

Abstract
The aim of this study was to develop a new drug substance with low toxicity and effective inhibitory activity against cisplatin-resistant oral cancer. The naturally produced pterostilbene was selected as the lead compound for design and synthesis of a series of bis(hydroxymethyl)propionate-based prodrugs. All derivatives were screened for antiproliferative effects against the cisplatin-resistant oral squamous (CAR) cell line and the results indicated that several compounds demonstrated superior inhibitory activity compared with pterostilbene and resveratrol. Among them, the most promising compound, 12, was evaluated for in vivo antitumor activity in a CAR xenograft nude mouse model. Obvious antitumor activity was observed at the lowest oral dose (25 mg/kg/day). Increasing the dose of 12 to 100 mg/kg/day reduced the tumor size to 22% of the control group. Based on these findings as well as the extremely low toxicity seen in the in vivo studies, we believe that compound 12 could serve as a new lead for further development.
Keywords: pterostilbene, resveratrol, cisplatin, oral cancer, prodrug, drug resistant
Graphical Abstract

1. Introduction
Although cancers of the head and neck comprise only approximately 6% of all cancer cases, they remain widespread and fatal.1 Oral cancers are classified as head and neck cancers and account for about 85% of these cancers. Based on the histological tumor type, most oral cancers (about 90%) are squamous cell carcinomas (SCCs); other types are salivary gland tumors, lymphomas and sarcomas.2 The identified risk factors related to the onset of oral cancer involve heavy tobacco and alcohol consumption, industrial inhalant exposure, and human papilloma virus (HPV) and Epstein-Barr virus (EBV) infections.3 To date, the drugs most commonly used to treat oral cancer, often in combination with radiation therapy, are cisplatin (Platinol, Platinol-AQ),4 5-fluorouracil (Adrucil, Efudex, Fluoroplex)5 and cetuximab (Erbitux).6 Other chemotherapeutic drugs include docetaxel (Taxotere), paclitaxel (Taxol), gemcitabine, methotrexate (Abitrexate, Folex, Folex PFS, Mexate, Mexate-AQ) and bleomycin (Blenoxane).7,8 Treatment with a combination of cetuximab and platinum-based chemotherapy involving cisplatin or carboplatin exhibits superior survival benefits and is the most powerful and commonly used first-line treatment for recurrent or metastatic oral cancer.9 However, severe adverse effects and drug resistance often retard the use of platinum-based drugs.
Based on clinical observations, cisplatin is connected with renal failure, gastrointestinal adverse effects, hematological toxicity, neurotoxicity, nephrotoxicity and ototoxicity.10 Compared with cisplatin, carboplatin causes less toxicity, but more myelosuppression.11 Despite continuous improvement in the treatment of oral cancer, a low survival rate is found among oral cancer patients after receiving standard therapeutic regimens due to fast disease progression, drug resistance and a high rate of recurrence/metastasis. Therefore, efficacious drugs with fewer side effects and better safety profiles are still needed for the treatment of oral cancer.
Pterostilbene (4-[(E)-2-(3,5-dimethoxyphenyl)ethenyl]phenol) is a naturally occurring phytoalexin predominantly found in blueberries, grapes and wood of various trees.12 Pterostilbene exhibits anticancer effects via various molecular mechanisms including modulation of signal transduction pathways,13 cell cycle regulatory genes,14 cell differentiation genes,15 oncogenes and tumor suppressor genes.16 Pterostilbene is a low- or non-toxic substance to humans. Experimental evidence showed that pterostilbene has potential for the prevention and treatment of various cancers, such as colon,17 liver,18 pancreatic,19 lung20 and breast.21 However, the antitumor activity of pterostilbene could be improved. The current study is aimed at the development of new pterostilbene derivatives for the treatment of cisplatin-resistant oral squamous (CAR) cell carcinoma. Thirteen new bis(hydroxymethyl)propionate analogs of pterostilbene (12–18, 20, 22, 23, 24, 25 and 27) were designed and synthesized as target compounds.
2. Results and discussion
2.1 Design and synthesis
The rational design of these analogs is described below using compound 12 as a model substrate. The phenolic group (pKa 9.6)22 on the 4′-position of pterostilbene is easily metabolized via transferase-catalyzed conjugation to yield water soluble glucuronide and sulfate metabolites, which are rapidly excreted.23 Therefore, the half-life of pterostilbene is relatively short (ca. 105 mins).24 We have demonstrated previously that, compared with phenolic OH groups, aliphatic OH groups have higher pKa values25 and therefore are more stable toward Phase II glucuronidation and sulfonation. Therefore, our target compound 12 was designed by incorporating a bulky bis(hydroxymethyl) alkanoate group on the 4′-position of pterostilbene. The pKa of 12 (13.6) is higher than the pKa of its parent compound pterostilbene, most likely making 12 less vulnerable than pterostilbene to metabolic conjugation, although the aliphatic OH group on 12 could still be capable of glucuronidation or sulfonation. In addition, compound 12 was designed to be a prodrug. The parent pterostilbene would be generated after in vivo hydrolysis of the 4′-position ester functionality of 12 by esterases. The pharmaceutical efficacy of target compound 12 is expected to be better than that of pterostilbene. To validate our postulates, various target compounds were synthesized and evaluated initially for antiproliferative activity against CAR cells.
