

Abstract
Selective clearance of senescent cells (SCs) has emerged as a potential therapeutic approach for age-related diseases, as well as chemotherapy- and radiotherapy-induced adverse effects. Through a cell-based phenotypic screening approach, we recently identified piperlongumine (PL), a dietary natural product, as a novel senolytic agent, referring to small molecules that can selectively kill SCs over normal or non-senescent cells. In an effort to establish the structure-senolytic activity relationships of PL analogues, we performed a series of structural modifications on the trimethoxyphenyl and the α,β-unsaturated δ-valerolactam rings of PL. We show that modifications on the trimethoxyphenyl ring are well tolerated, while the Michael acceptor on the lactam ring is critical for the senolytic activity. Replacing the endocyclic C2-C3 olefin with an exocyclic methylene at C2 render PL analogues 47-49 with increased senolytic activity. These α-methylene containing analogues are also more potent than PL in inducing ROS production in WI-38 SCs. Similar to PL, 47-49 reduce the protein levels of oxidation resistance 1 (OXR1), an important oxidative stress response protein that regulates the expression of a variety of antioxidant enzymes, in cells. This study represents a useful starting point toward the discovery of senolytic agents for therapeutic uses.
Keywords: Piperlongumine, Senescent cell, Senolytic agent, ROS, OXR1
Graphical Abstract
To create your abstract, type over the instructions in the template box below.
Fonts or abstract dimensions should not be changed or altered.

1. Introduction
Cellular senescence was termed by Hayflick and Moorhead on the basis of their observation that normal cells have a limited proliferative capacity and eventually enter a state of permanent growth arrest following extended culture.1,2 This phenomenon, also known as replicative senescence, is the result of telomere shortening. Cells can also undergo senescence in response to cellular stresses that are independent of telomere length, such as exposure to genotoxic and oncogenic stress.3,4 In this context, the senescence response acts as an effective barrier to tumorigenesis by eliminating aged or damaged cells.3–9 However, senescent cells (SCs) are metabolically and transcriptionally active. They secrete various pro-inflammatory cytokines, chemokines, growth factors, and extracellular matrix proteases, a senescent phenotype termed the senescence-associated secretory phenotype (SASP).3 The SASP factors can recruit immune cells to clear damaged cells and SCs, as well as stimulate the proliferation and differentiation of stem and progenitor cells to repopulate the damaged tissue.3,10–12 Thus, transient induction of senescence is considered beneficial in tissue repair and remodeling. However, SCs can accumulate if their production exceeds their elimination by the immune system or if the immune system is unable to efficiently clear these cells due to immunosenescence.13 Accumulation of SCs and the associated SASP over time lead to deterioration in local tissue environment by inducing chronic oxidative stress and inflammation, which plays a significant role in aging and the development of age-related diseases, including cancer.3,10,14–16
Recent studies have shown that genetic clearance of p16Ink4a-positive SCs via INK-ATTAC, which functions as a SC suicide transgene, coupled with administration of a fusion protein dimerizer, AP20187, prolonged lifespan of mice and delayed the onset of a number of age-related diseases and disorders in both progeroid and naturally aged mice.17,18 These findings indicate that pharmacological clearance of SCs is a potential “anti-aging” approach to prolong healthspan, the average numbers of years of living without disability or other impairment. The great therapeutic potential of targeting SCs has encouraged us and others to search “senolytic agents”, refer to small molecules that can selectively kill SCs.19–26 We carried out a screening campaign against a library of structurally diverse, rationally selected small molecules that target pathways predicted to be important for SC survival by titrating their cytotoxicity against normal or nonsenescent human WI-38 fibroblasts (WI-38 NCs) and ionizing radiation (IR)-induced senescent WI-38 fibroblasts (WI-38 IRSCs).20 As a result, we identified two senolytic agents. The first one is ABT-263 (navitoclax), an inhibitor of the anti-apoptotic Bcl-2 family proteins currently in Phase II clinical trials for cancer treatment.27 We found that ABT-263 was highly specific in promoting apoptosis in SCs.20 Oral administration of ABT-263 effectively depletes IR-induced SCs in various tissues, including senescent epithelial cells in the lungs and senescent hematopoietic stem cells (HSCs) in bone marrow; and importantly, elimination of senescent HSCs in bone marrow by ABT-263 functionally rejuvenates HSCs in both sublethally irradiated (6 Gy) and naturally-aged mice.20 In addition, clearance of SCs in mice with ABT-263 reverses IR-induced pulmonary fibrosis,28 attenuates doxorubicin caused heart dysfunction and fatigue,29 and reduces atherogenesis onset.30 Thus, senolytic agents have the potential to be developed into drugs that can extend healthspan or treat age-related diseases and cancer treatment-induced adverse effects.
Through the targeted screen effort, we identified another senolytic agent piperlongumine (PL, Figure 1), a natural dietary product isolated from Piper longum L. with a variety of bioactivities including anticancer.31,32 PL has been proposed to exert its selective killing of cancer cells by inhibiting oxidative stress response proteins which are important for cancer cell survival under elevated reactive oxygen species (ROS) levels.33 The rationale of selecting PL for senolytic screening is that compared with NCs, SCs are also under higher oxidative stress,34 thus may share common antioxidant survival pathways with cancer cells. PL preferentially kill senescent WI-38 cells, induced by IR exposure, oncogene transfection, or extensive replication, over WI-38 NCs.23 PL appears to be very safe, and its maximum tolerated oral dose in mice is >1 g/kg, while the effective therapeutic doses for cancer can be as low as 2.4 mg/kg by oral dosing.33 Thus, PL was considered as a novel lead for the development of senolytic agents.23 Although a large variety of PL analogues have been reported as anticancer agents,35–50 it is not clear how senolytic activity of PL analogues correlate with their anticancer activity. Previously, we have synthesized several PL analogues, including PL-7 (1), PL-FPh (2), and PL-DI (3) (Figure 1), that have been reported as potent anticancer agents.23 A good correlation of their cytotoxicity against cancer cells and WI-38 IR-SCs was observed. However, the selectivity between NCs and SCs, the more important component of the senolytic activity, appeared to be unpredictable (Table 1). Hence, in this report, we carried out an exploration of the structure-senolytic activity relationships (SAR) of PL analogues. We sought to investigate the effects of modifying the trimethoxyphenyl ring and the α,βunsaturated δ-valerolactam ring of PL.
Fig. 1.
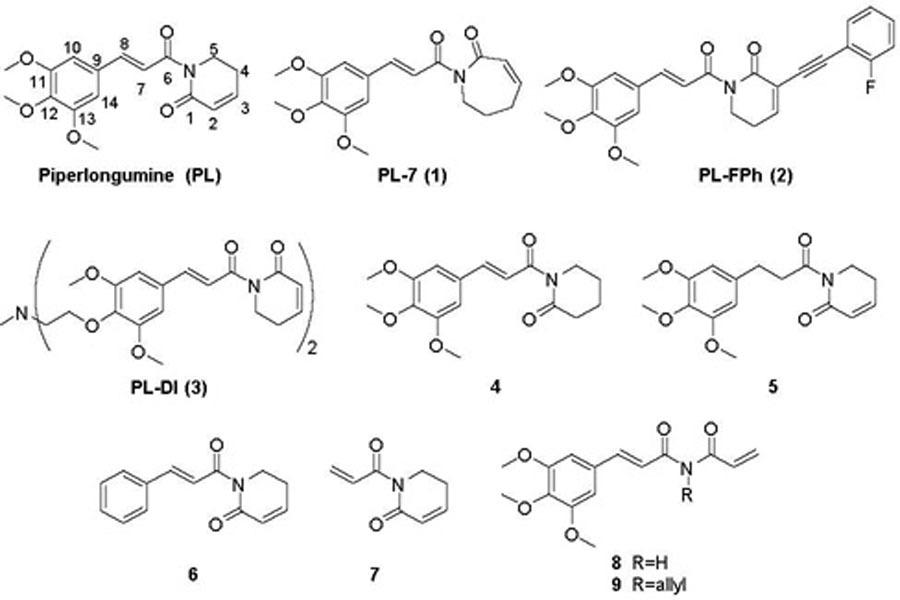
Structures of piperlongumine (PL) and analogues 1–9.
Table 1.
Senolytic Activity of PL and Analogues 1–9.
| compd | EC50 (µM)a NCsb |
IR-SCsc | EC50 ratio (NCs/SCs) |
|---|---|---|---|
| PLd | 20.3 | 8.0 | 2.5 |
| 1d | 13.0 | 8.9 | 1.5 |
| 2d | 5.9 | 1.1 | 5.4 |
| 3d | 1.5 | 0.7 | 2.1 |
| 4d | 170 | 208 | 0.8 |
| 5d | 126 | 46 | 2.7 |
| 6 | 10.2 | 2.7 | 3.8 |
| 7d | 35.7 | 9.7 | 3.7 |
| 8 | 4.1 | 4.9 | 0.8 |
| 9 | 15.3 | 10.5 | 1.5 |
a n ≥ 3.
b WI-38 non-senescent cells (NCs).
c ionizing radiation induced WI-38 senescent cells (IR-SCs).
d data from reference 23.
2. Results and discussion
2.1. Senolytic activities of PL analogues
All PL analogues were evaluated for their senolytic activity against WI-38 IR-SCs and NCs, by using a flow cytometry–based cell viability assay as previously described.23 The results of these studies are summarized in Tables 1–3. We first conducted an SAR study to understand the basic structure required to mediate the senolytic activity of PL. Previously, we found that compounds 4 and 5 (Figure 1), in which the C2–C3 or C7–C8 double bond of PL was replaced by a single bond, respectively, had a substantial reduction in senolytic activities, indicating that the integrity of the two-electrophile system is important for the senolytic activity of PL.23 In addition, both compound 6, in which the methoxy substituents of PL were removed, and 7,23 an abbreviated PL analogue with the trimethoxyphenyl ring removed, demonstrated retained or improved senolytic activity compared with PL, indicating that modification on the trimethoxyphenyl group can be tolerated. Further, retaining the lactam ring structure might be important for the selectivity of PL, as compounds 8 and 9 (Figure 1) showed diminished selectivity for WI-38 IR-SCs over NCs (Table 1).
Table 3.
