comprehensive set of fragment ions required to assign all variable features, or because they lead to decomposition products that confound the analysis [8, 31]. For example, fragmentation can be accompanied by decomposition of sulfo modifications, producing peaks that are reduced in mass by multiples of 80 mass units, but match the mass of standard glycosidic fragments of their counterparts with fewer sulfate modifications [28, 32]. If one does not recognize the peaks that arise from such decomposition, incorrect structural assignments will result. Common de novo strategies that have been successful for protein sequencing [25, 33–35] will inevitably be exposed to substantially more false positives due to the high-likelihood of SO3 loss fragments in GAG MS and MS2. Na+/H+ exchange has been shown to decrease SO3 loss and makes characterization of highly sulfate species possible [30], however SO3 loss is almost always observed in MS2 spectra.
An alternative to the above approach to interpretation is to generate a list of possible fragment peaks for a candidate structure, and to score the match with the experimental data. This process can be repeated for all possible isomers having a given elemental composition. Comparison of the experimental MS2 against the theoretical fragment list allows us to rank each permutation based on closeness-of-fit to the experimental results. This method becomes impractical to perform manually when the number of possible permutations for a composition exceeds the capability to examine the data. For example, Arixtra, a heparin with 5 monosaccharides, is the largest highly sulfated GAG to have complete mass spectral characterization [30]. The number of total possible permutations for a GAG scale logarithmically with the respect to chain length. For both chondroitin/dermatan sulfate and heparan sulfate/heparin, the number of permutations based on chain length and number of modifications is calculated as n-choose-k combinations, where n is the number of possible modifiable sites and k is the number of modifications:
| (eq.1) |
| (eq.2) |
Tools for comparison of user-input structures with fragment peaks from tandem MS have been developed[12, 36, 37], but the requirement for a known starting structure limit applicability for high-throughput analysis.
To address this bottleneck for high-throughput sequencing of GAGs, efforts in computer-assisted methods look to improve upon the speed of analysis and to reduce the amount of user-input and supervision. Several software packages have been developed to overcome modern challenges in GAG analysis although a few require addition steps at the experimental level for optimal software performance. HOST [38] is a computational tool designed for sequencing heparin/HS oligosaccharides using enzymatic digestion combined with ESI-MSn. The method scores and returns the best matching sequences of GAGs based on disaccharide composition analysis, yielding predicted compositions and calculating expected fragmentation patterns in silico. Comparisons of theoretical fragments can then be compared to fragmentation of heparin/HS oligosaccharide MSn data and is scored to return the most likely sequence. However, disaccharide analysis requires complete enzymatic digestion of the GAG using heparin lyase I, II and III over multiple hours of incubation (16 h), limiting the method’s overall speed and applicability in a high-throughput GAG analysis platform.
Another piece of software known as GAG-ID [39] has been shown to discriminate and identify 21 synthetic tetrasaccharides eluted from LC-MS/MS using a scoring system based on peak intensities. It is the first of its kind to automated the interpretation of mixtures when coupled to LC-MS/MS but require complete chemical derivatization of the GAG by replacing all labile sulfate modifications with more stable acetyl groups. Much like HOST, derivatization may not be a viable option for universal GAG analysis.
HS-SEQ [40] is a de novo GAG sequencing computation framework that has been used to automate the structural identification of HS of dp4, 5, 6, 8 and 15. The method determines a precursor sequence (unmodified GAG backbone) and uses information from the tandem MS to best assign possible sulfate and acetate modifications. Assignments are made based on confidence values and are used to generate a list of top candidates. This is the first GAG software that requires only the tandem MS for sequence information. While certainly a high-throughput option, the structural assignment conflicts can arise in the form of sulfate loss fragment, internal fragments or random matches. The authors of HS-SEQ note that the software removes the assignments with lower-confidence to resolve conflicting assignments but also believe this may produce false hits when examining samples extracted from biological sources.
The software developed in our laboratory is designed to sequence GAGs of indefinite length by comparing fragments of theoretical structures (in silico) against experimental data without the need for construction of a database, instead using a genetic algorithm optimization technique to limit the number of permutations while keeping analysis time to a maximum of a few minutes. The method assigns structures based on greatest likelihood using fragment ion products as a critical parameter for the genetic algorithm fitness criterion. Fragments that are in direct conflict with the highest scoring structure(s) are not discarded but reviewed again for possible additional components. We have tested this approach on MS2 data from intact CS chains released from the proteoglycan, bikunin. These chains vary in length from 27–43 saccharide residues, and vary in the degree of O-sulfo modification from 4 to 7, and thus represent a challenging test of this automated procedure.