As shown in Scheme 1, the commercially available starting materials, stilbenes 1–5, variously substituted with OMe and OH groups at the 3,5,4′-position (5 = resveratrol), were reacted with 6 in the presence of Et3N to produce the corresponding esters 7–11. HCl-promoted hydrolysis of these esters gave the corresponding targets 12–16. Next, as shown in Scheme 2, when treated with BCl3 ·SMe in refluxing 1,2-dichloroethane for 5 h, compound 7 underwent partial demethylation to produce the desired monomethoxy 17 in 40% yield. The diol moiety of 17 was protected as a 1,3-dioxane under acidic conditions and subsequently the phenolic OH group was acetylated to produce intermediate 19. Hydrolysis of 19 with HCl gave 20 in 63% yield. When compound 7 was treated with BCl3 ·SMe2 for an extended time (16 h), compound 18 with two phenolic OH groups was obtained as the major product in 35% yield. Compound 18 was readily converted to 22 via a three-step sequence of acetal formation, acetylation and acid-promoted hydrolysis. Treatment of compound 8 with excess BCl3 ·SMe2 gave 23 in 62% yield. A similar process was used for the synthesis of 24 from 10. Finally, treatment of 14 with BCl3 ·SMe2 produced 25, which underwent acetal formation and acetylation to give 26. Acid-promoted hydrolysis of 26 produced 27 in 85% yield.
Scheme 1.

Scheme 2.

2.2 Biological evaluation
The synthesized target compounds and positive controls (pterostilbene, resveratrol and cisplatin) were screened against CAR cell lines. The results are summarized in Table 1. CAR cells were proven to be resistant to cisplatin (IC50 > 100 μM). When the phenolic OH groups on 1–4 were esterified with bis(hydroxymethyl)propanoic (BHMP) acid, the resulting derivatives 12–15 were all more potent than pterostilbene. Among them, compound 12 (IC50 = 32.58 μM) with a 4′-BHMP ester and 3,5-OCH3 was superior to 13–15 (IC50 = 59.81~70.75 μM) and about three times more potent than pterostilbene (IC50 = 98.29 μM). However, compound 16 (IC50 = 124.1 μM) with BHMP esters at all three positions (4′,3,5) was less potent than the corresponding parent triol (resveratrol, IC50 = 88.26 μM).
Table 1.
Antiproliferative effects of target compounds against CAR cell line

|
|||||
|---|---|---|---|---|---|
|
| |||||
| Compound | R3 | R4′ | R5 | IC50 (μM)a,b
|
|
| 48 h | 72 h | ||||
| 12 | OCH3 |

|
OCH3 | 73.25±4.20 | 32.58±2.39 |
| 13 |
|
OCH3 | OCH3 | 64.17±2.43 | 67.32±2.55 |
| 14 |
|
|
OCH3 | 56.45±1.98 | 59.81±1.99 |
| 15 |
|
OCH3 |
|
73.93±3.25 | 70.75±3.96 |
| 16 |
|
|
|
149.0±4.45 | 124.1±3.45 |
| 17 | OH |
|
OCH3 | 67.31±3.26 | 63.96±2.44 |
| 18 | OH |
|
OH | 168.6±3.19 | 132.7±2.45 |
| 20 | OAc |
|
OCH3 | 51.99±2.60 | 50.30±1.98 |
| 22 | OAc |
|
OAc | 103.2±4.58 | 115.8±3.96 |
| 23 |
|
OH | OH | 54.90±2.59 | 113.5±3.25 |
| 24 |
|
OH |
|
90.44±3.65 | 76.92±3.22 |
| 25 |
|
|
OH | 105.2±1.98 | 113.3±2.77 |
| 27 |
|
|
OAc | 122.1±2.69 | 119.3±2.19 |
| Pterostilbene | OCH3 | OH | OCH3 | 100.3±4.78 | 98.29±5.21 |
| Resveratrol | OH | OH | OH | 152.7±6.30 | 88.26±4.20 |
| Cisplatin | >100 | >100 | |||
Data are presented as IC50 (μM, concentration for 50% antiproliferative effect).
Most potent IC50 values are highlighted.
Furthermore, the replacement of one OCH3 of 12 with a hydrophilic OH led to reduced antiproliferative activity [17 (3-OH, 5-OCH3), IC50 = 63.96 μM] and replacement of both groups significantly reduced the potency [18 (3,5-OH), IC50 = 132.7 μM]. Mono-acetylated 20 (IC50 = 50.30 μM) and di-acetylated 22 (IC50 = 115.8 μM) were more potent than their hydroxylated parent compounds 17 and 18, respectively.
When the phenolic 3-OH of resveratrol was converted to a BHMP ester, the resulting compound 23 was less potent (IC50 = 113.5 μM) than resveratrol. Di-esterified compound 24 with BHMP at both position-3 and -5 exhibited greater potency (IC50 = 76.99 μM) than non-esterified resveratrol and mono-esterified 23 (based on data at 72 h). However, di-esterified compound 25 with BHMP at position-3 and −4′ was less potent (IC50 = 113.3 μM) than 24 and equipotent to 23. Acetylation of the 5-OH of 25 did not have a significant effect on potency (27, IC50 = 119.3 μM). As mentioned above, alteration of the OCH3 groups of active compounds 12–15 to OH groups resulted in diminished antiproliferative activity (compare 12 with 18, 13 with 23, 14 with 25, 15 with 24).
We expected that the target compounds would act as prodrugs and generally not exhibit better activity than their parent compounds. However, unexpectedly, seven target compounds (12–15, 17, 20 and 24) demonstrated better inhibitory activity than pterostilbene and resveratrol. The most potent compound, 12, was further evaluated for its antiproliferative effect against two normal human cell lines, gingival fibroblast (HGF) and oral keratinocyte (OK). No significant effects on cell viability were observed in these studies, even at a concentration of 12 higher than 100 μM. Among all tested compounds, compound 12 was produced easily in a high synthetic yield, exhibited the best inhibitory activity against oral cancer cells, showed low toxicity toward normal human cells and, thus, was selected for further evaluation in vivo.