Structures and Senolytic Activity of PL Analogues 46–57.
| compd | structure | EC50 (μM)a |
EC50 ratio (NCs/IR-SCs)d |
|
|---|---|---|---|---|
| NCsb | IR-SCsc | |||
| PL | 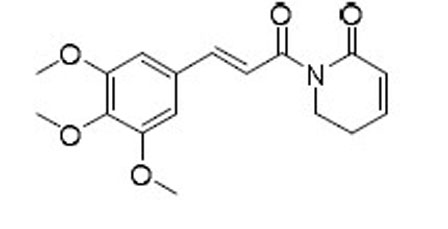 |
20.3 | 8.0 | 2.5 |
| 46 | 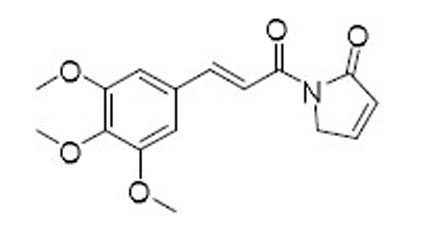 |
12.9 | 8.8 | 1.5 |
| 1 | 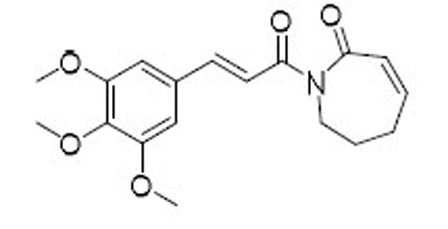 |
13.0 | 8.9 | 1.5 |
| 47 | 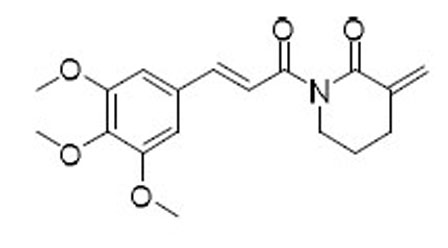 |
12.1 | 2.1 | 5.8 |
| 48 | 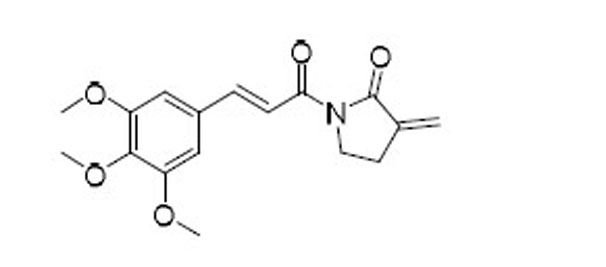 |
5.6 | 2.0 | 2.8 |
| 49 | 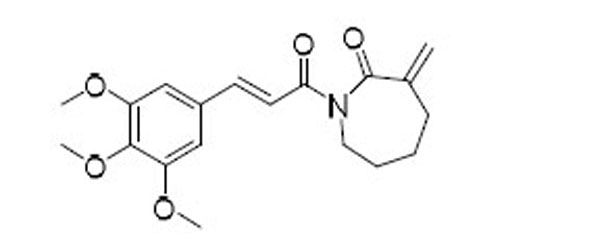 |
2.0 | 0.67 | 3.0 |
| 50 | 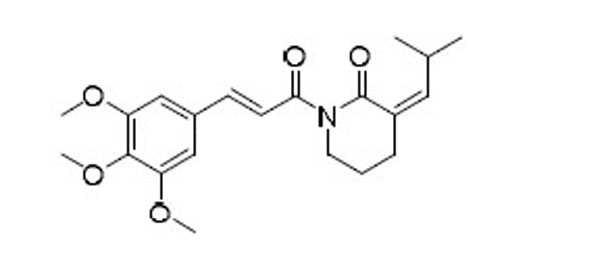 |
>40 | >40 | - |
| 51 | 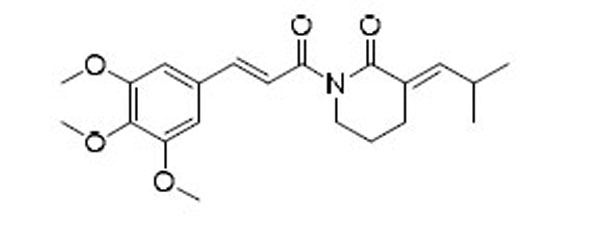 |
>40 | >40 | - |
| 52 | 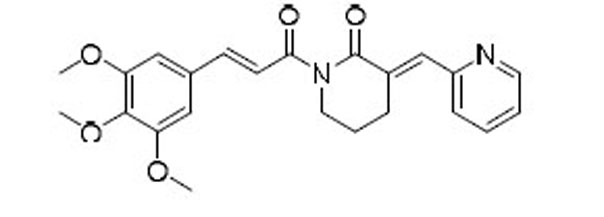 |
>40 | >40 | - |
| 53 | 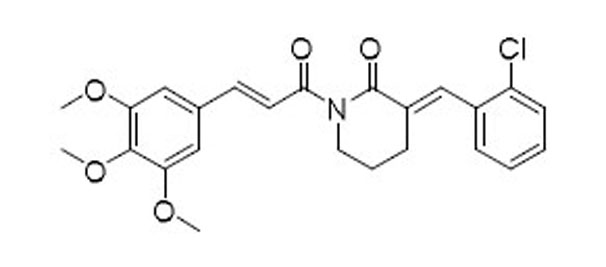 |
>40 | >40 | - |
| 54 | 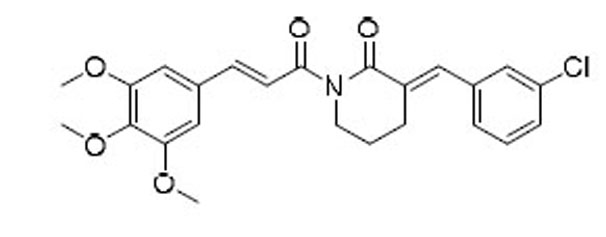 |
>40 | >40 | - |
| 55 | 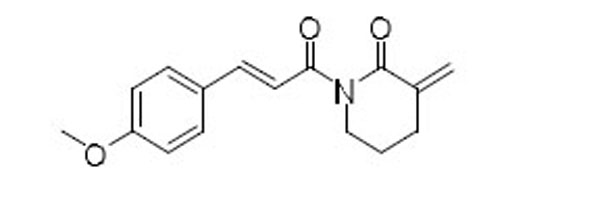 |
15.8 | 9.0 | 1.8 |
| 56 | 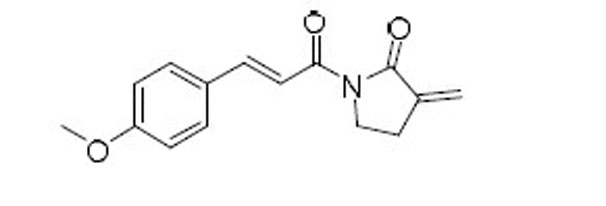 |
7.4 | 3.9 | 1.9 |
| 57 | 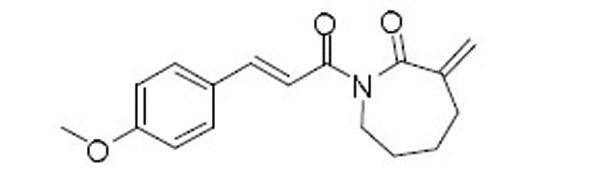 |
7.6 | 4.3 | 1.8 |
a n ≥ 3
b WI-38 non-senescent cells (NCs).
c ionizing radiation induced WI-38 senescent cells (IR-SCs).
Based on the initial SAR findings, a series of analogues were synthesized by varying the substituents on the phenyl ring of PL or replacing the trimethoxyphenyl ring with a heteroaromatic ring while holding the rest of the molecule constant (Table 2). Although well tolerated, the SAR obtained from this part of modifications was surprisingly shallow. Most of the analogues had comparable senolytic activity (i.e., potency and selectivity) to PL but no apparent SAR conclusions can be drawn. Compound 18, in which the two meta-methoxy groups on the phenyl ring of PL were removed, exhibited the best senolytic activity in this series with an EC50 of 1.3 µM in killing WI-38 IRSCs and 3.6-fold selectivity over NCs. Replacing the 4-methoxy in 18 with another para-substitution group, including hydroxyl group (compound 19), nitro group (compound 26), amino group (compounds 27–29), and halogens (compounds 30–32) resulted in reduced senolytic activity. One important finding was that the para-position of the phenyl ring is a suitable site to attach an affinity tag on PL. We synthesized probes (compounds 42-45, Table 2) with a “clickable” tag, a terminal alkyne or azide that can react with the corresponding immobilized azides or alkynes, respectively, through click chemistry.51 One probe, 43, which has similar senolytic activity to PL, was selected for our target-protein identification study.51
Table 2.
Structures and Senolytic Activity of PL Analogues 10–45.
| compd | R | EC50 (µM)a |
Ec50
ratiod |
compd | R | EC50 (µM)a |
Ec50
ratiod |
||
|---|---|---|---|---|---|---|---|---|---|
| NCsb | IR-SCsc | NCsb | IR-SCsc | ||||||
| 10 | 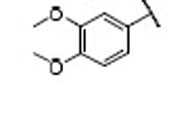 |
22.9 | 8.3 | 2.8 | 28 | 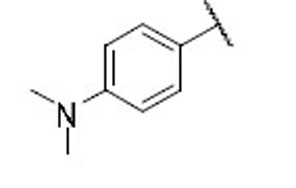 |
15.8 | 12.9 | 1.2 |
| 11 | 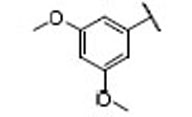 |
14.4 | 7.9 | 1.8 | 29 | 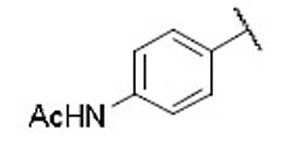 |
12.7 | 5.3 | 2.4 |
| 12 | 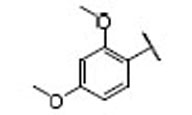 |
3.7 | 5.1 | 0.7 | 30 | 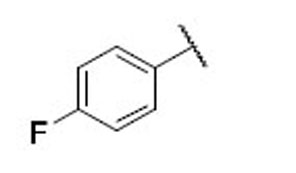 |
19.9 | 6.8 | 2.9 |
| 13 | 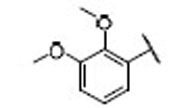 |
21.3 | 7.1 | 3.0 | 31 | 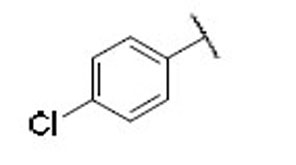 |
7.3 | 3.8 | 1.9 |
| 14 | 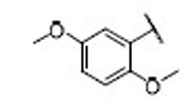 |
17.5 | 6.4 | 2.7 | 32 | 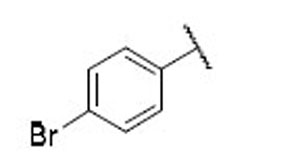 |
9.0 | 5.4 | 1.7 |
| 15 | 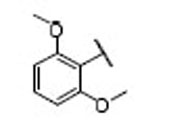 |
13.5 | 6.5 | 2.1 | 33 | 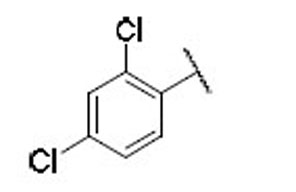 |
4.8 | 4.0 | 1.2 |
| 16 | 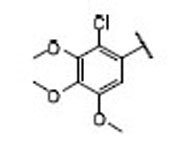 |
22.4 | 6.6 | 3.4 | 34 | 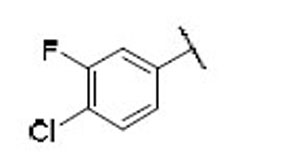 |
8.5 | 6.1 | 1.4 |
| 17 | 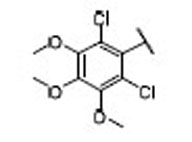 |
15.7 | 5.4 | 2.9 | 35 | 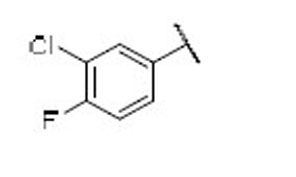 |
8.6 | 5.9 | 1.5 |
| 18 | 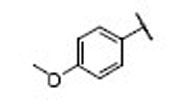 |
4.8 | 1.3 | 3.6 | 36 | 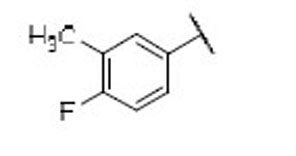 |
12.4 | 9.5 | 1.3 |
| 19 | 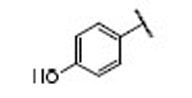 |
7.4 | 11.0 | 0.7 | 37 | 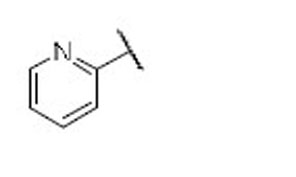 |
11.4 | 10.2 | 1.1 |
| 20 | 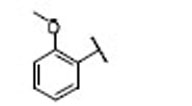 |
7.7 | 7.1 | 1.1 | 38 | 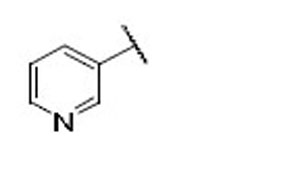 |
26.0 | 10.0 | 2.6 |
| 21 | 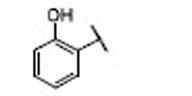 |
9.5 | 5.2 | 1.8 | 39 | 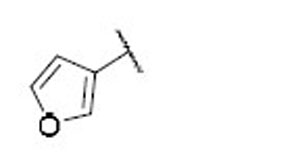 |
24.5 | 18.1 | 1.4 |
| 22 | 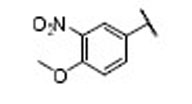 |
15.4 | 6.7 | 2.3 | 40 | 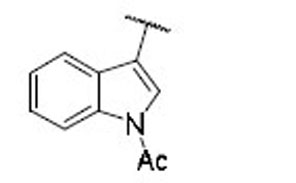 |
22.1 | 17.0 | 1.3 |
| 23 | 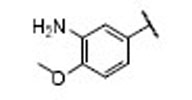 |
24.2 | 19.0 | 1.3 | 41 | 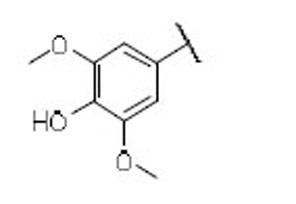 |
34.3 | 16.1 | 2.1 |
| 24 | 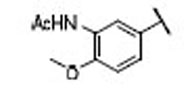 |
40.3 | 18.6 | 2.2 | 42 | 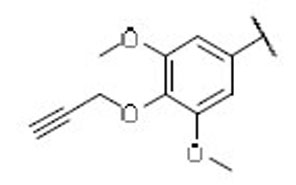 |
18.1 | 6.2 | 2.9 |
| 25 | 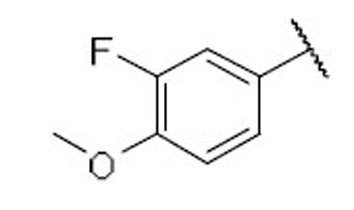 |
8.7 | 5.2 | 1.7 | 43 | 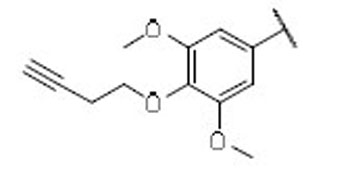 |
12.7 | 3.6 | 3.5 |
| 26 | 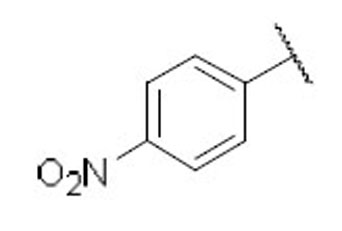 |
15.4 | 6.7 | 2.3 | 44 | 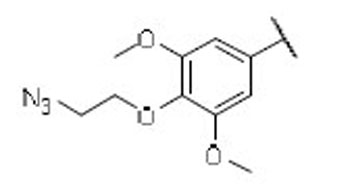 |
15.2 | 9.5 | 1.6 |
| 27 | 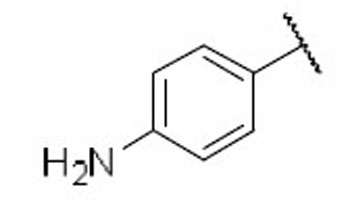 |
24.2 | 19.0 | 1.3 | 45 | 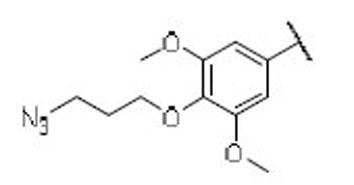 |
19.7 | 8.5 | 2.3 |
a n ≥ 3
b WI-38 non-senescent cells (NCs)