EXPERIMENTAL METHODS
Mass spectrometry analysis. Bikunin GAG MS and MS2 data reported in [41] was used as a proof-of-principle data set for the purposes of testing genetic algorithm efficacy. The monoisotopic peaks were selected via the SNAP algorithm from Bruker DataAnalysis software. Analysis of the MS2 was performed with the software alone and with no user supervision or assistance.
Computational methods. MS1 analysis of parent ion mass is performed using a composition assignment software module written in the MATLAB coding environment. Monoisotopic peaks and charge states are acquired from Bruker DataAnalysis and deconvoluted to a neutral mass. A composition is derived from one or more neutral mass(es) by searching a data matrix of possible chain lengths, degrees of sulfation, deacetylation, and sodium/hydrogen exchange. The user input also includes the possibility of reducing end modifications, and nonreducing ends that can terminate in unsaturated uronic acids, as is common in enzymatically produced GAG oligomers. Theoretical neutral masses in the spreadsheet are compared against user specified masses with a user-defined mass tolerance. The sequences that match are then used for performing the MS2 analysis.
For MS2 assignment, we implement a genetic algorithm based on fundamental aspects common to all genetic algorithms[42–44]. For MS2 analysis, the software uses a binary vector to represent glycan structures where on-bits denote an occupied site of SO3 modification. The first step generates two glycan structures at random that fit the expected composition (initialization step) and then proceeds to “breed” these structures into a new generation of candidates (crossover step). The new generation also is subject to potential mutations in their structure in the form of exchanges between their on and off-bits (mutation step) in an effort to avoid converging upon a local maximum. Theoretical structures created in the crossover and mutation steps are then tested against the experimental MS2 data where the score of each structure is determined based on a closeness-of-fit paradigm (fitness). The scoring system is subject to various factors that will be discussed in detail in future papers. In the case of bikunin, the score of a structure is a naïve model that determines the top candidate based on the number of matching glyocosidic fragments. The primary three steps (crossover, mutation and fitness) are iterated until the maximum fitness value does not change after numerous cycles. The number of iterations required before termination of the algorithm can be defined by the user but is defaulted at a value of 3. The structure(s) containing the highest scores are then examined using additional data interpretation tools that assign fragment peak masses alongside their charge, intensity and mass error (in ppm).
Experimental MS2 data collected by FT-ICR is extracted from Bruker Apex user interface software using the SNAP peak-picking algorithm. Monoisotopic peak masses and intensities are extracted in the form of comma-separate value (.csv) files. MATLAB software prompts the user for a .csv file containing mass-to-charge in column 1 and intensity in column 2, with mass-to-charge sorted in ascending order. Parent ion mass and charge must be provided by the user as well as mass information pertaining to a linker region mass on the reducing end. Composition details (chain length and numbers of: sulfation, n-acetylation, Na-H exchange) are calculated from a composition calculation module and then given to the software in the preliminary step before initializing the genetic algorithm.
For bikunin proteoglycan a linker mass of 641.1473 (Gal4S-Gal-Xyl-Serine) was used with the remainder of the bikunin chain length represented as a binary vector.
Software integrates separate functional modules to perform mass calculations of theoretical fragment ions, performing standard genetic algorithm features, and scoring theoretical structures against experimental data.
RESULTS AND DISCUSSION
As GAG chain length and modification increases, the number of possible structural permutations exceeds a value suitable for practical, computationally efficient search methods. For the chondroitin sulfate oligomers studied here, the number of structural possibilities is as large as 3.7E22 for an oligomer of length 50 (eq. 2). The number of possibilities is narrowed down when composition can be assigned and the number of known sulfate modifications is determined. While the paradigm for comparing theoretical structures against experimental data can differ, a minimum number of elements such as fragment type, fragment intensity and sequence coverage must be considered for complete GAG characterization [45]. Thus, instead of trying to shortcut these facets of analysis, we chose an approach that reduces the total search space. Hundreds of millions of structures may exist for a specific GAG composition but for a pure sample only one of these structures is a valid assignment. The impracticality of searching through a massive number of incorrect structures is reduced dramatically when a genetic algorithm search heuristic is applied [44].