The antitumor activity of compound 12 was evaluated in a CAR xenograft nude mouse model. Propane-1,2-diol was used as vehicle. Doses of 25, 50 and 100 mg/kg of body weight were orally administrated to all experimental groups once daily for 30 consecutive days, whereas control mice were fed orally with vehicle. As shown in Fig. 1–3, compound 12 exhibited dose- and time-dependent inhibitory effects on CAR tumor size and weight.
Fig. 1.

Effect of compound 12 in CAR xenograft nude mouse model
Fig. 3.
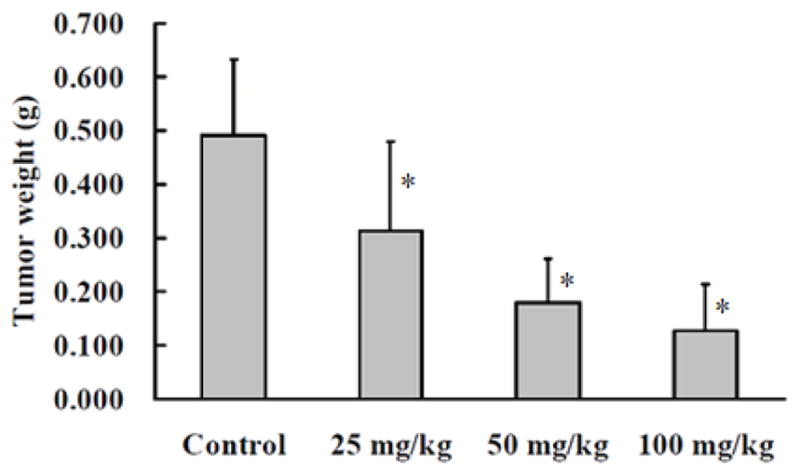
In vivo antitumor activity of compound 12. Mean tumor weight-dose profiles in CAR xenograft nude mice (n = 6) following PO dosing of 12 at 25, 50 and 100 mg/kg/day. *P<0.05 compared with untreated control.
Tumor weight in mice treated with 12 at the lowest dose (25 mg/kg) was 60% of that in control mice. At 50 and 100 mg/kg doses, the weight of CAR tumor was reduced to 47% and 22% of the control, respectively (Fig. 3). During the animal study, no obvious changes in body weight (Fig. 4), food consumption and behavior of tested mice were observed between the 12-treated and untreated control groups.
Fig. 4.

Mean body weight-time profile of 12 in CAR xenograft nude mice model
At the study’s end point, the mice were sacrificed and blood samples were collected to quantify total protein, albumin, creatinine, blood urea nitrogen, uric acid and glucose. According to the blood analysis listed in Table 2, no significant difference was observed between 12-fed groups and the control group. The above results indicated that 12 is an effective and low-toxicity drug substance for treating CAR.
Table 2.
Blood biochemical analyses of BALB/c nude mice with subcutaneously implanted CAR cell xenografts administered 12 once per day for 30 days (QD×30, oral)
| Group | TP (g/dL) | Alb (g/dL) | BUN (mg/dL) | Creatinine (mg/dL) | UA (mg/dL) | Glucose (mg/dL) |
|---|---|---|---|---|---|---|
| Control | 5.600±0.283 | 3.450±0.138 | 27.700±4.116 | 1.000±0.180 | 4.650±0.864 | 180.000±10.392 |
| 25 mg/kg | 5.383±0.117 | 3.666±0.052 | 28.200±5.784 | 1.133±0.109 | 4.567±0.784 | 181.333±32.067 |
| 50 mg/kg | 5.583±0.376 | 3.417±0.075 | 30.950±1.265 | 0.910±0.077 | 5.450±0.878 | 172.500± 6.504 |
| 100 mg/kg | 5.283±0.194 | 3.400±0.089 | 29.250±3.855 | 0.830±0.128 | 5.050±1.882 | 196.500± 8.643 |
TP: Total protein; Alb: Albumin; BUN: blood urea nitrogen; UA: Uric acid
Results were performed as mean ± S.E.M. at least five samples from each group.
To validate the assumption that compound 12 is a prodrug of pterostilbene, a preliminary pharmacokinetic study was performed with 12 in male SD rats. Compound 12 was administered in a single oral (100 mg/kg) dose using two dosage forms, PO-1 formulation (HPβCD : PEG300 : H2O = 20 : 10 : 70; 10 mg/mL) and PO-2 formulation (TPGS : PEG400 : H2O = 15 : 35 : 50; 10 mg/mL). Blood samples (about 250 μL) were then collected at 0.5, 1.0, 1.5, 2.0, 3.0, 4.0, 5.0, 6.0, 8.0, and 24.0 h after oral administration. The mean plasma concentration–time profiles of 12 and one major metabolite, pterostilbene-4′-O-sulfate, are shown in Fig. 5. Compound 12 was not detected in all blood samples. However, pterostilbene-4′-O-sulfate was detected by LC-MS and reached the maximum concentration at 2.0 h after oral administration (for PO-1 formulation). The results suggest that compound 12 was quickly hydrolyzed into the parent compound pterostilbene and then metabolized by phase II enzymes into pterostilbene-4′-O-sulfate in vivo. A detailed pharmacokinetic study of 12 is underway and the results will be published in due course.
Fig. 5.

Whole blood concentrations of pterostilbene-4′-O-sulfate in male SD rats (N = 3) after a single oral administration of compound 12 (100 mg/kg). PO-1: 10 mg compound 12 was dissolved in 1 mL solution (HPβCD : PEG300 : H2O = 20: 10 : 70). PO-2: 10 mg compound 12 was dissolved in 1 mL solution (TPGS : PEG400 : H2O = 15 : 35 : 50). Compound 12 was not detected at all time intervals.