c ionizing radiation induced WI-38 senescent cells (IR-SCs).
d EC50 ratio: NCs/IR-SCs.
As indicated by comparing the senolytic activity of 4 and 5 (Table 1), the C2–C3 double bond is more critical than the C7–C8 double bond for the senolytic activity of PL. It was also reported that the lactam olefin was important for the cytotoxicity and ROS induction in cancer cells.35 We therefore speculated that tuning the reactivity of the Michael acceptor on the α,β-unsaturated δ-valerolactam ring of PL would produce analogues with improved senolytic activity. In addition to the ring-expanded ε-caprolactam analogue (1),23 we also synthesized the ring-contracted γ-butyrolactam analogue (46) (Table 3).38 However, no improvement in terms of senolytic potency and selectivity was observed with the change of the lactam ring size. Compounds 47, 48, and 49 (Table 3), which contain lactams bearing an exocyclic double bond at C2, were then synthesized. Compared to their endocyclic double bond–containing counterparts with the same lactam size (i.e., PL, 46, and 1, respectively), 47–49 had 4 to 13-fold increases in senolytic potency against WI-38 IR-SCs and had moderate increases in senolytic selectivity for SCs over NCs. Compound 49 had the most potent senolytic activity in this series with an EC50 of 0.67 µM in killing WI-38 IR-SCs. The increased potency could be attributed to improved accessibility of the exocyclic methylene unit by nucleophiles on the target protein(s) and/or increased reactivity of the Michael acceptor. As indirect evidence, increasing the steric hindrance of the Michael acceptor in compound 47 by introducing an isopropyl group (50 and 51, Table 3) on the exocyclic methylene abolished senolytic activity. In addition, introduction of a pyridinyl group (52) or a chlorophenyl group (53 and 54) on the exocyclic methylene also resulted in complete loss of senolytic activity, likely due to the steric hindrance or conjugation effect of these substitutions, which impair the electrophilicity of the Michael acceptor.
Finally, we incorporated the exocyclic methylene containing lactams into the best modification from Table 2, i.e., compound 18, with the intent of further improving senolytic activity. However, replacement of the trimethoxyphenyl in 47, 48, and 49 with a para-methoxyphenyl ring (compounds 55, 56, and 57, respectively) resulted in a decrease in both potency and selectivity in killing WI-38 IRSCs, demonstrating the unpredictable nature of these types of molecules where their bioactivities are likely derived from covalent interaction with biological targets.
2.2 Induction of OXR1 degradation in senescent cells
Consistent with our hypothesis that PL targets the oxidative stress response proteins in SCs, we recently identified oxidation resistance 1 (OXR1) as a senolytic target of PL.51 PL binds OXR1 directly and induces its degradation through the ubiquitin-proteasome system.51 Compounds 47, 48, and 49 were selected to test their effects on OXR1. WI-38 IR-SCs and NCs were treated with 5 µM of 47, 48, or 49 for 24 h and cell lysates were analyzed by Western blotting. As expected, all three analogues selectively reduced the levels of OXR1 in SCs, indicating that they share a similar mechanism of action with PL (Figure 2).
Fig. 2.
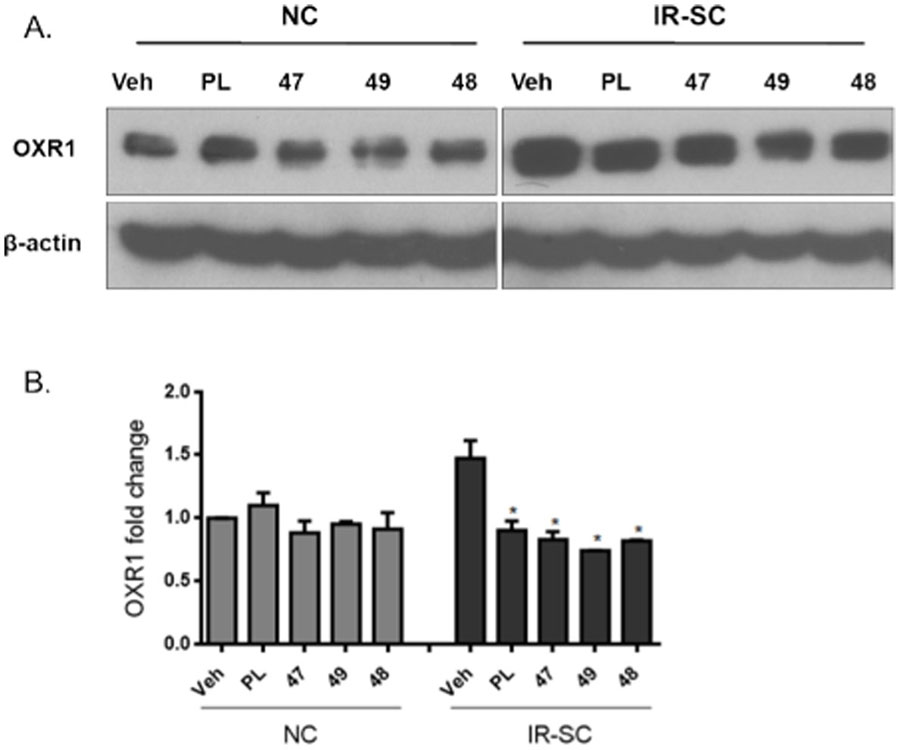
Compounds PL, 47, 49, and 48 abrogated the upregulation of OXR1 in WI-38 senescent cells induced by irradiation: (A) representative western blot analysis of OXR1 and β‐actin in WI‐38 normal or nonsenescent cells (NCs) and ionizing radiation-induced WI-38 senescent cells (IR‐SCs) 24 h after incubation with vehicle (Veh) or 5 µM of compound PL, 47, 49, or 48; (B) quantitative analysis of OXR1 levels in WI‐38 NCs and IR‐SCs 24 h after incubation with Veh or 5 µM of compound PL, 47, 49, or 48. Data in bar graphs are presented as mean ± SEM, n = 3, *p < 0.05 indicates significant difference comparing with IR-SCs with vehicle treatment.
2.3 ROS production in senescent cells
We evaluated compounds 47, 48, and 49 for their ability to induce ROS in SCs using dihydrorhodamine 123 (DHR 123) as a probe, which can passively diffuse across membranes where it is oxidized to green fluorescent rhodamine 123 in the presence of ROS. The fluorescence intensity was measured by flow cytometry and was proportional to the cytosolic ROS levels. The corresponding endocyclic double bond–containing analogues, PL, 46, and 1, respectively, were also evaluated in this assay for comparison. WI-38 IR-SCs were treated with various concentrations of analogues for 1.5 h and the changes in ROS levels were assessed by flow cytometry after treating with DHR 123 for 30 min. As shown in Figure 3, PL caused a moderate increase of the ROS levels in WI-38 IR-SCs. However, compound 46 did not affect ROS levels in SCs while compound 1 slightly inhibited ROS production at the same concentration range. Significant increases in ROS production were observed when IR-SCs were treated with compounds 47, 48, and 49. Although the ability of PL analogues to elevate ROS did not correlate with their cell killing effects in SCs and cancer cells, the largely elevated ROS levels caused by compounds 47-49 could be the basis of their increased toxicity to SCs. However, further studies are needed to determine whether induction of oxidative stress alone is sufficient to kill SCs.
Fig. 3.