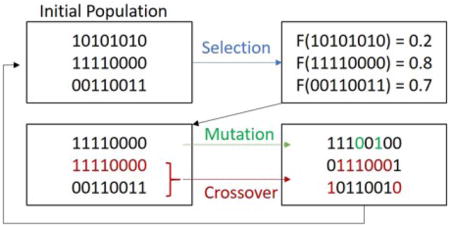
The genetic algorithm is an optimization tool that has been used for a wide variety of applications[46–51]. It mimics the evolutionary process, by using a survival of the fittest mechanism that quickly eliminates large groups of candidates from a pool if they share a feature that does not meet a specific set of criteria [44]. Here we examine the application of this approach to GAG MS2 analysis. We have developed software in the MATLAB coding environment that utilizes the genetic algorithm. GAG sequences are expressed as a binary code where on-bits (1’s) and off-bits (0’s) represent the presence or absence of modifications, respectively and can be applied to both CS/DS and HS/Hp GAG classes, Figure 1 [42, 43]. The binary sequence is shortened or lengthened to accommodate the appropriate composition calculated from the parent-ion mass. The number of on and off bits in the genome is also adjusted based on the number of modifications observed. The final structure is determined via a genetic algorithm, the workflow for which is shown in Figure 2.
Figure 1.

4-bit binary representation for both CS and HS/Hp glycan disaccharides. Each bit is turned on (assigned 1) if a modification is present and off (assigned 0) if the R-group is a hydrogen. Bit 2 represents R2 which has an acetyl modification instead of a hydrogen for an off-bit assignment. In the case of HS where the free-amine is possible, a different numeral can be used to represent the absence of SO3 and acetylation. Additional bits can be introduced so serve as negative control bits as well as a representation for the uronic sugar stereochemistry.
Figure 2.

2a. Workflow for our MATLAB software. User is asked to input three pieces of information for the software: parent ion mass, mass list from MS2 (charge state deconvolution will be automated), and desired mass accuracy for composition assignment and fragment matching (in ppm). The software automates the remaining steps and calculates compositions from the parent ion mass and generates a list of optimized structures using a genetic algorithm. (User provided information is highlighted in the green box. Automated features are highlighted in blue. Software output is shown in purple.)
2b. A demonstration of how genetic operators work on glycan structures. Child candidate modification positions are limited to the modification position of their parents. Mutations, however, and not dependent on parent candidate structure.
Improvements in analysis time and search space reduction can be observed using CID MS2 data from several fractions of intact CS chains for the proteoglycan bikunin [41]. The advantage of using these data is threefold. First, the mass spectra are rich in structurally informative fragments. Structural assignment of bikunin from MS2 was done previously with manual de novo analysis of these fragments. Software suitable for analysis should make the same assignments using these fragments without any user supervision. A second advantage is that modifications are limited to a single sulfate group per disaccharide. Sulfate modifications have been shown to only occur on the 4-O position of the amino sugar using enzymatic disaccharide analysis. Reducing the total number of possible modification diminishes the search space dramatically. For example, a CS dp43 with 5 sulfate groups has 20,349 possible structures when only examining the occupancy of the 4-O position but 5,949,147 possible structures when every sulfate position (2-O, 4-O, 6-O) is taken into consideration. A simplified search space allows us to demonstrate proof of principle while still maintaining computational efficiency. Finally, the structures of bikunin fractions have been manually verified and reported in the literature [41]. A common motif among bikunin fractions was observed after manual sequence analysis. We were particularly interested to see if the unsupervised approach with our software also yielded these same patterns. Candidate structures of bikunin GAGs produced in the genetic algorithm cycles are assigned scores based on the number of matched glycosidic fragments in the experimental data. The fitness of a candidate structure is determined using three separate tiers of scoring:
| (eq.3) |
| (eq.4) |
| (eq.5) |
Unambiguous mass tags such as the linker region dictate that greater emphasis should be placed on the reducing end (Y and Z fragments) and provide a more valid structural assignment. The primary fitness of a score is therefore based on its calculated f1 value, which considers the number of glycosidic fragments from the reducing end (NRE) that are matched in the experimental data. The software then checks to see if any match is potentially a sulfate decomposition peak by adding the mass of an SO3-H exchange (79.9568 Da) and searches the experimental data again for a matching mass. The value of f1 is then reduced by the number of peaks determined to be a product of sulfate decomposition (NRE+SO3).
If the value of f1 is tied among multiple structures, a secondary ranking is then determined with f2, the value of which is based on the number of glycosidic matches from the non-reducing end (B and C fragments). In similar fashion to calculating f1, considerations for potential sulfate decompositions are considered. Non-reducing end fragments are a tier below reducing end fragments since they could potentially match internal fragments due to the lack of an unambiguous mass tag. Incorrect assignment of internal fragments as non-reducing end fragments limits the validity of assignment.