3. Conclusion
In conclusion, in the search for new chemical entities with low toxicity and effective inhibitory activity against CAR, novel bis(hydroxymethyl)propionate-based prodrugs were synthesized. Unexpectedly, several target compounds demonstrated inhibitory effects superior to those of pterostilbene and resveratrol. Further evaluation showed that 12 is a low toxic substance with significant antitumor activity against CAR. Our previous data indicated that compound 12 induced autophagic cell death by regulating PI3 kinase class III/beclin 1/Atg-related proteins in CAR cells;26 however, the molecular target of 12 must still be identified. Based on these facts, we conclude that 12 could serve as a new lead compound for further development as a safe drug for CAR treatment. Currently, we are investigating the combination of 12 with chemotherapeutic agents for the treatment of drug resistant oral cancer.
4. Materials and Methods
4.1. General Synthetic Methods
The reactions were performed under an air atmosphere unless otherwise stated. All solvents and reagents were employed as received. Analytical thin layer chromatography was performed on SiO2 60 F-254 plates and flash column chromatography was carried out using SiO2 60 (particle size 0.040–0.055 mm, 230–400 mesh), both of which are available from E. Merck. Visualization was performed under UV irradiation at 254 nm followed by staining with aqueous potassium permanganate [KMnO4 (3 g) and K2CO3 (20 g) in 300 mL of H2O containing 5 mL of an aqueous solution of NaOH (5%, w/v)] and charring by heat gun. 1H and 13C NMR spectra were recorded on Bruker 500 FT NMR. Chloroform-d, DMSO-d and methanol-d were used as solvents and TMS (δ = 0.00 ppm) as an internal standard. Chemical shifts are reported as δ values in ppm as referenced to TMS. Multiplicities are recorded as s (singlet), d (doublet), t (triplet), q (quartet), quint (quintet), sext (sextet), sept (septet), dd (doublet of doublets), dt (doublet of triplets), br (broad), m (multiplet). Coupling constants (J) are expressed in Hz.
4.2. General procedure for the synthesis of compounds 12–16
The general procedure is illustrated immediately below with compound 12 as a specific example. To a solution of compound 7 (1.100 g, 4.30 mmol) in CH2Cl2 (10 mL) were slowly added 2,2,5-trimethyl-1,3-dioxane-5-carbonyl chloride (913 mg, 4.70 mmol) and triethylamine (0.9 mL, 6.50 mmol). The resulting mixture was stirred at rt for 1.0 h. Then saturated NH4Cl solution (3 mL) and water (5 mL) were added to quench the reaction. The aqueous layer was separated and extracted with CH2Cl2 (2 × 10 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered and concentrated to give the crude residue, which was purified by flash chromatography on silical gel to provide compound 7 (1.503 g, 85% yield). To a stirred solution of compound 7 (0.928 g, 2.40 mmol) in THF (15 mL) was added 12 N HCl (0.2 mL) at rt. The reaction mixture was stirred at the same temperature for 1 h and then concentrated to give the crude product, which was then purified by flash chromatography on silical gel (EtOAc:n-hexane = 2:1) to give compound 12 (0.752 g, 90% yield) as a white solid.
4.2.1. 4-(3,5-Dimethoxystyryl)phenyl-3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate (12)
Mp = 108.0–109.5 °C; 1H NMR (CDCl3, 500 MHz) δ 7.50 (d, J = 8.6 Hz, 2H), 7.19-6.99 (m, 4H), 6.64–6.63 (m, 2H), 6.38 (s, 1H), 4.05 (d, J = 10.0 Hz, 2H), 3.85-3.80 (m, 8H), 2.89 (br s, 2H), 1.21 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 174.7, 160.9, 149.9, 139.1, 135.2, 129.0, 128.0, 127.5, 121.8, 104.5, 100.1, 68.7, 55.3, 49.5, 17.0; LRMS [EI]+ calculated for C21H25O6: 373.1 [M + H]+; found: 372.4.
4.2.2. (E)-3-Methoxy-5-(4-methoxystyryl)phenyl-3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate (13)
Yield: 86%; Mp = 101.0–102.5 °C; 1H NMR (DMSO-d6, 500 MHz) δ 7.54 (d, J = 8.6 Hz, 2H), 7.19–6.94 (m, 5H), 6.87 (s, 1H), 6.53 (s, 1H), 4.92–4.85 (m, 2H), 3.78 (s, 6H), 3.89–3.68 (m, 2H), 1.20 (s, 3H); 13C NMR (DMSO-d6, 125 MHz) δ 171.7, 160.6, 150.3, 140.0, 129.6, 129.3, 129.2, 127.9, 125.5, 114.1, 111.5, 110.0, 106.1, 68.2, 55.3, 49.5, 17.0; LRMS [EI]+ calculated for C21H25O6: 373.1 [M + H]+; found: 372.9.
4.2.3. (E)-4-(3-((3-Hydroxy-2-(hydroxymethyl)-2-methylpropanoyl)oxy)-5-methoxystyryl) phenyl 3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate (14)
Yield: 83%; Mp = 168.0–169.5 °C; 1H NMR (DMSO-d6, 500 MHz) δ 7.63 (d, J = 8.6 Hz, 2H), 7.29–7.18 (m, 2H), 7.06–7.09 (m, 3H ), 6.92 (s, 1H), 6.57 (s, 1H), 4.93 (d, J = 10.0 Hz, 4H), 3.79 (s, 3H), 3.70–3.65 (m, 4H), 1.20 (s, 6H); 13C NMR (DMSO-d6, 125 MHz) δ 174.1, 173.9, 160.6, 152.6, 150.9, 139.5, 134.7, 129.1, 128.1, 127.9, 122.6, 112.5, 109.8, 107.6, 64.4, 55.9, 51.3, 17.3; LRMS [EI]+ calculated for C25H31O9: 475.1 [M + H]+; found: 475.4.