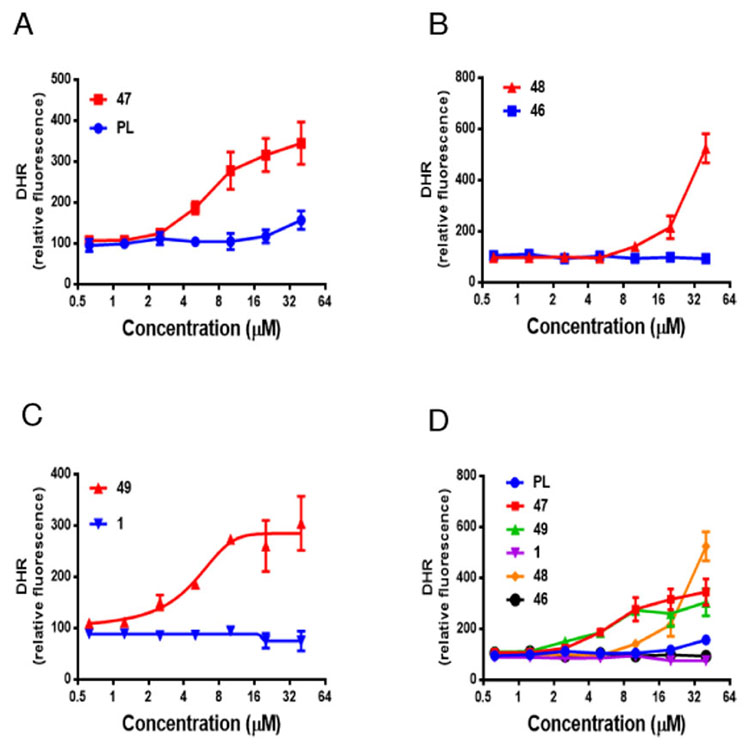
Comparison of ROS production in IR-induced WI-38 senescent cells 1.5 h after incubation with compounds 47, 48, and 49 and their corresponding endocyclic double bond–containing analogues PL, 46, and 1, respectively: (A) compound 47 vs. PL; (B) compound 48 vs 46; (C) compound 49 vs 1.; (D) Comparison of 47, PL, 49, 1, 48, and 46 in one graph. Data are presented as mean ± SEM, n = 3. Compounds concentration used corresponds to their EC50s against IR-induced WI-38 senescent cells.
2.4. Induction of apoptosis in senescent cells
PL induces apoptosis in cancer cells33 and SCs23. To determine if compound 49, the most potent PL analogue in this series, killed SCs by inducing apoptosis, we used FITC labeled Annexin V and propidium iodide, which stain phosphatidylserine (PS) residues and DNA, respectively, to detect apoptosis in WI-38 SCs by fluorescence-activated cell sorting analysis. As shown in Figure 4A, treatment with 1 µM of 49 increased the number of Annexin-V-positive cells in SCs by 3.6-fold when compared to the vehicle group. To further confirm that 49 killed SCs by apoptosis, we treated WI-38 IRSCs with the pan-caspase inhibitor Q-VD-OPh (QVD) to inhibit apoptosis. As shown in Figure 4B and 4C, QVD significantly reduced apoptosis and partially rescued SCs from 49-induced death (Fig. 4B, C), confirming the apoptotic cell-death mechanism.
Fig. 4.
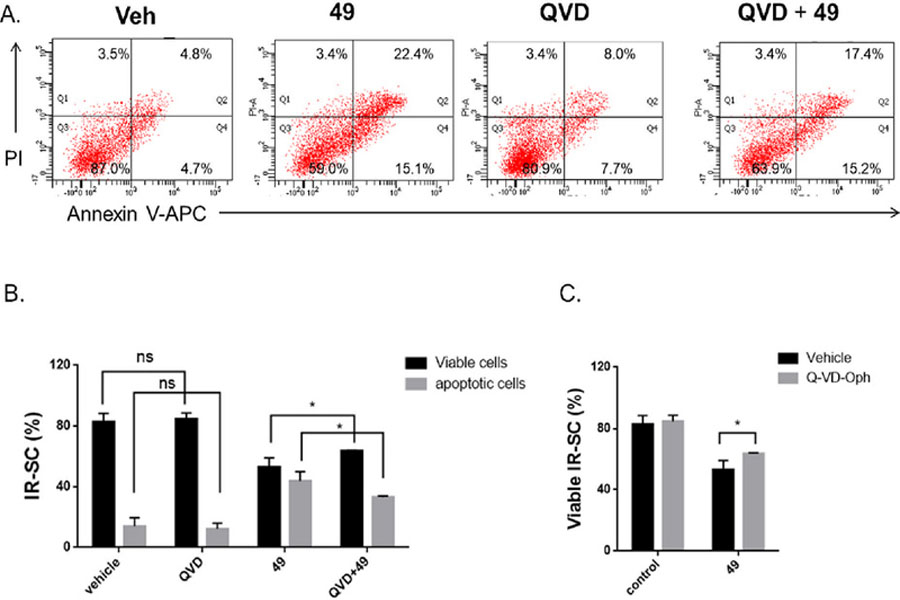
Compound 49 induced apoptosis in IR-induced WI-38 senescent cells (IR-SCs): (A) representative flow cytometric plots to measure apoptotic WI‐38 IR‐SCs at 48 h after treatment with vehicle (Veh), 49 (1 µM), Q-VD-Oph (QVD) (10 µM), or the combination of 49 (1 µM) and QVD (10 µM); (B) quantification of the percentage of viable and apoptotic WI-38 IR-SCs after 48 h treatment with vehicle, QVD (10 µM), 49 (1 µM), or the combination of 49 (1 µM) and QVD (10 µM); (C) quantification of the percentage of viable WI-38 IR-SCs 72 h after treatment with vehicle with or without QVD (10 µM) or with 49 (1 µM) with or without QVD (10 µM). Data in bar graphs are presented as mean ± SD, n = 3, *p < 0.05 indicates significant difference.
2.5. Chemistry
PL and its analogues 6, 10-18, 20, 22, 25, 26, 28, and 30-40 with modifications on the trimethoxyphenyl ring were synthesized via a one-pot reaction by coupling α,β-unsaturated δ-lactam 61 with appropriate cinnamic acid derivative 59, obtained commercially or prepared via Knoevenagel condensation of aldehyde 58 with malonic acid. Acids 59 were pre-activated by forming mixed anhydrides 60 with pivaloyl chloride (Scheme 1).36 Lactam 61 was prepared using a previously reported procedure.36 Compounds 19 and 21 were synthesized by initial treatment of 4- (62) and 2-hydroxycinnamic acid (63), respectively, with Boc2O in the presence of DMAP and triethylamine, followed by coupling with 61 and removal of the O-Boc group (Scheme 1). Reduction of the nitro group of 22 by treatment with Fe/NH4Cl in EtOH/H2O afforded amine 23, which was then N-acetylated to form compound 24 (Scheme 1). Similarly, compounds 27 and 29 were prepared from 26. Compound 41 was synthesized by de-methylation of PL.35 Compounds 42-45 were prepared from 41 as we reported recently.51
Scheme 1.
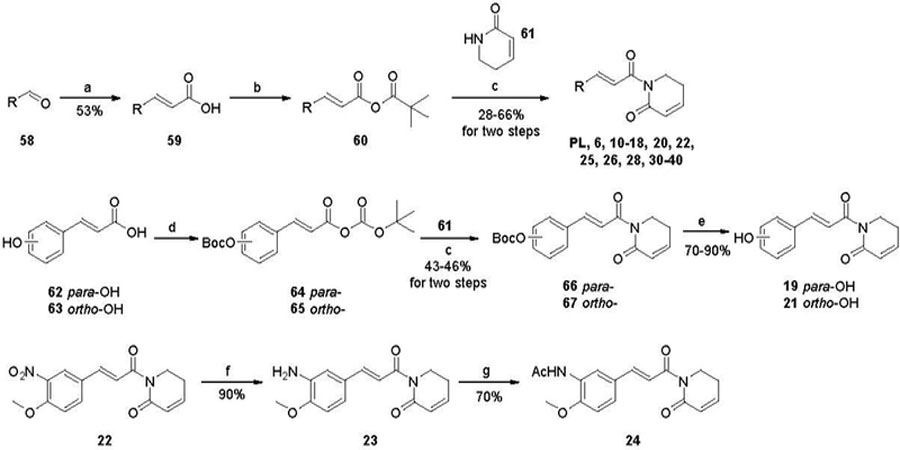
Reagents and conditions: (a) malonic acid, piperidine, pyridine, reflux; (b) trimethylacety 1 chloride, Et3N, THF, 0 ºC; (c) n-BuLi, THF, −78 ⁰C; (d) Boc2O, DMAP, Et3N, CH2Cl2, −15 ⁰C; (e) Trifluoroacetic acid, CH2Cl2, rt; (f) Fe, NH4Cl, EtOH/H2O, 40–50 ⁰C; (g) Ac2O, Et3N, CH2Cl2, rt.
PL analogues 1 and 46-51 (Table 2) with modifications on the lactam ring were synthesized in a similar manner to PL via coupling of 3,4,5-trimethoxycinnamic pivalic anhydride (68) with the appropriate lactam (70-76) (Scheme 2). Similarly, analogues 55-57 were synthesized by coupling 4-methoxycinnamic pivalic anhydride (69) with lactam 72, 73, and 74, respectively (Scheme 2). The coupling reaction between 68 and lactam 7138 resulted in a complex mixture, from which we failed to isolate compound 46 with sufficient purity. Alternatively, 46 was prepared by Grubbs ring closing metathesis reaction of compound 9. The synthesis of 9 was accomplished by a three-step process started with conversion of 3,4,5-trimethoxycinnamic acid (77) to the corresponding acid chloride, followed by coupling with allylamine and then acylation with acryloyl chloride (Scheme 3).
Scheme 2.
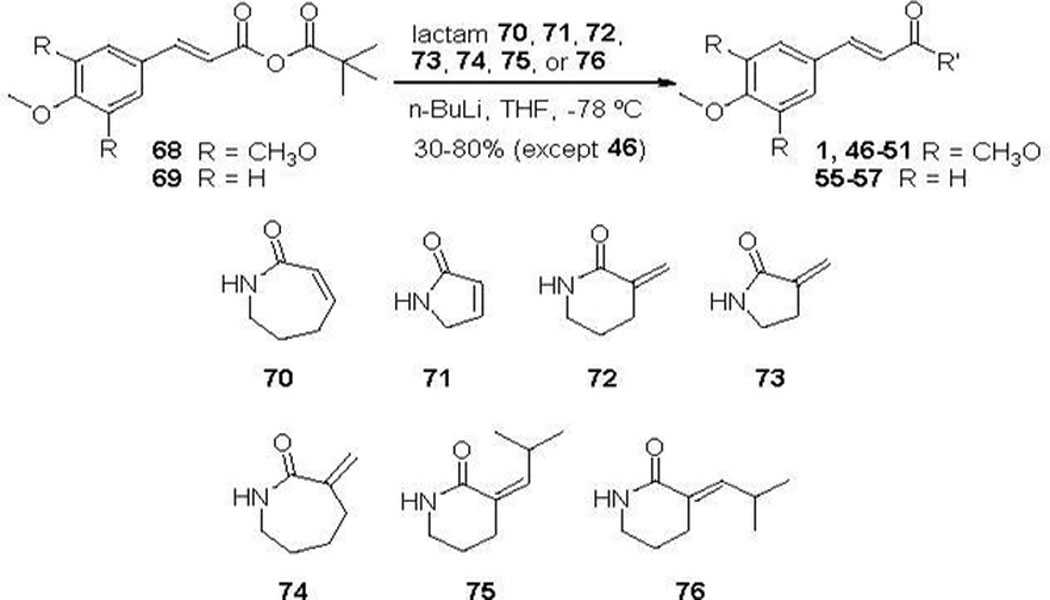
Synthesis of Compounds 1, 46–51, and 55–57.
Scheme 3.