A tertiary score f3 is used after matching glycosidic fragments from both reducing and non-reducing ends. Typically, a small selection of candidate structures (2–4) may end up with equal f1 and f2 values, in which case the summation of the intensities of all matched glycosidic fragments is the tiebreaker. This simple algorithm can and should be continuously fine-tuned for other purposes as software development continues but is sufficient for proof-of-principle purposes.
11 bikunin samples of different compositions were tested using the genetic algorithm. Of these 11, the single highest scoring candidate of the genetic algorithm for 9 of these samples matched the structures reported in literature. Without user supervision, the genetic algorithm results also reaffirm the common bikunin motif reported in literature [41], figure 3. For the remaining 2 samples, the genetic algorithm software reported multiple top-scoring candidates. MS2 data for these two samples could not unambiguously differentiate these structures; however, the structures reported in literature for these samples were present among the top-scoring candidates. This highlights the importance of data quality for optimal software performance. A lack of informative fragmentation peaks can result in structural ambiguities, but information-rich mass spectra can be interpreted with minimal trouble. However, a genetic algorithm approach has no theoretical minimum for data quality. Spectra not containing sufficient fragmentation for complete glycan characterization can still be interpreted based on available fragment ions and a partial sequence can be generated. Although the spectral quality of bikunin GAG tandem MS are high, more complex and longer chain intact GAGs of proteoglycans may yield less than the full suite of fragments necessary for complete sequencing. In this event, our approach can still be used to determine some portion of the overall glycan structure, as has been done recently for decorin glycans [52].
Figure 3.

A list of the highest scoring structures for all MS2 collected on FT-ICR using the genetic algorithm. The structures provided by the genetic algorithm match ones reported in literature. The conserved sulfation pattern of bikunin is also observed. For structures dp43–5S and dp43–6S, three structures are tied for highest scores. Alternate structures for these chain lengths are shown in figure.
In addition to matching previously reported structures, a closer examination of other high-scoring candidate structures among samples shows a consistent motif across compositions. Additional structural motifs shown in Figure 4 consistently score within the top 5 structures of the genetic algorithm. These alternate structures are ones consisting of similar f1 and f2 scores and but have low intensity values for some of their fragment matches (affecting the value of f3). The high degree of similarity between the primary component identified in literature and the alternate structures may be a result of A) our scoring method being favored towards reducing end fragments, B) assigning low intensity noise peaks as glycosidic fragments or C) the possibility of a mixture containing some minor components.
Figure 4.

The highest scoring structure assigned to the all bikunin compositions (except d35–7S) provided, where the bracketed region is a variable stretch of unmodified disaccharides is outlined in blue. Two alternative structures are also frequently observed and outlined in black. The structures appear in the top 5 highest-scoring candidates for all compositions. For chain length dp43 (both 5SO3 and 6SO3), the highest score is tied amongst all three structures. Diagnostic fragments to confidently differentiate between these differences is absent.
The speed of analysis between using the genetic algorithm versus the exhaustive search of every possible permutation of a composition is shown in Figure 5. Here we see that the genetic algorithm has found the correct answer within a small fraction of the time (0.9–2.5% on average) required to examine every possible structure with the assumption that sulfation only occurs on the 4-O position of the N-acetylgalactosamine. Decrease in search time is primarily due to a reduction in the frequency in which unlikely features are eliminated from the genetic algorithm gene pool. As reported [41], bikunin’s sulfation occurs near the reducing end. Isomeric structures that contain sulfate groups in the non-reducing end ranked lowest in the scoring process, resulting in rapid elimination of a test structure and all structures of similar sulfation patterns with one single iteration. A greater number of iterations were spent refining high-scoring structures once poorly scored structures have been eliminated from consideration. The algorithm is designed to rerun the entire genetic process from scratch multiple times in order to avoid plateauing at local maxima. Convergence upon the same highest scoring structure 5 times was the baseline criterion for an acceptable structural assignment. The repetition number is a user-adjustable parameter, as well.
Figure 5.

Speed comparison between the genetic algorithm and exhaustive search method. The bar graph shows the amount of time in hours (left y-axis) it requires for a standard desktop PC (2.4 GHz processor, 4GB ram) to exhaustively search through all possible combinations of a specific composition. The line plot shows the percentage of time (right y-axis) that is required for the genetic algorithm to arrive at the correct answer. Overall search space is reduced dramatically as the number of permutations per composition increases.