4.2.4. (E)-5-(4-Methoxystyryl)-1,3-phenylene bis(3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate) (15)
Yield: 81%; Mp = 141.0–142.5 °C; 1H NMR (DMSO-d6, 500 MHz) δ 7.55 (d, J = 8.4 Hz, 2H), 7.17-6.95 (m, 6H), 6.73 (s, 1H), 4.93 (d, J = 10.0 Hz, 4H), 3.80-3.75 (m, 4H), 3.77 (s, 3H), 1.21 (s, 6H); 13C NMR (DMSO-d6, 125 MHz) δ 173.8, 159.7, 151.9, 140.0, 130.3, 129.6, 128.5, 124.9, 117.1, 115.0, 114.7, 64.4, 55.6, 51.4, 17.3; LRMS [EI]+ calculated for C25H31O9: 475.1 [M + H]+; found: 475.0.
4.2.5. (E)-5-(4-((3-Hydroxy-2-(hydroxymethyl)-2-methylpropanoyl)oxy)styryl)-1,3-phenylene bis(3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate) (16)
Yield: 65%; Mp = 228.0–229.5 °C; 1H NMR (DMSO-d6, 500 MHz) δ 7.66 (d, J = 8.6 Hz, 2H), 7.27-7.09 (m, 6H), 6.78 (s, 1H), 4.96-4.90 (m, 4H), 3.70-3.65 (m, 4H), 3.55-3.52 (m, 4H), 1.21 (s, 9H); 13C NMR (DMSO-d6, 125 MHz) δ 177.0, 174.1, 173.8, 151.9, 129.7, 128.2, 128.0, 122.6, 117.5, 109.9, 104.7, 64.4, 51.4, 49.9, 17.4, 17.3; LRMS [EI]+ calculated for C29H37O12: 577.2 [M + H]+; found: 577.0.
4.3. General procedure for the synthesis of compounds 17, 18, 23, 24 and 25
The general procedure is illustrated immediately below with compound 17 as a specific example. To a stirred solution of compound 7 (412 mg, 1.0 mmol) in 1,2-dichloroethane (10 mL) was added BCl3 ·SMe2 (2.0M in CH2Cl2, 1.5 mL, 3.0 mmol) The reaction mixture was stirred under reflux for 5 h. Then saturated NaHCO3 solution (2 mL) and water (5 mL) were added to quench the reaction. The aqueous layer was separated and extracted with CH2Cl2 (2 × 10 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered and concentrated to give the crude residue, which was purified by flash chromatography on silical gel to provide compound 17 (138 mg, 40% yield).
4.3.1. (E)-4-(3-Hydroxy-5-methoxystyryl)phenyl-3-hydroxy-2-(hydroxymethyl)-2-methyl propanoate (17)
Mp = 129.0–131.0 °C; 1H NMR (DMSO-d6, 500 MHz) δ 7.61 (d, J = 8.6 Hz, 2H), 7.17–7.06 (m, 4H), 6.63–6.57 (m, 2H), 6.26 (s, 1H), 4.92–4.85 (m, 2H), 3.73 (s, 3H), 3.67–3.60 (m, 2H), 1.20 (s, 3H); 13C NMR (DMSO-d6, 125 MHz) δ 174.1, 161.0, 159.0, 150.7, 139.3, 134.9, 129.1, 127.9, 127.8, 122.5, 106.7, 103.3, 101.4, 64.4, 55.4, 51.3, 17.3; LRMS [EI]+ calculated for C20H23O6: 359.1 [M + H]+; found: 359.2.
4.3.2. (E)-4-(3-Acetoxy-5-methoxystyryl)phenyl 3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate (18)
Mp = 109.0–111.0 °C; 1H NMR (DMSO-d6, 500 MHz) δ 7.63 (d, J = 8.5 Hz, 2H), 7.30–7.08 (m, 5H), 6.98 (s, 1H), 6.64 (s, 1H), 4.93–4.86 (m, 2H), 3.79 (s, 3H), 3.67–3.60 (m, 2H), 2.27 (s, 3H), 1.20 (s, 3H); 13C NMR (DMSO-d6, 125 MHz) δ 174.1, 169.5, 160.7, 152.2, 150.9, 139.6, 134.6, 129.3, 128.0, 127.9, 122.6, 112.3, 110.0, 107.6, 64.4, 55.9, 51.3, 21.3, 17.3; LRMS [EI]+ calculated for C22H25O7: 401.1 [M + H]+; found: 401.1.
4.3.3. (E)-5-(4-((3-Hydroxy-2-(hydroxymethyl)-2-methylpropanoyl)oxy)styryl)-1,3-phenylene diacetate (23)
Yield: 62%; Mp = 135.6–135.9 °C; 1H NMR (DMSO-d6, 500 MHz) δ 7.41 (d, J = 8.6 Hz, 2H), 7.02–6.78 (m, 5H), 6.70 (s, 1H), 6.35 (s, 1H), 4.90–4.86 (m, 2H), 3.65–3.60 (m, 2H), 1.19 (s, 3H); 13C NMR (DMSO-d6, 125 MHz) δ 174.0, 158.1, 157.1, 152.1, 140.1, 129.1, 128.7, 127.5, 124.5, 115.1, 110.2, 107.4, 64.5, 50.6, 15.9; LRMS [EI]+ calculated for C19H21O6: 344.1 [M + H]+; found: 345.0.