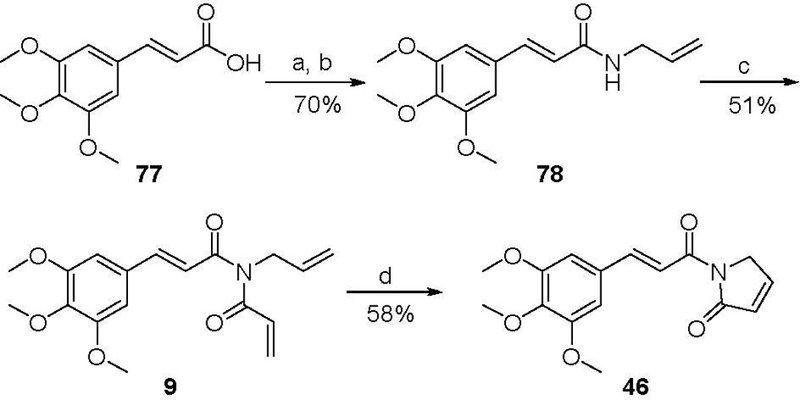
Reagents and conditions: (a) oxalyl chloride, DMF (cat.), CH2Cl2, rt; (b) allylamine, Et2O, rt; (c) acryloyl chloride, NaH, THF, 0 ºC; (d) Grubbs 2nd generation catalyst, CH2Cl2, reflux.
Lactam 70 was synthesized according to previously published method,52 with minor modifications (Scheme 4). Exocyclic double bond containing δ- and γ-lactams (72 and 73) were synthesized with ethyl 2-oxopiperidine-3-carboxylate (83) and ethyl 2-oxopyrrolidine-3-carboxylate (84), respectively, as starting materials. Reduction of the ester group of 83 and 84 with NaBH4/CaCl2 gave the corresponding alcohols 85 and 86, which underwent dehydration in the presence of N, N-dicyclohexylcarbodiimide (DCC) and catalytic amount of CuI to furnish the target lactams 72 and 73, respectively (Scheme 4B).53 For the preparation of α-methylene-ε-caprolactam (74), we initially tried the same method used for the synthesis of 72 and 73. To synthesize the requisite starting material methyl 2-oxoazepane-3-carboxylate 88, methyl carboxylate was first introduced to the α-position of N-Boc protected ε-caprolactam (80). However, the resulting N-Boc protected methyl 2-oxoazepane-3-carboxylate 87 was unstable under various de-Boc conditions; the carboxylate group was also removed to afford ε-caprolactam (79) (Scheme 4C, method 1). An alternative route, which started with reduction of 87 to alcohol 89, also failed at the de-Boc step (Scheme 4C, method 2). Forming the N-Boc protected αmethylene-ε-caprolactam 91 by dehydration of 89 with DCC/CuI or through forming mesylate was also unsuccessful (Scheme 4C, method 3). Finally, 91 was obtained by treating 87 with paraformaldehyde, 18-crown-6, and K2CO3 (Scheme 4C, method 4).54 Removal of the Boc protection of 91 under acidic condition resulted in the desired lactam 74.
Scheme 4.
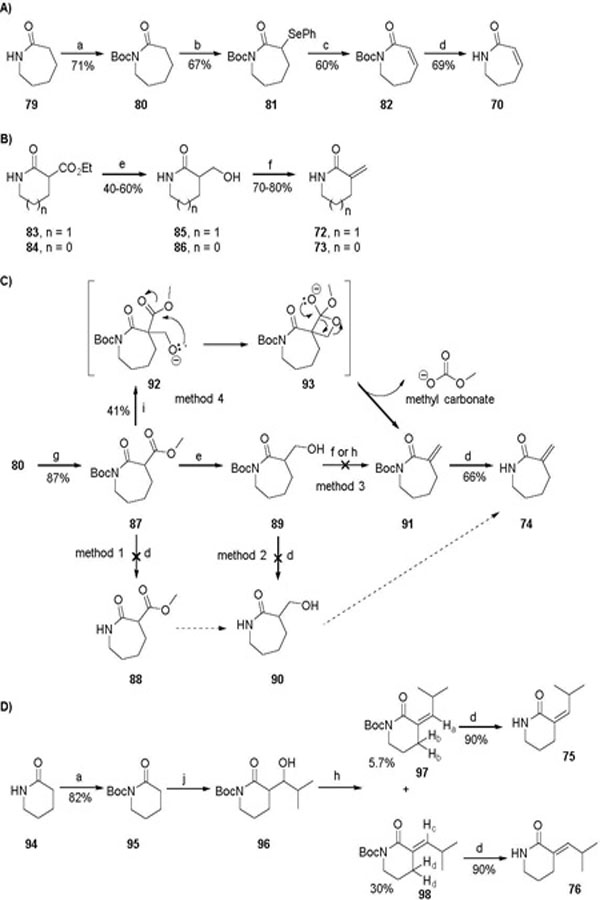
Reagents and conditions: (a) (Boc)2O, Et3N, DMAP, CH2Cl2; (b) LiHMDS, PhSeCl, THF, −78 ºC; (c) 30% H2O2, THF, 0 ºC to rt; (d) TFA, CH2Cl2, 0 ºC; (e) NaBH4, CaCl2, EtOH, 0 ºC to rt; (f) DCC, CuI, toluene, reflux; (g) 1.0 M LiHMDS, methyl chloroformate, THF, −78 ºC; (h) 1) CH3SO2Cl, Et3N, toluene, 0 ºC; 2) Et3N, toluene, reflux; (i) paraformaldehyde, 18-crown-6, K2CO3, toluene, reflux; (j) LiHMDS, isobutyraldehyde, THF, −78 ºC.
Lactams 75 and 76 that bear an isopropyl substituent at the olefin were prepared via a four-step transformation starting with N-Boc protection of δ-valerolactam (94) to form compound 95. Aldol condensation of 95 and isobutyraldehyde afforded 96, a mixture of four possible diastereomers. Using a standard dehydration method that involved the formation of mesylate intermediates, this isomeric mixture was converted into a mixture of Z- and E-isomers, 97 and 98 (1:5.2 ratio), which were separated in pure form by silica gel chromatography. The Z/E configurations were assigned based on NOE (NOE enhancement was observed for Hb when Ha of compound 97 was irradiated, while little NOE enhancement was observed for Hd when Hc of compound 98 was irradiated). Subsequently, removal of the N-Boc group of 97 and 98 with TFA/CH2Cl2 led to the corresponding lactam 75 and 76, respectively. Utilizing a similar procedure for the preparation of 97/98, analogues 52–54 were synthesized from compound 4. Under the same dehydration conditions, the aldol condensation product 99, formed via reacting 4 with an appropriate aromatic aldehyde, gave almost exclusively the E-isomer (Scheme 5).
Scheme 5.
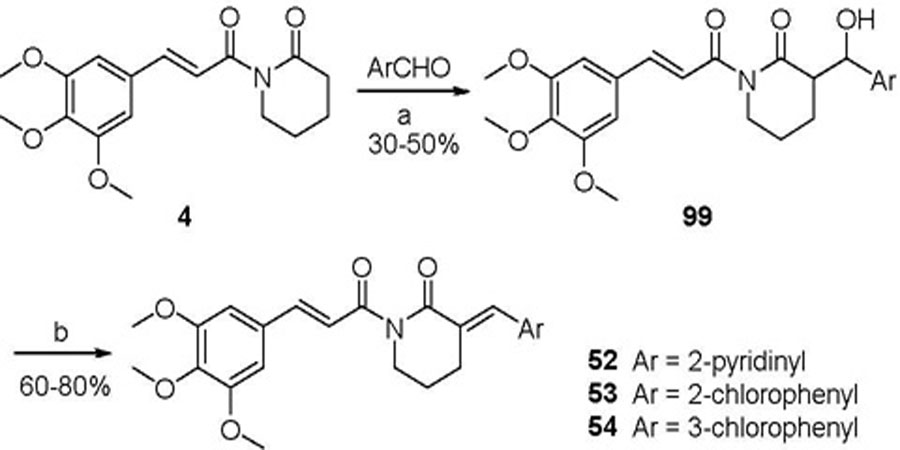
Reagents and conditions: (a) LiHMDS, THF, 78 ºC; (b) 1) CH3SO2Cl, Et3N, toluene, 0 ºC; 2) Et3N, toluene, reflux.
3. Conclusion
In this report, we explored a series of structural modifications on the trimethoxyphenyl and α,β-unsaturated δ-valerolactam moieties of PL. These PL analogues were evaluated for their senolytic activity using WI-38 IR-SCs and NCs. One important finding from our initial SAR studies is that structural modifications on the trimethoxyphenyl ring are well tolerated, which allows us to design probe molecules with a “clickable” tag on the phenyl ring to pull down target proteins in live cells, as well as to introduce structural moieties on the phenyl ring to potentially optimize the physiochemical properties of PL. Our SAR studies also reveal that the Michael acceptor on the lactam ring is critical for the senolytic activity. While changing the lactam ring size has minimal effect on the toxicity of PL analogues to SCs, introducing an exocyclic methylene at C2 of the lactam ring offers PL analogues, 47-49, with increased senolytic activity. These α-methylene containing PL analogues strongly induce ROS production in SCs, which could be partially attributed to their ability of lowering the protein levels of OXR1, a potential target of PL and an important oxidative stress response protein that regulates the expression of a variety of antioxidant enzymes. In addition, lead compound 49, which has a 12-fold increase in potency when compared with PL, selectively induces apoptosis in SCs. Further analogue development and in vitro and in vivo characterization of novel analogues are in progress.
4. Experimental
4.1. Chemistry
4.1.1. General methods
THF, CH2Cl2, toluene, and DMF were obtained via a solvent purification system by filtering through two columns packed with activated alumina and 4 Å molecular sieve, respectively. All other reagents and solvents obtained from commercial sources were used without further purification. If dry and air-free conditions were required, reactions were performed in oven-dried glassware (130 °C) under a positive pressure of argon. Flash chromatography was performed using silica gel (230–400 mesh) as the stationary phase. Reaction progress was monitored by thin layer chromatography (silica-coated glass plates) and visualized by UV light, and by GCMS or TLC-CMS. NMR spectra were recorded in CDCl3 at 400 MHz for 1H, and 100 MHz for 13C NMR. Chemical shifts δ are given in ppm using tetramethylsilane as an internal standard. Multiplicities of NMR signals are designated as singlet (s), broad singlet (br s), doublet (d), doublet of doublets (dd), triplet (t), quartet (q), and multiplet (m). All final compounds for biological testing were of ≥ 95% purity as analyzed by LC-MS, performed on a Waters 2965 system using a Thermo, betabasic-18 column (3 µm, 150 × 4.6 mm) at ambient temperature. Gradient elution was used for HPLC with a mobile phase of acetonitrile (ACN) and water containing 0.1% formic acid (0 min, ACN/0.1% formic acid in water = 40/60; 5 min, ACN/0.1% formic acid in water = 60/40; 10 min, ACN/0.1% formic acid in water = 70/30; 13 min, ACN/0.1% formic acid in water = 90/10; 15 min, ACN/0.1% formic acid in water = 40/60).
4.1.2. (E)-1-(3-(3,4,5-Trimethoxyphenyl)acryloyl)-1,5-dihydro-2H-pyrrol-2-one (46).