Of particular significance, the efficiency of this approach is found to increase as the total number of permutations increases. For a pure sample, only a single structure can be assigned to the MS2 spectrum, but the number of structures with drastically different modification patterns increases with respect to chain length. An increase in chain length also increases the number of GAG structures that could potentially share a feature not observed in the MS2. Structures containing these features drop out of the algorithm as possible options once a single structure of that particular type is scored.
Calculations shown here are run on a 2.4 GHz dual-core processor with 4GB of RAM, a standard laptop or desktop computer. Speed of calculations can increase with more powerful processors such as a GPU workstation or computer cluster. It’s important to note that the genetic algorithm in MATLAB is operated with separate function calls at each step of the algorithm’s cycle. Parallelization of these function calls is particularly attractive for samples of higher chain length and, in theory, could make spectra interpretation no longer the bottleneck for structural elucidation of GAGs. Additional GAG structures determined using this genetic-algorithm based GAG analysis software have been reported [53].
CONCLUSIONS
The software performance is limited by two factors: 1) the quality of the MS2 data and 2) the specificity of the fitness function. The former limitation can be reduced by using a high-performance instrument such as FTICR or Orbitrap mass spectrometers. Some fragment mass values differ by less than 1 Da, increasing the possibility of ambiguity in low performance instruments. High resolution mass spectra with single digit or lower ppm mass error minimize margins for incorrect assignment. Acquisition condition must also be optimized for glycan fragmentation and ideally limits production of confounding fragments such as SO3 loss or internal cleavages.
The latter factor, specificity of the fitness function in the genetic algorithm, is one that can be fine-tuned to GAG analysis by tandem mass spectrometry. The fitness function presented in this paper is simple, arbitrary and based on the basics of glycan analysis. This approach works for the examples selected here because only glycosidic bond cleavage was assigned. Higher level structure analysis based on cross-ring cleavages requires a more sophisticated fitness function. A more complete and non-arbitrary scoring algorithm is being developed that assigns statistical weights and importance factors to various fragment peaks. Additional, peak intensity, while not considered heavily in this iteration of the code, can also signify important characteristics in GAG structure. Details for creating an optimized scoring algorithm will be discussed in future work.
Peak picking for GAG fragmentation is not discussed in this paper but is an important consideration moving forward. Bikunin fragment peaks were selected by the SNAP algorithm using averagine and manually validated; this approach is practical for lowly sulfated samples but averagine is insufficiently for highly sulfated compounds due to contributions of sulfur to the A+2 isotope peak. A fully-automated and GAG-specific peak picking system is current in development.
The software is applicable for GAGs that are both lowly sulfated such as bikunin and moderate and highly sulfated samples for both CS/DS and HS/Hp samples. Short chain HS with more than one SO3 modification per disaccharide and long chain chondroitin sulfate such as decorin with approximate 1 SO3 per disaccharide have been determined using our software [52, 53].
The uronic sugar stereochemistry is a variable modification in GAGs that is difficult to observe using just mass spectrometry. EDD data of heparin and heparan sulfate GAGs has produced a small subset of diagnostic fragments capable of distinguishing between glucuronic and iduronic acid epimers [22]. Chemometric applications has yielded a diagnostic fragment ratio that can definitively determine the C5 stereochemistry [54]. Application of this ratio can be integrated into the software after basic structural features have been assigned using the approach presented here.
Acknowledgments
The authors gratefully acknowledge funding from the National Institute of Health, grants P41GM103390 and R21HL136271.