4.3.4. (E)-5-(4-Hydroxystyryl)-1,3-phenylene bis(3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate) (24)
Yield: 65%; Mp = 195.0–197.0 °C; 1H NMR (DMSO-d6, 500 MHz) δ 7.45 (d, J = 8.6 Hz, 2H), 7.15–7.03 (m, 4H), 6.77–6.75 (m, 2H), 6.71 (s, 1H), 4.95–4.89 (m, 4H), 3.68–3.60 (m, 4H), 1.20 (s, 6H); 13C NMR (DMSO-d6, 125 MHz) δ 173.8, 158.1, 151.9, 140.2, 130.6, 128.6, 128.0, 123.9, 117.0, 116.0, 114.8, 64.4, 51.4, 17.3; LRMS [EI]+ calculated for C24H29O9: 461.1 [M + H]+; found: 461.1.
4.3.5. (E)-4-(3-Hydroxy-5-((3-hydroxy-2-(hydroxymethyl)-2-methylpropanoyl)oxy) styryl)phenyl 3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate (25)
Yield: 52%; Mp = 146.0–148.0 °C; 1H NMR (MeOD, 500 MHz) δ 7.58 (d, J = 8.0 Hz, 2H), 7.20–7.05 (m, 4H), 6.87–6.84 (m, 2H), 6.49 (s, 1H), 3.87 (d, J = 11.0 Hz, 4H), 3.77 (d, J = 11.0 Hz, 4H), 1.32 (s, 6H); 13C NMR (MeOD, 125 MHz): δ172.5, 158.2, 152.2, 150.6, 139.4, 134.9, 128.2, 127.1, 121.7, 110.5, 108.0, 64.5, 50.8, 50.7, 50.6, 16.0, 15.9; LRMS [EI]+ calculated for C24H29O9: 461.1 [M + H]+; found: 461.2.
4.4. General procedure for the synthesis of compounds 20, 22, and 27
The general procedure is illustrated immediately below with compound 20 as a specific example. A solution of compound 17 (716 mg, 2 mmol) and catalytic amount of p-TSA in 2,2-dimethoxypropane (10 mL) was stirred at rt for 1.0 h. The reaction mixture was then added with NaHCO3 and further stirred for 15 min. The solution was concentrated under reduced pressure to remove 2,2-dimethoxypropane, and then quenched with water and extracted with CH2Cl2. The organic layer was washed with brine, dried over MgSO4 and concentrated under reduced pressure. The residue was dissolved in pyridine (4.0 mL), added with acetic anhydride (4.0 mL) and stirred at rt for 2.0 h. The reaction mixture was quenched with water and extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4 and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel to provide compound 19. To a solution of compound 19 in THF (10 mL), 2N HCl (aq) (10 mL) was slowly added and then stirred at rt for 1.0 h. The solution was diluted with water and extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4 and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel to provide compound 20 (504 mg, 1.4 mmol) in 63% yield.
4.4.1. (E)-4-(3,5-Dihydroxystyryl)phenyl 3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate (20)
Yield: 35%; Mp = 138.0–139.5 °C; 1H NMR (CDCl3, 500 MHz) δ 7.65 (d, J = 8.6 Hz, 2H), 7.23–7.07 (m, 4H), 6.64–6.63 (m, 2H), 6.35 (s, 1H), 3.98 (d, J = 10.0 Hz, 2H), 3.87 (d, J = 10.0 Hz, 2H), 1.43 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 172.5, 156.8, 148.8, 137.7, 133.6, 127.3, 125.5, 125.4, 120.2, 103.2, 100.3, 63.0, 49.2, 14.4; LRMS [EI]+ calculated for C19H20NaO6: 367.1 [M + Na]+; found: 367.1.
4.4.2. (E)-5-(4-((3-Hydroxy-2-(hydroxymethyl)-2-methylpropanoyl)oxy)styryl)-1,3-phenylene diacetate (22)
Mp = 121.0–122.5 °C; 1H NMR (DMSO-d6, 500 MHz) δ 7.63 (d, J = 8.5 Hz, 2H), 7.30–7.10 (m, 6H), 6.90 (s, 1H), 4.93–4.86 (m, 2H), 3.68–3.60 (m, 2H), 2.29 (s, 3H), 1.20 (s, 3H); 13C NMR (DMSO-d6, 125 MHz) δ 174.1, 169.4, 151.6, 151.1, 139.8, 134.4, 130.0, 128.0, 127.1, 122.6, 117.5, 115.3, 64.4, 51.3, 21.3, 17.3; LRMS [EI]+ calculated for C23H25O8: 401.1 [M + H]+; found: 429.1.
4.4.3. (E)-4-(3-Acetoxy-5-((3-hydroxy-2-(hydroxymethyl)-2-methylpropanoyl)oxy) styryl)phenyl 3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate (27)
Mp = 161.0–163.0 °C; 1H NMR (DMSO-d6, 500 MHz) δ 7.63 (d, J = 8.4 Hz, 2H), 7.32–7.07 (d, 6H), 6.82 (s, 1H), 4.93–4.86 (m, 4H), 3.66–3.60 (m, 4H), 2.27 (s, 3H), 1.21 (s, 3H), 1.19 (s, 3H); 13C NMR (DMSO-d6, 125 MHz) δ 174.1, 173.8, 169.4, 152.0, 151.6, 151.1, 139.7, 134.5, 129.9, 128.0, 127.2, 122.6, 117.6, 117.3, 115.4, 64.4, 51.4, 21.2, 17.3;; LRMS [EI]+ calculated for C26H31O10: 503.1 [M + H]+; found: 503.5.