Step 1: Preparation of 3,4,5-trimethoxycinnamoyl chloride. To a solution of 3,4,5-trimethoxycinnamic acid (77) (5 g, 21 mmol) in CH2Cl2 (100 mL) was added one drop of DMF and then oxalyl chloride (7.2 mL, 84 mmol) dropwise at 0 °C. The resulting mixture was allowed to stir at room temperature overnight and condensed under vacuum to give the chloride as a yellow solid which was used in the next step without further purification. Step 2: Preparation of (E)-N-allyl-3-(3,4,5-trimethoxyphenyl) acrylamide (78). To a stirring solution of the above chloride (0.50 g, 1.95 mmol) in Et2O (5 mL) was added allylamine (1.10 g, 19.50 mmol) at 0 °C. The resulting mixture was stirred at 0 °C for 10 min. The precipitated solid was filtered and washed with hexanes to give 78 (0.38 g, 70%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.53 (d, J = 14.6 Hz, 1H), 6.70 (s, 2H), 6.37 (d, J = 14.8 Hz, 1H), 6.05 (s, 1H), 5.87 (s, 1H), 5.30–5.00 (m, 2H), 4.00 (s, 2H), 3.83 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 165.89, 153.44, 141.19, 139.60, 134.17, 130.48, 120.02, 116.63, 105.01, 61.02, 56.18, 42.26. MS (EI) m/z 277.4 (M+). Step 3: Preparation of (E)-N-acryloyl-N-allyl-3-(3,4,5-trimethoxyphenyl)acrylamide (9). A solution of 78 (0.15 g, 0.54 mmol) in THF (1 mL) was added dropwise to a stirred suspension of NaH (60% in mineral oil, 43 mg, 1.10 mmol) in THF (3 mL) at room temperature. The mixture was stirred for 1 h at room temperature, cooled to 0 °C, and acrylyl chloride (53 µL, 0.65 mmol) was added slowly. The reaction was allowed to stir at 40 °C for 2 h before quenched with aqueous NH4Cl solution. The resulting mixture was extracted with EtOAc (10 mL × 3), and the combined organic phases were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (hexanes/EtOAc 1:1) to afford 9 (94 mg, yield 51%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.69 (d, J = 15.4 Hz, 1H), 6.96 (d, J = 15.4 Hz, 1H), 6.79–6.68 (m, 3H), 6.48 (d, J = 16.5 Hz, 1H), 5.92 (ddd, J = 16.5, 10.3, 5.0 Hz, 1H), 5.83 (d, J = 10.3 Hz, 1H), 5.28–5.16 (m, 2H), 4.56–4.43 (m, 2H), 3.89 (m 9H); 13C NMR (100 MHz, CDCl3) δ 168.93, 168.79, 153.58, 145.35, 140.56, 133.09, 130.72, 130.24, 130.20, 119.66, 116.92, 105.68, 61.13, 56.34, 46.76. MS (EI) m/z 331.4 (M+). Step 4: Ring-closing metathesis of 9. To a solution of 9 (15 mg, 0.045 mmol) in CH2Cl2 (8 mL) under N2 protection was added Grubbs 2nd generation catalyst (2 mg, 0.0022 mmol). The resulting mixture was heated under reflux for 4 h. Upon completion, the reaction mixture was cooled to room temperature, and solvent was removed under vacuum. The crude product was purified by silica gel column chromatography (hexanes/EtOAc 2:1) to afford 46 (8 mg, yield 58%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 15.7 Hz, 1H), 7.83 (d, J = 15.7 Hz, 1H), 7.36 (d, J = 6.1 Hz, 1H), 6.87 (s, 2H), 6.22 (d, J = 6.1 Hz, 1H), 4.55 (s, 2H), 3.92 (s, 6H), 3.89 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.38, 165.32, 153.54, 147.01, 146.29, 140.50, 130.52, 127.99, 117.95, 105.89, 61.14, 56.35, 51.27. MS (ESI) m/z 304.2 [M+H]+.
4.1.3. 3-(Hydroxymethyl)pyrrolidin-2-one (86).
To a suspension of NaBH4 (151 mg, 3.98 mmol) and CaCl2 (585 mg, 3.98 mmol) in EtOH (5 mL) was added ethyl 2oxopyrrolidine-3-carboxylate (84) (250 mg, 1.59 mmol) at 0 °C. The resulting mixture was stirred at room temperature overnight before quenched with HCl solution (1.0 M). The resulting mixture was extracted with CH2Cl2/ MeOH (10/1) 10–20 times. The combined organic phases were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (EtOAc/MeOH 10:1) to afford 86 (90 mg, yield 50%) as a white solid: 1H NMR (400MHz, CDCl3) δ 6.05 (br s, 1H), 3.94–3.84 (m, 1H), 3.80–3.70 (m, 1H), 3.433.35 (m, 2H), 2.68–2.55 (m, 1H), 2.30–2.19 (m, 1H), 2.02–1.88 (m, 1H). MS (APCI) m/z 116 [M+H]+.
4.1.4. 3-Methylenepyrrolidin-2-one (73).
To a solution of 86 (16 mg, 0.139 mmol) in toluene was added CuI (2.6 mg, 0.0139 mmol) and DCC (36 mg, 0.174 mmol). The resulting mixture was stirred under reflux for 3 h. After cooling down to room temperature, water was added, and the mixture was extracted with CH2Cl2 (× 20). The combined organic phases were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (CH2Cl2/MeOH 20:1) to afford 73 (11 mg, yield 82%): 1H NMR (400MHz, CDCl3) δ 6.20 (br s, 1H), 6.01 (s, 1H), 5.38 (s, 1H), 3.44 (t, J = 6.4, 2H), 2.91 (m, 2H). MS (APCI) m/z 98.2 [M+H]+.
4.1.5. 1-(t-Butyl) 3-methyl 2-oxoazepane-1,3-dicarboxylate (87).
To a solution of t-butyl 2-oxoazepane-1-carboxylate (80) (2.45 g, 11.5 mmol) in THF (50 mL) was added LiHMDS (1.0 M in THF, 13.2 mL, 13.2 mmol) dropwise at −78 °C. The resulting mixture was stirred at the same temperature for 45 min, and methyl chloroformate (1.0 mL, 12.6 mmol) was added. After stirring for an additional hour, the reaction was quenched with saturated aqueous NH4Cl solution, extracted with EtOAc (50 mL× 3). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and condensed under reduced pressure. The crude product was purified by silica gel column chromatography (hexanes/EtOAc 3:1) to afford 87 (2.7 g, yield 87%): 1H NMR (400MHz, CDCl3): 1H NMR (400 MHz, CDCl3) δ 5.36 (s, 1H), 3.81 (s, 3H), 3.58 (s, 2H), 2.12 (dd, J = 11.4, 6.6 Hz, 2H), 1.75 (dt, J = 11.3, 5.8 Hz, 2H), 1.60–1.49 (m, 2H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 154.06, 153.14, 145.54, 111.34, 81.05, 77.48, 77.16, 76.84, 55.10, 47.13, 29.39, 28.23, 24.52, 24.02. MS (APCI) m/z 272.1 [M+H]+.
4.1.6. t-Butyl 3-methylene-2-oxoazepane-1-carboxylate (91).
To a suspension of 87 (90 mg, 0.33 mmol), 18-crown-6 (26 mg, 0.1 mmol), and K2CO3 (137 mg, 1 mmol) in toluene (15 mL) was added paraformaldehyde (348 mg, 11.55 mmol). The resulting mixture was stirred under reflux for 5 h. After cooling down to room temperature, water was added, and the mixture was extracted with EtOAc (15 mL× 3). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and condensed under reduced pressure. The crude product was purified by silica gel column chromatography (hexanes/EtOAc 5:1) to afford 91 (30 mg, yield 41%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 5.76 (s, 1H), 5.38 (s, 1H), 3.70–3.63 (m, 2H), 2.48–2.36 (m, 2H), 1.84–1.65 (m, 4H), 1.52 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 172.64, 153.05, 147.29, 123.00, 82.74, 46.07, 32.59, 29.23, 28.22, 28.17; MS (APCI) m/z 226 [M+H]+.
4.1.7. 3-Methyleneazepan-2-one (74).
To a solution of 91 (30 mg, 0.133 mmol) in CH2Cl2 (2 mL) at 0 °C was added TFA (0.2 mL, 2.66 mmol). The resulting mixture was stirred at 0 °C for 2 h. Saturated aqueous NaHCO3 was added to adjust the pH to > 7, and the mixture was extracted with CH2Cl2 (10 mL × 3). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and condensed under reduced pressure. The crude product was purified by silica gel column chromatography (hexanes/EtOAc 2:1) to afford 74 (11 mg, yield 66 %) as a white solid: 1H NMR (400 MHz, CDCl3) δ 6.60 (s, 1H), 5.58 (s, 1H), 5.26 (s, 1H), 3.17 (dd, J = 9.9, 5.2 Hz, 2H), 2.42–2.25 (m, 2H), 1.81–1.58 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 176.03, 146.42, 120.37, 42.82, 32.77, 29.69, 28.98. MS (APCI) m/z 126.2 [M+H]+.
4.1.8. (Z)- (97) and (E)-t-Butyl 3-(2-methylpropylidene)-2oxopiperidine-1-carboxylate (98).
Step 1: To a solution of tbutyl 2-oxopiperidine-1-carboxylate (95) (800 mg, 4 mmol) in THF (15 mL) was added LiHDMS (1.60 M, 2.76 mL, 4.4 mmol) dropwise at −78 °C. After stirring at the same temperature for 1 h, isobutyraldehyde (0.44 mL, 4.8 mmol) was added dropwise. The resulting mixture was allowed to stir for an additional hour before quenched by slowly adding saturated aqueous NH4Cl solution. The mixture was extracted with EtOAc. The organic phase was washed with brine, and dried over anhydrous dried over anhydrous Na2SO4, filtered, and condensed under reduced pressure. The crude product was purified by silica gel column chromatography (hexanes/EtOAc 3:1) to afford t-butyl 3-(1-hydroxy-2-methylpropyl)-2oxopiperidine-1-carboxylate (96) as a mixture of four diasteromers, which was used directly in the next step. Step 2: To a solution of 96 (470 mg, 1.73 mmol) and Et3N (0.725 mL, 5.19 mmol) in toluene (10 mL) was added methanesulfonyl chloride (0.2 mL, 2.60 mmol) dropwise at 0 °C. After stirred at 0 °C for 1 h, additional Et3N (0.725 mL) was added. The resulting mixture was stirred under reflux for 6 h. Upon cooling to room temperature, the reaction was quenched with water, and extracted with EtOAc (10 mL × 3). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and condensed under reduced pressure. The crude product was purified by silica gel column chromatography (hexanes/EtOAc 10:1) to afford 97 (Z-isomer, 25 mg, 5.7%) and 98 (E-isomer, 130 mg, 30%). Compound 97: 1H NMR (400 MHz, CDCl3) δ 5.63 (dt, J = 9.7, 1.5 Hz, 1H), 3.69–3.58 (m, 2H), 3.49–3.30 (m, 1H), 2.41 (td, J = 6.8, 1.5 Hz, 2H), 1.84 (td, J = 12.4, 6.6 Hz, 2H), 1.53 (s, 9H), 0.99 (s, 3H), 0.98 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 166.41, 153.02, 151.06, 127.65, 82.71, 45.60, 30.62, 28.22, 28.07, 22.97, 22.83. MS (APCI) m/z 254.2 [M+H]+. Compound 98: 1H NMR (400 MHz, CDCl3) δ 6.72 (dt, J = 10.0, 2.0 Hz, 1H), 3.67–3.57 (m, 2H), 2.56–2.46 (m, 1H), 2.44–2.39 (m, 2H), 1.84–1.76 (m, 2H), 1.48 (s, 9H), 0.96 (s, 3H), 0.94 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 165.67, 153.31, 149.05, 127.70, 82.67, 45.86, 28.05, 27.56, 24.12, 22.35, 21.75. MS (APCI) m/z 254.2 [M+H]+.
4.1.9. (Z)-3-(2-Methylpropylidene)piperidin-2-one (75).