References
- 1.Xie B, Costello CE. Carbohydrate structure determination by mass spectrometry. Carbohydrate Chemistry, Biology and Medical Applications. 2008:29–57. [Google Scholar]
- 2.Gandhi NS, Mancera RL. The Structure of Glycosaminoglycans and their Interactions with Proteins. Chemical Biology & Drug Design. 2008;72:455–482. doi: 10.1111/j.1747-0285.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- 3.Rabenstein DL. Heparin and heparan sulfate: structure and function. Natural Product Reports. 2002;19:312–331. doi: 10.1039/b100916h. [DOI] [PubMed] [Google Scholar]
- 4.Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 5.Zhao YJ, Singh A, Li LY, Linhardt RJ, Xu YM, Liu J, Woods RJ, Amster IJ. Investigating changes in the gas-phase conformation of Antithrombin III upon binding of Arixtra using traveling wave ion mobility spectrometry (TWIMS) Analyst. 2015;14:6980–6989. doi: 10.1039/c5an00908a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao YJ, Singh A, Xu YM, Zong CL, Zhang FM, Boons GJ, Liu J, Linhardt RJ, Woods RJ, Amster IJ. Gas-phase analysis of the complex of fibroblast growthfactor 1 with heparan sulfate: a traveling wave ion mobility spectrometry (TWIMS) and molecular modeling study. Journal of the American Society for Mass Spectrometry. 2017;28:96–109. doi: 10.1007/s13361-016-1496-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thanawiroon C, Rice KG, Toida T, Linhardt RJ. Liquid chromatography/mass spectrometry sequencing approach for highly sulfated heparin-derived oligosaccharides. Journal of Biological Chemistry. 2004;279:2608–2615. doi: 10.1074/jbc.M304772200. [DOI] [PubMed] [Google Scholar]
- 8.Jones CJ, Beni S, Limtiaco JFK, Langeslay DJ, Larive CK. Heparin characterization: challenges and solutions. Annual Review of Analytical Chemistry. 2011;4:439–465. doi: 10.1146/annurev-anchem-061010-113911. [DOI] [PubMed] [Google Scholar]
- 9.Elias JE, Haas W, Faherty BK, Gygi SP. Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nat Methods. 2005;2:667–675. doi: 10.1038/nmeth785. [DOI] [PubMed] [Google Scholar]
- 10.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 11.Chi LL, Amster J, Linhardt RJ. Mass spectrometry for the analysis of highly charged sulfated carbohydrates. Current Analytical Chemistry. 2005;1:223–240. doi: 10.2174/157341105774573929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooper CA, Gasteiger E, Packer NH. GlycoMod - A software tool for determining glycosylation compositions from mass spectrometric data. Proteomics. 2001;1:340–349. doi: 10.1002/1615-9861(200102)1:2<340::AID-PROT340>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 13.Kailemia MJ, Patel AB, Johnson DT, Li LY, Linhardt RJ, Amster IJ. Differentiating chondroitin sulfate glycosaminoglycans using collision-induced dissociation; uronic acid cross-ring diagnostic fragments in a single stage of tandem mass spectrometry. European Journal of Mass Spectrometry. 2015;21:275–285. doi: 10.1255/ejms.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flangea C, Serb AF, Schiopu C, Tudor S, Sisu E, Seidler DG, Zamfir AD. Discrimination of GalNAc (4S/6S) sulfation sites in chondroitin sulfate disaccharides by chip-based nanoelectrospray multistage mass spectrometry. Central European Journal of Chemistry. 2009;7:752–759. [Google Scholar]
- 15.Huang RR, Pomin VH, Sharp JS. LC-MS (n) analysis of isomeric chondroitin sulfate oligosaccharides using a chemical derivatization strategy. Journal of the American Society for Mass Spectrometry. 2011;22:1577–1587. doi: 10.1007/s13361-011-0174-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leach FE, Xiao ZP, Laremore TN, Linhardt RJ, Amster IJ. Electron detachment dissociation and infrared multiphoton dissociation of heparin tetrasaccharides. Int J Mass Spectrom. 2011;308:253–259. doi: 10.1016/j.ijms.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bin Oh H, Leach FE, Arungundram S, Al-Mafraji K, Venot A, Boons GJ, Amster IJ. Multivariate analysis of electron detachment dissociation and infrared multiphoton dissociation mass spectra of heparan sulfate tetrasaccharides differing only in hexuronic acid stereochemistry. Journal of the American Society for Mass Spectrometry. 2011;22:582–590. doi: 10.1007/s13361-010-0047-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolff JJ, Laremore TN, Leach FE, Linhardt RJ, Amster IJ. Electron capture dissociation, electron detachment dissociation and infrared multiphoton dissociation of sucrose octasulfate. European Journal of Mass Spectrometry. 2009;15:275–281. doi: 10.1255/ejms.