4.5. Cell culture
The human head and neck carcinoma cell line CAL27 was obtained from American Tissue Culture Collection (ATCC). The cisplatin-resistant cell line CAR (CAL27-cisplatin resistance) was established by clonal selection of CAL27 using 10 cycles of 1 passage treatment with 10–80 μM of cisplatin (Sigma-Aldrich Corp. (St. Louis, MO, USA) followed by a recovery period of another passage. CAR cells were cultivated in DMEM supplemented with 10% FBS, 100 μg/mL streptomycin, 100 U/mL penicillin G, 2 mM L-glutamine (Gibco by Life Technologies (Carlsbad, CA, USA)) and 80 μM of cisplatin (Sigma-Aldrich Corp. (St. Louis, MO, USA).
4.6. MTT [3-(4,5-dimethylthiazaol-2-yl)-2,4-diphenyltetrazolium bromide] assay
Anti-proliferative activities of target compound were determined by an improved MTT assay. The CAR cells were individually plated at a density of 2×104 cells/well onto 96-well plates and treated with DMSO alone [0.5% (v/v) in media served as a vehicle control] and various concentrations (0, 25, 50, 75 and 100 μM) of target compounds for 48 and 72 h. Following treatments, the supernatant was discarded before a 100-μl solution of MTT (500 μg/ml) was added to each well for 4 h at 37 °C. After incubation, the medium was replaced by the addition of 200 μL DMSO to solubilize the violet formazan crystal produced from MTT. The absorbance of the dissolved formazan grained within the cells was measured at 570 nm by a microplate reader to calculate viability (data shown as % of control).27
4.7. In vivo antitumor activity assay
All animal experiments complied with institutional guidelines (Affidavit of Approval of Animal Use Protocol) approved by the Institutional Animal Care and Use Committee (IACUC) of China Medical University, Taiwan (permit number: 100–165-N). All pathogen-free four-week-old male BALB/c nude mice were purchased from the National Laboratory Animal Center (Taipei, Taiwan). These animals were housed at a constant rt with a regular 12-h light/12-h dark cycle and fed a standard rodent diet and water ad libitum. The CAR cells (1×107 cells/mouse) in 0.2 mL (1:1) culture medium and Matrigel (BD Biosciences) were subcutaneously injected into the flank of nude mice. When xenograft tumors reached approximately 50 mm3 (at day 22 after cell inoculation), 24 mice were randomly divided into four groups with eight mice in each group. Three groups of mice were given compound 12 orally at dosages of 25, 50 and 100 mg/kg body weight, respectively, with one dose each day for 30 days (dosing regimen: QD × 30, PO), whereas control mice were orally treated with 100 μL 1,2-propanediol throughout the experiment. The tumor size (mm3) from each mouse was determined by caliper measurement using the calculation 0.5 × length × width. At the end of treatment, all animals were anaesthetized with carbon dioxide (CO2) and sacrificed on day 31. The tumor tissues from each mouse were removed, measured and weighed individually as previously reported.28
4.8. Biochemical analysis: the levels of biochemical enzyme profiles and hematologic counts
All mice were monitored for the related toxicity of each group after the animals were sacrificed, and whole blood samples were drawn from the heart for biochemical measurements for evaluating the safety of 12. Briefly, blood was collected from each mouse and allowed to clot and centrifuged at 1000 × g for 10 min at rt to determine the further biochemical tests, including total protein, albumin, creatinine, blood urea nitrogen (BUN), uric acid and Glucose.
4.9. Preliminary pharmacokinetic study of compound 12 in rats
Male Sprague-Dawley rats (approximately 200–220 g) were obtained from the National Laboratory Animal Center (Taipei, Taiwan). All rats were housed in a regular 12-h light/12-h dark cycle, given clean water and fed commercial diet ad libitum in standard conditions of constant temperature and humidity. The experiment was started after the rats were kept under these conditions for at least one week. The rats were randomly divided into two groups with three rats in each group. All animal studies were conducted according to institutional guidelies (Affidavit of Approval of Animal Use Protocol, No. 100–165-N) which was approved by the Institutional Animal Care and Use Committee (IACUC) of China Medical University (Taichung, Taiwan). For each group, two dosage forms of compound 12, including PO-1 formulation (HPβCD : PEG300 : H2O = 20 : 10 : 70; 10 mg/mL) and PO-2 formulation (TPGS : PEG400 : H2O = 15 : 35 : 50; 10 mg/mL) were used for a single oral (100 mg/kg) administration. Disposable sterilized syringes were used for collecting blood samples from tail veins in the rats. Blood samples (about 250 μL) were immediately collected in heparinized 1.5 mL polythene tubes at 0.5, 1.0, 1.5, 2.0, 3.0, 4.0, 5.0, 6.0, 8.0, and 24.0 h after oral administration of compound 12. All blood samples were immediately centrifuged at 2000 rpm for 10 min for the separation of plasma. The plasma was immediately submitted for LC analysis. Analytical HPLC resolution was performed on a HPLC Shimadzu LC-30A system with a CBM-30A HPLC pump, FRC-20A autosampler, UV and PDA detectors, and LiChrospher-RP-18e (250 × 4.6 mm, 5 μm) column. Methanol and RO water = 65:35 as eluent. Flow rate: 1.0 mL/min.
Fig. 2.

Effect of compound 12 on tumor size in CAR xenograft nude mice model
Acknowledgments
We are grateful to China Medical University (CMU 102-N-03) and AnnCare Bio-Tech Center for financial support. Partial support was also provided by NIH grant CA177584 from the US National Cancer Institute awarded to K.H. L.