To a solution of 97 (130 mg, 0.514 mmol) in CH2Cl2 (8 mL) at 0 °C was added TFA (0.79 mL, 10.3 mmol). The resulting mixture was stirred at 0 °C for 2 h. Afterwards, the reaction was quenched with saturated aqueous NaHCO3 and extracted with CH2Cl2 (× 3). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and condensed under reduced pressure. The crude product was purified by silica gel column chromatography (hexanes/EtOAc 3:1) to afford 75 (70 mg, yield 90%): 1H NMR (400 MHz, CDCl3) δ 5.66 (s, 1H), 5.57 (dd, J = 9.6, 0.7 Hz, 1H), 3.73 (ddt, J = 13.3, 9.6, 6.6 Hz, 1H), 3.34–3.19 (m, 2H), 2.51–2.32 (m, 2H), 1.891.78 (m, 2H), 0.98 (s, 3H), 0.97 (s, 3H); 13CNMR (100 MHz, CDCl3) δ 167.03, 149.93, 125.65, 42.51, 32.40, 27.63, 23.61, 22.96. MS (APCI) m/z 154.3 [M+H]+.
4.1.10. (E)-3-(2-Methylpropylidene)piperidin-2-one (76).
By utilizing the same procedure to that described for the preparation of compound 75, compound 76 (13.5 mg, yield 90%) was prepared from 98: 1H NMR (400 MHz, CDCl3) δ 7.03 (s, 1H), 6.64 (dt, J = 9.9, 1.8 Hz, 1H), 3.45–3.21 (m, 2H), 2.64–2.51 (m, 1H), 2.50–2.42 (m, 2H), 1.87–1.75 (m, 2H), 1.00 (s, 3H), 0.98 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 167.32, 145.49, 126.48, 42.01, 27.07, 24.39, 22.92, 22.09. MS (APCI) m/z 254.3 [M+H]+.
4.1.11. General procedure for the preparation of 47–51 and 55–57.
Step 1: Preparation of t-butyl-carbonic (E)-3-(3,4,5-trimethoxyphenyl)acrylic anhydride (68) and t-butyl-carbonic (E)-3-(4-methoxyphenyl)acrylic anhydride (69). To a solution of 3,4,5-trimethoxycinnamic acid or 4-methoxycinnamic acid (1.0 eq.) in THF (~0.3 M) was added Et3N (1.3 eq.) and then trimethyl acetic chloride (1.2 eq.) dropwise at 0 °C. The resulting mixture was stirred at 0 °C for 30 min and filtered. The filtrate was used for the next step without further purification.
Step 2: To a solution of lactam 72, 73, 74, 75, or 76 (1.0 eq.) in THF (~0.3 M) was added n-BuLi (1.6 M in THF, 1.2 eq.) dropwise at −78 °C. After stirred at −78 °C for 45 min, the filtrate from last step (1.0 eq.) was added. The resulting mixture was allowed to stir at −78 °C until completion of the reaction (monitored by TLC). The reaction was then quenched with saturated aqueous NH4Cl and extracted with EtOAc (3 ×). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography.
4.1.11.1. (E)-3-Methylene-1-(3-(3,4,5-trimethoxyphenyl)acryloyl)piperidin-2-one (47).
Purified with hexanes/EtOAc (2.5:1) to afford 47 (53 mg, yield 35.8%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 15.5 Hz, 1H), 7.45 (d, J = 15.5 Hz, 1H), 6.79 (s, 2H), 6.39 (d, J = 1.6 Hz, 1H), 5.51 (dd, J = 3.3, 1.6 Hz, 1H), 3.88 (s, 6H), 3.86 (s, 3H), 3.84 (d, J = 6.0 Hz, 2H), 2.71–2.56 (m, 2H), 1.93 (dt, J = 15.4, 6.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 170.05, 166.96, 153.44, 143.64, 140.05, 138.78, 130.83, 125.86, 121.58, 105.61, 61.07, 56.31, 45.15, 29.36, 22.55; MS (ESI) m/z 332.1 [M+H]+; TR = 4.40 min.
4.1.11.2. (E)-3-Methylene-1-(3-(3,4,5-trimethoxyphenyl)acryloyl)pyrrolidin-2-one (48).
Purified with hexanes/EtOAc (2:1) to afford 48 (20 mg, yield 44%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 15.7 Hz, 1H), 7.79 (d, J = 15.7 Hz, 1H), 6.85 (s, 2H), 6.27 (t, J = 2.8 Hz, 1H), 5.59 (t, J = 2.4 Hz, 1H), 3.85–3.95 (m, 11H), 2.922.67 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 167.89, 167.00, 153.53, 145.92, 140.46, 140.22, 130.56, 120.97, 118.34, 105.82, 61.13, 56.34, 42.56, 23.08; MS (ESI) m/z 318.5 [M+H]+; TR = 3.95 min.
4.1.11.3. (E)-3-Methylene-1-(3-(3,4,5-trimethoxyphenyl)acryloyl)azepan-2-one (49).
Purified with hexanes/EtOAc (3:1) to afford 49 (22 mg, yield 50%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 15.5 Hz, 1H), 7.28 (d, J = 15.5 Hz, 1H), 6.79 (s, 2H), 5.85 (d, J = 1.1 Hz, 1H), 5.50 (d, J = 1.1 Hz, 1H), 3.92–3.88 (m, 11H), 2.56–2.44 (m, 2H), 1.89–1.71 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 174.94, 167.68, 153.50, 147.13, 144.33, 140.10, 130.71, 123.91, 119.84, 105.56, 61.11, 56.33, 44.18, 32.83, 29.44, 28.12; MS (EI) m/z 345.2 (M+); TR = 5.00 min.
4.1.11.4. (Z)-3-(2-Methylpropylidene)-1-((E)-3-(3,4,5-trimethoxyphenyl)acryloyl)piperidin-2-one (50).
Purified with hexanes/EtOAc (5:1) to afford 50 (13 mg, yield 53%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 15.5 Hz, 1H), 7.39 (d, J = 15.5 Hz, 1H), 6.80 (s, 2H), 5.78 (dd, J = 9.7, 1.4 Hz, 1H), 3.90 (s, 6H), 3.88 (s, 3H), 3.83–3.77 (m, 2H), 3.42 (ddt, J = 13.2, 9.7, 6.7 Hz, 1H), 2.58–2.44 (m, 2H), 1.991.85 (m, 2H), 1.04 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 169.62, 168.76, 153.45, 152.29, 143.31, 140.02, 130.96, 127.62, 121.67, 105.68, 61.12, 56.35, 43.53, 30.37, 28.27, 22.84, 22.62; MS (EI) m/z 373.3 (M+); TR = 8.24 min.
4.1.11.5. (E)-3-(2-Methylpropylidene)-1-((E)-3-(3,4,5-trimethoxyphenyl)acryloyl)piperidin-2-one (51).
Purified with hexanes/EtOAc (5:1) to afford 51 (12 mg, yield 50%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 15.5 Hz, 1H), 7.50 (d, J = 15.5 Hz, 1H), 6.86 (d, J = 10.0 Hz, 1H), 6.80 (s, 2H), 3.90 (s, 6H), 3.88 (s, 3H), 3.86–3.81 (m, 2H), 2.70–2.59 (m, 1H), 2.55 (dd, J = 9.1, 4.0 Hz, 2H), 1.98–1.82 (m, 2H), 1.07 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 170.05, 168.01, 153.46, 150.42, 143.36, 139.98, 131.01, 127.97, 121.91, 105.60, 61.11, 56.35, 44.22, 27.96, 24.30, 22.39, 21.88; MS (EI) m/z 373.2 (M+); TR = 7.79 min.
4.1.11.6. (E)-1-(3-(4-Methoxyphenyl)acryloyl)-3methylenepiperidin-2-one (55).
Purified with hexanes/EtOAc (2:1) to afford 55 (41 mg, yield 55%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.71 (d, J = 15.6 Hz, 1H), 7.54 (d, J = 8.5 Hz, 2H), 7.46 (d, J = 15.6 Hz, 1H), 6.89 (d, J = 8.5 Hz, 2H), 6.40 (s, 1H), 5.51 (s, 1H), 3.97–3.76 (m, 5H), 2.65 (t, J = 6.3 Hz, 2H), 1.97–1.84 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 170.36, 166.98, 161.36, 143.53, 138.86, 130.15, 128.06, 125.73, 119.91, 114.32, 55.50, 45.18, 29.46, 22.62; MS (EI) m/z 271.1 (M+); TR = 5.34 min.
4.1.11.7. (E)-1-(3-(4-Methoxyphenyl)acryloyl)-3methylenepyrrolidin-2-one (56).
Purified with hexanes/EtOAc (2:1) to afford 56 (47 mg, yield 68%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 15.7 Hz, 1H), 7.83 (d, J = 15.7 Hz, 1H), 7.59 (d, J = 8.7 Hz, 2H), 6.90 (d, J = 8.7 Hz, 2H), 6.25 (t, J = 2.6 Hz, 1H), 5.57 (t, J = 2.1 Hz, 1H), 3.89 (t, J = 7.3 Hz, 2H), 3.84 (s, 3H), 2.81 (t, J = 7.3 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 167.74, 167.24, 161.67, 145.53, 140.28, 130.39, 127.81, 120.69, 116.69, 114.37, 55.49, 42.47, 23.00; MS (ESI) [M+H]+ m/z 258; TR = 4.54 min.
4.1.11.8. (E)-1-(3-(4-Methoxyphenyl)acryloyl)-3methyleneazepan-2-one (57).
Purified with hexanes/EtOAc (3:1) to afford 57 (49 mg, yield 72%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 15.5 Hz, 1H), 7.52 (d, J = 8.6 Hz, 2H), 7.30–7.21 (m, 1H), 6.89 (d, J = 8.7 Hz, 2H), 5.84 (s, 1H), 5.48 (s, 1H), 3.97–3.87 (m, 2H), 3.83 (s, 3H), 2.55–2.39 (m, 2H), 1.91–1.70 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 174.83, 167.96, 161.36, 147.12, 143.99, 130.09, 127.94, 123.78, 118.23, 114.33, 55.48, 44.09, 32.79, 29.39, 28.09; MS (EI) m/z 285.1 (M+); TR = 6.02 min.
4.2. Senolytic activity assay on normal and senescent WI-38 cells
Human WI-38 fibroblasts (WI-38, catalog no. CCL-75, American Type Culture Collection, Manassas, VA) were cultured in a complete cell culture medium (CM) (Dulbecco’s Modified Eagle Medium) supplemented with 10% Fetal Bovine Serum (FBS, catalog no. 16000044, Thermo Fisher Scientific, Waltham, MA), 100 U/mL penicillin and 100 µg/mL streptomycin (purchased from Atlanta Biologicals, Norcross, GA) in a 37 °C, humidified incubator with 5% CO2. Low passaged WI-38 (< 25 passages) cells were used as controls or for the induction of senescence. Senescent WI-38 cells were prepared from normal WI-38 cells by exposing lowpassage WI-38 cells to 15 Gy of IR at a dose rate of 1.080 Gy/min in a J.L. Shepherd Model Mark I 137Cesium γ-irradiator (J.L. Shepherd, Glendale, CA). Cell viability was measured by flow cytometry as previously described [20,23]. Dose-response curves were generated, and the half-maximal effective concentrations (EC50 values) were calculated with GraphPad Prism 6 software.
4.3 ROS assay
Both normal WI-38 cells and senescent WI-38 cells were cultured overnight in a 37 °C, humidified incubator with 5% CO2. Cells were then treated with various concentrations of PL or PL analogues for 1.5 h. The medium was then aspirated and replaced with pre-warmed new culture medium containing 1 µM dihydrorhodamine 123 (DHR 123, catalog no. D632, Thermo Fisher Scientific). After incubating at 37 °C for 30 min, the culture medium was removed and the cells were washed with PBS and lifted with trypsin. Culture medium was then added to stop the reaction and cell suspensions were transferred to plastic tubes. Mean fluorescence intensity (MFI) of DHR 123 was determined with a BD LSR II flow cytometer (BD Biosciences, San Jose, CA).