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolff JJ, Laremore TN, Busch AM, Linhardt RJ, Amster IJ. Influence of charge state and sodium cationization on the electron detachment dissociation and infrared multiphoton dissociation of glycosaminoglycan oligosaccharides. Journal of the American Society for Mass Spectrometry. 2008;19:790–798. doi: 10.1016/j.jasms.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leach FE, Ly M, Laremore TN, Wolff JJ, Perlow J, Linhardt RJ, Amster IJ. Hexuronic acid stereochemistry determination in chondroitin sulfate glycosaminoglycan oligosaccharides by electron detachment dissociation. Journal of the American Society for Mass Spectrometry. 2012;23:1488–1497. doi: 10.1007/s13361-012-0428-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leach FE, Wolff JJ, Laremore TN, Linhardt RJ, Amster IJ. Evaluation of the experimental parameters which control electron detachment dissociation, and their effect on the fragmentation efficiency of glycosaminoglycan carbohydrates. Int J Mass Spectrom. 2008;276:110–115. doi: 10.1016/j.ijms.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolff JJ, Chi LL, Linhardt RJ, Amster IJ. Distinguishing glucuronic from iduronic acid in glycosaminoglycan tetrasaccharides by using electron detachment dissociation. Analytical Chemistry. 2007;79:2015–2022. doi: 10.1021/ac061636x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolff JJ, Laremore TN, Aslam H, Linhardt RJ, Amster IJ. Electron-Induced Dissociation of Glycosaminoglycan Tetrasaccharides. Journal of the American Society for Mass Spectrometry. 2008;19:1449–1458. doi: 10.1016/j.jasms.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolff JJ, Laremore TN, Busch AM, Linhardt RJ, Amster IJ. Electron detachment dissociation of dermatan sulfate oligosaccharides. Journal of the American Society for Mass Spectrometry. 2008;19:294–304. doi: 10.1016/j.jasms.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Y, Yu X, Mao Y, Costello CE, Zaia J, Lin C. De Novo Sequencing of Heparan Sulfate Oligosaccharides by Electron-Activated Dissociation. Analytical Chemistry. 2013;85:11979–11986. doi: 10.1021/ac402931j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leach FE, Riley NM, Westphall MS, Coon JJ, Amster IJ. Negative electron transfer dissociation sequencing of increasingly sulfated glycosaminoglycan oligosaccharides on an orbitrap mass spectrometer. Journal of the American Society for Mass Spectrometry. 2017;28:1844–1854. doi: 10.1007/s13361-017-1709-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolff JJ, Leach FE, Laremore TN, Kaplan DA, Easterling ML, Linhardt RJ, Amster IJ. Negative Electron Transfer Dissociation of Glycosaminoglycans. Analytical Chemistry. 2010;82:3460–3466. doi: 10.1021/ac100554a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolff JJ, Amster IJ, Chi L, Linhardt RJ. Electron detachment dissociation of glycosaminoglycan tetrasaccharides. Journal of the American Society for Mass Spectrometry. 2007;18:234–244. doi: 10.1016/j.jasms.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Domon B, Costello CE. a systematic nomenclature for carbohydrate fragmentations in fab-ms ms spectra of glycoconjugates. Glycoconjugate J. 1988;5:397–409. [Google Scholar]
- 30.Kailemia MJ, Li LY, Ly M, Linhardt RJ, Amster IJ. Complete Mass Spectral Characterization of a synthetic ultralow-molecular-weight heparin using collision-induced dissociation. Analytical Chemistry. 2012;84:5475–5478. doi: 10.1021/ac3015824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kailemia MJ, Ruhaak LR, Lebrilla CB, Amster IJ. Oligosaccharide Analysis by Mass Spectrometry: A Review of Recent Developments. Analytical Chemistry. 2014;86:196–212. doi: 10.1021/ac403969n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zaia J, Costello CE. Tandem mass Spectrometry of sulfated heparin-like glycosaminoglycan oligosaccharides. Analytical Chemistry. 2003;75:2445–2455. doi: 10.1021/ac0263418. [DOI] [PubMed] [Google Scholar]
- 33.Dancik V, Addona TA, Clauser KR, Vath JE, Pevzner PA. De novo peptide sequencing via tandem mass spectrometry. Journal of Computational Biology. 1999;6:327–342. doi: 10.1089/106652799318300. [DOI] [PubMed] [Google Scholar]
- 34.Ma B, Zhang KZ, Hendrie C, Liang CZ, Li M, Doherty-Kirby A, Lajoie G. PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Communications in Mass Spectrometry. 2003;17:2337–2342. doi: 10.1002/rcm.1196. [DOI] [PubMed] [Google Scholar]