Footnotes
Conflicts of Interest: The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vigneswaran N, Williams MD. Oral Maxillofac Surg Clin North Am. 2014;56:123. doi: 10.1016/j.coms.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Backes C, Bier H, Knopf AT. Oncotarget. 2017;8:84320. doi: 10.18632/oncotarget.21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Argiris A, Karamouzis MV, Raben D, Ferris RL. Lancet. 2008;371:1695. doi: 10.1016/S0140-6736(08)60728-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strojan P, Vermorken JB, Beitler JJ, Saba NF, Haigentz M, Bossi P, Worden FP, Langendijk JA, Eisbruch A, Mendenhall WM, Lee AWM, Harrison LB, Bradford CR, Smee R, Silver CE, Rinaldo A, Ferlito A. Head Neck. 2016;38:E2151. doi: 10.1002/hed.24026. [DOI] [PubMed] [Google Scholar]
- 5.Chen CC, Lin JC, Chen KW. Oral Oncol. 2017;69:11. doi: 10.1016/j.oraloncology.2017.03.017. [DOI] [PubMed] [Google Scholar]
- 6.Reeves TD, Hill EG, Armeson KE, Gillespie MB. Otolaryngol Head Neck Surg. 2011;144:676. doi: 10.1177/0194599811399559. [DOI] [PubMed] [Google Scholar]
- 7.Argiris A, Li Y, Murphy BA, Langer CJ, Forastiere AA. J Clin Oncol. 2004;22:262. doi: 10.1200/JCO.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 8.Vermorken JB, Specenier P. Ann Oncol. 2010;21:vii252. doi: 10.1093/annonc/mdq453. [DOI] [PubMed] [Google Scholar]
- 9.Vermorken J, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol D, Peyrade F, Benasso M, Vynnychenko I, De Raucourt D, Bokemeyer C, Schueler A, Amellal N, Hitt R. N Engl J Med. 2008;359:1116. doi: 10.1056/NEJMoa0802656. [DOI] [PubMed] [Google Scholar]
- 10.Florea AM, Büsselberg D. Cancers. 2011;3:1351. doi: 10.3390/cancers3011351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hah SS, Stivers KM, White RWDV, Henderson PT. Chem Res Toxicol. 2006;5:622. doi: 10.1021/tx060058c. [DOI] [PubMed] [Google Scholar]
- 12.Baur JA, Sinclair DA. Nat Rev Drug Discov. 2006;6:493. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 13.Pan M-H, Chiou Y-S, Chen W-J, Wang J-M, Badmaev V, Ho C-T. Carcinogenesis. 2009;30:1234. doi: 10.1093/carcin/bgp121. [DOI] [PubMed] [Google Scholar]
- 14.Pan MH, Chang YH, Badmaev V, Nagabhushanam K, Ho CT. J Agric Food Chem. 2007;55:7777. doi: 10.1021/jf071520h. [DOI] [PubMed] [Google Scholar]
- 15.Chakraborty A, Bodipati N, Demonacos MK, Peddinti R, Ghosh K, Roy P. Mol Cell Endocrinol. 2012;355:25. doi: 10.1016/j.mce.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 16.Pan MH, Lai CS, Wu J-C, Ho CT. Mol Nutr Food Res. 2011;55:32. doi: 10.1002/mnfr.201000412. [DOI] [PubMed] [Google Scholar]
- 17.Nutakul W, Sobers HS, Qiu P, Dong P, Decker EA, McClements DJ, Xiao H. J Agric Food Chem. 2011;59:10964. doi: 10.1021/jf202846b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo L, Tan K, Wang H, Zhang X. Oncol Rep. 2016;36:3233. doi: 10.3892/or.2016.5151. [DOI] [PubMed] [Google Scholar]
- 19.Mannal PW, Alosi JA, Schneider JG, McDonald DE, McFadden DW. J Gastrointest Surg. 2010;14:873. doi: 10.1007/s11605-010-1164-4. [DOI] [PubMed] [Google Scholar]
- 20.Schneider JG, Alosi JA, McDonald DE, McFadden DW. J Surg Res. 2010;161:18. doi: 10.1016/j.jss.2009.06.027. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Ding L, Wang X, Zhang J, Han W, Feng L, Sun J, Jin H, Wang XJ. Am J Transl Res. 2012;4:44. [PMC free article] [PubMed] [Google Scholar]
- 22.PKa values of the tested compounds were calculated by ChemBioDraw Ultra 14.0.
- 23.Shao X, Chen X, Badmaev V, Ho CT, Sang S. Rapid Commun Mass Spectrom. 2010;24:1770. doi: 10.1002/rcm.4579. [DOI] [PubMed] [Google Scholar]
- 24.Yeo SC, Ho PC, Lin HS. Mol Nutr Food Res. 2013;57:1015. doi: 10.1002/mnfr.201200651. [DOI] [PubMed] [Google Scholar]
- 25.Hsieh MT, Chang LC, Hung HY, Lin HY, Shih MH, Tsai CH, Kuo SC, Lee KH. Eur J Med Chem. 2017;131:141. doi: 10.1016/j.ejmech.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 26.Hsieh MT, Chen HP, Lu CC, Chiang JH, Wu TS, Kuo DH, Huang LJ, Kuo SC, Yang JS. Int J Oncol. 2014;45:782. doi: 10.3892/ijo.2014.2478. [DOI] [PubMed] [Google Scholar]
- 27.Yang JS, Hour MJ, Huang WW, Lin KL, Kuo SC, Chung JG. J Pharmacol Exp Ther. 2010;334:477. doi: 10.1124/jpet.109.165415. [DOI] [PubMed] [Google Scholar]
- 28.Ho YT, Yang JS, Lu CC, Chiang JH, Li TC, Lin JJ, Lai K. Phytomedicine. 2009;16:887. doi: 10.1016/j.phymed.2009.02.015. [DOI] [PubMed] [Google Scholar]