4.4. Apoptosis assay
WI-38 cells were first treated with 10 µM QVD-Oph (QVD, catalog no. A1901 APExBIO, Houston, TX) or vehicle for 4 h, and then incubate with 1 µM of compound 49 for 48 h. The cells were harvested and washed twice with Annexin V binding buffer and then stained with Alexa Fluor 647-Annexin V (1: 50, catalog no. 640912, BioLegend, San Diego, CA) and propidium iodide (PI, 10 µg/ml, catalog no. P4170, Sigma-Aldrich) following the manufacturer’s instructions (Biotium, Hayward, CA). All of the stained cells were analyzed with the BD LSR II flow cytometer.
4.5. OXR1 degradation assay using western blot
Cells were treated with 5 µM PL or PL analogs for 24 h and then lysed in RIPA buffer with EDTA and EGTA for protein extraction (catalog no. BP-115DG, Boston BioProducts, Ashland, MA), supplemented with 1% Phosphatase Inhibitor Cocktail 3 (catalog no. P0044, Sigma-Aldrich) and 1% Protease Inhibitor Cocktail (catalog no. P8340, Sigma-Aldrich). Proteins were quantified with BCA assay kit (catalog no. 23225, pierce, Thermofisher Scientific, Waltham, MA) and an equal amount of protein (30 µg/lane) from each cell extract was resolved on a 4–20% Mini-PROTEAN® TGX™ Precast Protein Gels (Bio-rad, Hercules, CA). Proteins were transferred to a NOVEX PVDF membrane (catalog no. LC2002, Life Technologies). The membranes were blocked with 5% nonfat milk in TBST blocking buffer and probed with primary antibodies.at a predetermined optimal concentration overnight at 4 °C, included OXR1 (catalog no. A302–035A, Bethyl Laboratories Inc, Montgomery, TX), β-actin (catalog no. SC-1615, Santa Cruz Biotechnology, Dallas, TX). The secondary antibodies (anti-rabbit or anti-goat) (CST, USA) were conjugated to horseradish peroxidase. Signals were detected using the ECL system (catalog no. WBKLS0100, EMD Millipore, Newmarket, Suffolk, UK) and recorded with autoradiography (Pierce Biotech, Rockford, IL, USA).
Supplementary Material
Acknowledgement
This work was supported in part by grants from the U.S. National Institutes of Health (R01CA122023 and R01CA211963 to D.Z. and P20GM109005 and R56AG056372 to D.Z. and G.Z.) and a sponsored research agreement between UNITY Biotechnology and the University of Arkansas for Medical Sciences. Y.W., J.C., X.L., G.Z., and D.Z. filed a patent application for the use of PL and PL analogs as antiaging agents. A potential royalty stream to Y.W., J.C., X.L., G.Z., and D.Z. may occur consistent with University of Arkansas for Medical Sciences policy. G.Z. is a consultant and D.Z. is a co-founder and advisor of UNITY Biotechnology that develops senolytic drugs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supplementary data associated with this article can be found, in the online version, at .
References
- 1.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 1961;25:585–621. [DOI] [PubMed] [Google Scholar]
- 2.Hayflick L The Limited in vitro lifetime of human diploid cell strains. Exp Cell Res 1965;37: 614–636 [DOI] [PubMed] [Google Scholar]
- 3.Campisi J Aging, cellular senescence, and cancer. Annu. Rev Physiol 2013;75: 685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aravinthan A Cellular senescence: a Hitchhiker’s guide. Hum Cell 2015;28:51–64. [DOI] [PubMed] [Google Scholar]
- 5.Braig M, Lee S, Loddenkemper C, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005;436:660–665. [DOI] [PubMed] [Google Scholar]
- 6.Chen Z, Trotman LC, Shaffer D, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005;436:725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campisi J, Fagagna F. Cellular senescence: when bad things happen to good cells Nat Rev Mol Cell Biol 2007;8:729–740. [DOI] [PubMed] [Google Scholar]
- 8.Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol 2008;20:150–155. [DOI] [PubMed] [Google Scholar]
- 9.Kuilman T, Michaloglou C, Mooi WJ, et al. The essence of senescence. Genes Dev 2010;24:2463–24:24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohtani N, Takahashi A, Mann D, et al. Cellular senescence: a double-edged sword in the fight against cancer. Exp Dermatol 2012;21:1–4. [DOI] [PubMed] [Google Scholar]
- 11.Childs BG, Durik M, Baker DJ, et al. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 2015;21:1424–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 2014;15:482–496. [DOI] [PubMed] [Google Scholar]
- 13.Augert A, Bernard D. Immunosenescence and senescence immunosurveillance: one of the possible links explaining the cancer incidence in ageing population, senescence and senescence-related disorders, Wang Z, Inuzuka H, Eds. InTech, 2013:87–111. [Google Scholar]
- 14.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol 2011;192:547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tchkonia T, Zhu Y, Deursen JV, et al. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 2013;123:966–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salama R, Sadaie M, Hoare M, et al. Cellular senescence and its effector programs. Genes & Dev 2014;28:99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011;479:232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baker DJ, Childs BG, Durik M, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016;530:184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu Y, Tchkonia T, Pirtskhalava T, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging cell 2015;4:644–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang J, Wang Y, Shao L, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med 2016;22:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of antiapoptotic factors. Aging cell 2016;15:428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yosef R, Pilpel N, Tokarsky-Amiel R, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun 2016;7:11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Chang J, Liu X, et al. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging 2016;8:2915–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baar MP, Brandt RM, Putavet DA, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 2017;169:132–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu Y, Doornebal EJ, Pirtskhalava T, et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017;9:955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuhrmann-Stroissnigg H, Ling YY, Zhao J, et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun 2017;8:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudin CM, Hann CL, Garon EB, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res 2012;18:3163–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan J, Li D, Xu Y, et al. Inhibition of Bcl-2/xl with ABT-263 selectively kills senescent yype II pneumocytes and reverses persistent pulmonary fibrosis induced by ionizing radiation in mice. Int. J Radiat Oncol Biol Phys 2017; 99:353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Demaria M, O’Leary MN, Chang J, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer discovery 2017;7:165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Childs BG, Baker DJ, Wijshake T, et al. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016;354:472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bezerra DP, Pessoa C, de Moraes MO, et al. Overview of the therapeutic potential of piplartine (piperlongumine). Eur J Pharm Sci 2013;48:453–463. [DOI] [PubMed] [Google Scholar]
- 32.Prasad S, Tyagi AK. Historical spice as a future drug: Therapeutic potential of piperlongumine. Curr Pharm Des 2016;22:4151–4159. [DOI] [PubMed] [Google Scholar]
- 33.Raj L, Ide T, Gurkar AU, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 2011;475:231–234. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Chandrasekaran A, Idelchik M, Melendez JA. Redox control of senescence and age-related disease. Redox Biol 2017;11:91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams DJ, Dai MJ, Pellegrino G, et al. Synthesis, cellular evaluation, and mechanism of action of piperlongumine analogs. Pro Natl Aca. Sci U.S.A 2012;109:15115–15120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rao VR, Muthenna P, Shankaraiah G, et al. Synthesis and biological evaluation of new piplartine analogues as potent aldose reductase inhibitors (ARIs). Eur J Med Chem 2012;57:344–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boskovic ZV, Hussain MM, Adams DJ, et al. Synthesis of piperlogs and analysis of their effects on cells. Tetrahedron 2013;69:7559–7567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seo YH, Kim JK, Jun JG. Synthesis and biological evaluation of piperlongumine derivatives as potent anti-inflammatory agents. Bioorg Med Chem Lett 2014;24:5727–5730. [DOI] [PubMed] [Google Scholar]
- 39.Wu YL, Min X, Zhuang CL, et al. Design, synthesis and biological activity of piperlongumine derivatives as selective anticancer agents. Eur J Med Chem 2014;82:545–551. [DOI] [PubMed] [Google Scholar]
- 40.Peng SJ, Zhang BX, Meng XK, et al. Synthesis of piperlongumine analogues and discovery of nuclear factor erythroid 2-related factor 2(Nrf2) activators as potential neuroprotective agents. J Med Chem 2015;58:5242–5255. [DOI] [PubMed] [Google Scholar]
- 41.Sun LD, Wang F, Dai F, et al. Development and mechanism investigation of a new piperlongumine derivative as a potent antiinflammatory agent. Biochem Pharmacol 2015;95:156–169. [DOI] [PubMed] [Google Scholar]
- 42.Liao Y, Niu X, Chen B, et al. Synthesis and antileukemic activities of piperlongumine and HDAC inhibitor hybrids against acute myeloid leukemia cells. J Med Chem 2016;59:7974–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Wang J, Li J, et al. Design, synthesis and pharmacological evaluation of novel piperlongumine derivatives as potential antiplatelet aggregation candidate. Chem Biol Drug Des 2016;87:833–840. [DOI] [PubMed] [Google Scholar]
- 44.Yan WJ, Wang Q, Yuan CH, et al. Designing piperlongumine-directed anticancer agents by an electrophilicity-based prooxidant strategy: A mechanistic investigation. Free Radical Biol Med 2016;97:109–123. [DOI] [PubMed] [Google Scholar]
- 45.Lad NP, Kulkarni S, Sharma R, et al. Synthesis and in vitro exploration for therapeutic potential against HeLa cancer cell lines. Eur J Med Chem 2017;126:870–878. [DOI] [PubMed] [Google Scholar]
- 46.Meegan MJ, Nathwani S, Twamley B, et al. Piperlongumine (piplartine) and analogues: Antiproliferative microtubule-destabilising agents. Eur J Med Chem. 2017;125:453–463. [DOI] [PubMed] [Google Scholar]
- 47.Wang HB, Jin XL, Zheng JF, et al. Developing piperlonguminedirected glutathione S-transferase inhibitors by an electrophilicitybased strategy. Eur J Med Chem 2017;126:517–525. [DOI] [PubMed] [Google Scholar]
- 48.Xu X, Fang X, Wang J, Zhu H. Identification of novel ROS inducer by merging the fragments of piperlongumine and dicoumarol. Bioorg Med Chem Lett 2017;27:1325–1328. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y, Ma H, Wu Y, et al. Novel non-trimethoxylphenyl piperlongumine derivatives selectively kill cancer cells. Bioorg Med Chem Lett 2017;27:2308–2312. [DOI] [PubMed] [Google Scholar]
- 50.Zou Y, Yan C, Zhang H, et al. Synthesis and evaluation of Nheteroaromatic ring-based analogs of piperlongumine as potent anticancer agents. Eur J Med Chem 2017;138:313–319. [DOI] [PubMed] [Google Scholar]
- 51.Zhang X, Zhang S, Liu X, et al. Oxidation resistance 1 is a novel senolytic target. Aging Cell 2018, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alvarez S, Dominguez G, Gradillas A, et al. Unusual skeletal rearrangement of unsaturated seven-membered lactams into fused pyrrolidinolactones. Eur J Org Chem 2013;3094–3102. [Google Scholar]
- 53.Zachariassen ZG, Thiele S, Berg EA, et al. Design, synthesis, and biological evaluation of scaffold-based tripeptidomimetic antagonists for CXC chemokine receptor 4 (CXCR4). Bioorg Med Chem 2014;22:4759–4769. [DOI] [PubMed] [Google Scholar]
- 54.Riofski MV, John JP, Zheng MM, et al. Exploiting the facile release of trifluoroacetate for the α-methylenation of the sterically hindered carbonyl groups on (+)-sclareolide and (−)-eburnamonine. J Org Chem 2011;76:3676–3683. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.