- 35.Taylor JA, Johnson RS. Implementation and uses of automated de novo peptide sequencing by tandem mass spectrometry. Analytical Chemistry. 2001;73:2594–2604. doi: 10.1021/ac001196o. [DOI] [PubMed] [Google Scholar]
- 36.Campbell MP, Hayes CA, Struwe WB, Wilkins MR, Aoki-Kinoshita KF, Harvey DJ, Rudd PM, Kolarich D, Lisacek F, Karlsson NG, Packer NH. UniCarbKB: Putting the pieces together for glycomics research. Proteomics. 2011;11:4117–4121. doi: 10.1002/pmic.201100302. [DOI] [PubMed] [Google Scholar]
- 37.Maxwell E, Tan Y, Tan Y, Hu H, Benson G, Aizikov K, Conley S, Staples GO, Slysz GW, Smith RD, Zaia J. GlycReSoft: A software package for automated recognition of glycans from LC/MS data. Plos One. 2012;7 doi: 10.1371/journal.pone.0045474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saad OM, Leary JA. Heparin sequencing using enzymatic digestion and ESI-MSn with HOST: A heparin/HS oligosaccharide sequencing tool. Analytical Chemistry. 2005;77:5902–5911. doi: 10.1021/ac050793d. [DOI] [PubMed] [Google Scholar]
- 39.Chiu YL, Huang RR, Orlando R, Sharp JS. GAG-ID: Heparan Sulfate (HS) and Heparin Glycosaminoglycan High-Throughput Identification Software. Mol Cell Proteomics. 2015;14:1720–1730. doi: 10.1074/mcp.M114.045856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu H, Huang Y, Mao Y, Yu X, Xu YM, Liu J, Zong CL, Boons GJ, Lin C, Xia Y, Zaia J. A Computational Framework for Heparan Sulfate Sequencing Using High-resolution Tandem Mass Spectra. Mol Cell Proteomics. 2014;13:2490–2502. doi: 10.1074/mcp.M114.039560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ly M, Leach FE, III, Laremore TN, Toida T, Amster IJ, Linhardt RJ. The proteoglycan bikunin has a defined sequence. Nature Chemical Biology. 2011;7:827–833. doi: 10.1038/nchembio.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baeck T, Schwefel HP. An Overview of Evolutionary Algorithms for Parameter Optimization. Evolutionary Computation. 1993;1:1–23. doi: 10.1162/evco_a_00325. [DOI] [PubMed] [Google Scholar]
- 43.Fogel LJ, Owens AJ, Walsh MJ. Artificial intelligence through a simulation of evolution. Proceedings of the Second Cybernetic Sciences Symposium: Biophysics and cybernetic systems. 1965:131–155. [Google Scholar]
- 44.Forrest S. Genetic algorithms - principles of natural-selection applied to computation. Science. 1993;261:872–878. doi: 10.1126/science.8346439. [DOI] [PubMed] [Google Scholar]
- 45.Han L, Costello CE. Mass spectrometry of glycans. Biochemistry-Moscow. 2013;78:710–720. doi: 10.1134/S0006297913070031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kilgour DPA, Neal MJ, Soulby AJ, O’Connor PB. Improved optimization of the Fourier transform ion cyclotron resonance mass spectrometry phase correction function using a genetic algorithm. Rapid Communications in Mass Spectrometry. 2013;27:1977–1982. doi: 10.1002/rcm.6658. [DOI] [PubMed] [Google Scholar]
- 47.Das S, Suganthan PN. Differential evolution: a survey of the state-of-the-art. IEEE Trans Evol Comput. 2011;15:4–31. [Google Scholar]
- 48.Knowles JD, Corne DW. Approximating the nondominated front using the Pareto archived evolution strategy. Evolutionary Computation. 2000;8:149–172. doi: 10.1162/106365600568167. [DOI] [PubMed] [Google Scholar]
- 49.Phillips SJ, Anderson RP, Schapire RE. Maximum entropy modeling of species geographic distributions. Ecological Modelling. 2006;190:231–259. [Google Scholar]
- 50.Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM. Systematic determination of genetic network architecture. Nature Genetics. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- 51.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins-Structure Function and Genetics. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 52.Yu YL, Duan JN, Leach FE, Toida T, Higashi K, Zhang H, Zhang FM, Amster IJ, Linhardt RJ. Sequencing the dermatan sulfate chain of decorin. J Am Chem Soc. 2017;139:16986–16995. doi: 10.1021/jacs.7b10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singh A, Kett WC, Severin IC, Agyekum I, Duan JN, Amster IJ, Proudfoot AEI, Coombe DR, Woods RJ. The interaction of heparin tetrasaccharides with chemokine CCL5 is modulated by sulfation pattern and pH. Journal of Biological Chemistry. 2015;290:15421–15436. doi: 10.1074/jbc.M115.655845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Agyekum I, Patel AB, Zong CL, Boons GJ, Amster IJ. Assignment of hexuronic acid stereochemistry in synthetic heparan sulfate tetrasaccharides with 2-O-sulfo uronic acids using electron detachment dissociation. Int J Mass Spectrom. 2015;390:163–169. doi: 10.1016/j.ijms.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]