

Abstract
Cytotoxic T lymphocyte (CTL)–based immunotherapies have had remarkable success at generating objective clinical responses in patients with advanced metastatic melanoma. Although the melanocyte differentiation antigens (MDA) MART-1, PMEL, and tyrosinase were among the first melanoma tumor-associated antigens identified and targeted with immunotherapy, expression within normal melanocytes of the eye and inner ear can elicit serious autoimmune side effects, thus limiting their clinical potential as CTL targets. Using a tandem mass spectrometry (MS) approach to analyze the immunopeptidomes of 55 melanoma patient–derived cell lines, we identified a number of shared HLA class I–bound peptides derived from the melanocyte-specific transporter protein SLC45A2. Antigen-specific CTLs generated against HLA-A*0201- and HLA-A*2402–restricted SLC45A2 peptides effectively killed a majority of HLA-matched cutaneous, uveal, and mucosal melanoma cell lines tested (18/25). CTLs specific for SLC45A2 showed significantly reduced recognition of HLA-matched primary melanocytes that were, conversely, robustly killed by MART1- and PMEL-specific T cells. Transcriptome analysis revealed that SLC45A2 mRNA expression in normal melanocytes was less than 2% that of other MDAs, therefore providing a more favorable melanoma-to-melanocyte expression ratio. Expression of SLC45A2 and CTL sensitivity could be further upregulated in BRAF(V600E)-mutant melanoma cells upon treatment with BRAF or MEK inhibitors, similarly to other MDAs. Taken together, our study demonstrates the feasibility of using tandem MS as a means of discovering shared immunogenic tumor-associated epitopes and identifies SLC45A2 as a promising immunotherapeutic target for melanoma with high tumor selectivity and reduced potential for autoimmune toxicity.
Introduction
Proteins with expression patterns restricted to melanoma have been identified as potential targets for immunotherapy. Immune-based strategies targeting melanocyte differentiation antigens (MDA), such as MART-1, PMEL, and tyrosinase, which are overexpressed in malignant cells, have been used as effective treatments for patients with refractory disease (1, 2). However, when large numbers of MDA-specific cytotoxic T lymphocytes (CTL) are infused for adoptive cell transfer therapy, destruction of normal melanocytes can be observed, which manifests in the skin as vitiligo (3–5). Efforts to augment antitumor efficacy through the use of high-dose lymphodepletion and IL2, and/or the use of engineered T cells with greater avidity for these MDA-associated epitopes, have resulted in more serious autoimmune manifestations, such as uveitis and inner ear toxicities from melanocyte destruction within these tissues, leading to significant morbidity (6–8). Therefore, a means to target melanoma tumor cells without inducing serious autoimmune toxicities is highly desirable.
SLC45A2 (solute carrier family 45, member 2) is an MDA protein localized within the melanosome membrane whose function is linked to processing and trafficking of tyrosinase to the melanosome and/or pH maintenance within the melanosomes (9–11). SLC45A2 (also referred to as AIM1 or MATP) is associated with dark skin, hair, and eye pigmentation. In humans, a pathogenic mutation of SLC45A2 leads to type IV oculocutaneous albinism (OCA4; refs. 9, 12–14). SLC45A2 variants have been associated with an increased risk for melanoma. SLC45A2 has been proposed as a melanoma susceptibility gene in light-skinned populations, and the encoded protein can elicit immune recognition (15, 16). SLC45A2 expression is restricted to the melanocyte lineage, and according to the The Cancer Genome Atlas Research Network (TCGA) database, it is expressed by approximately 80% of cutaneous melanomas (17).
In the current study, we performed mass spectrometry (MS) analysis on 55 melanoma patient–derived tumor cell lines and identified a number of shared HLA class I–bound peptides derived from SLC45A2. We demonstrate the immunogenicity of HLA-A*0201- and HLA-A*2402–restricted SLC45A2 epitopes by generating peptide-specific CTLs that recognize endogenously presented targets on SLC45A2+ cutaneous, uveal, and mucosal melanoma cell lines. Although SLC45A2 is a melanocyte-associated protein, its expression in mature normal melanocytes was found to be less than 2% that of other MDAs, such as MART-1 and PMEL, resulting in a significantly improved melanoma-to-melanocyte CTL killing index. We show here that an MDA can serve as an effective melanoma CTL target, with high tumor selectivity and minimal potential for autoimmune toxicity. This study also provides a clear example wherein tandem MS/MS tumor profiling has yielded immunogenic, and potentially therapeutic, endogenously presented peptide epitopes that could elicit robust antitumor CTL responses in multiple donors.
Materials and Methods
Peptide elution and tandem MS
Human melanoma tumor cell lines were expanded to approximately 108 cells (10 × 10 cm confluent plates), then lysed using Triton X-100. Cell lysates were incubated overnight at 4°C with gentle agitation with 1 μg HLA-A,B,C–specific mAb W6/32 for every 10 mg of protein. Protein A/G Ultralink resin beads were used to immunoprecipitate HLA molecules, which were then directly eluted along with tumor-associated peptides using 0.1 N acetic acid in five consecutive 1-mL eluates. Purification of HLA was confirmed by Western blot analysis, and HLA-positive elutes were pooled and analyzed by tandem MS (MS/MS), as described below.
For discovery phase MS/MS, eluted HLA-bound peptides were injected onto a high-sensitivity HPLC system (Dionex 3000 RSLC), separated by reverse-phase chromatography in 0.1% formic acid water–acetonitrile on 1.8 μm C18 (Agilent Technologies) and analyzed on an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific) using data-dependent acquisition. The Mascot algorithm was employed to search acquired MS/MS spectra against the SwissProt complete human protein database using 10 ppm parent mass tolerance, 0.8 d fragment ion tolerance, Met oxidation, no enzyme selectivity. Search results were cross-referenced with the appropriate MHC-binding specificities using NetMHC 3.4 (18). On the basis of the results of Discovery MS/MS, next-generation sequencing, and bioinformatics analysis considering target gene expression in normal tissues [GTex RNA sequencing (RNA-seq) databases] and human tumors (TCGA RNA-seq database), high-confidence peptides of interest (corresponding to high Mascot ion scores) were synthesized and used as standards in a more sensitive targeted MS/MS analyses (19, 20). In this analysis, retention-time windows for the synthetic peptide standards of interest were predetermined by MS analysis of the synthetic peptides, then targeted methods for searching tumor-associated peptides were constructed using mass windows of 3 Da around each m/z.
Cell lines and blood donors
Melanoma tumor lines were established from metastatic melanoma samples from patients enrolled on the IRB-approved adoptive T-cell therapy study for the use of tumor-infiltrating lymphocytes (2004-0069). Briefly, a single-cell suspension was obtained through manual dissection of the tumor specimen, followed by incubation with an enzymatic digestion cocktail (0.375% collagenase type I, 75 μg/mL hyaluronidase and 250 U/mL DNAse I in RPMI1640) in a humidified incubator at 37°C with 5% CO2 and with gentle rotation for 2 to 3 hours. The digested material was then filtered through a 70-μm filter, washed, resuspended in fresh media [RPMI1640 (with GlutaMAX), 10% FBS, 10 mmol/L HEPES, 1× penicillin/streptomycin (20 μg/mL), 50 μmol/L 2-mercaptoethanol, insulin–selenium–transferrin supplement (5 μg/mL, Invitrogen)], and plated in one well of a 6-well culture plate. The next day, the nonadherent cells were washed out and fresh media were added. Cultures were deemed established when the expanded cells stained positive for a melanoma tumor marker and negative for a fibroblast marker (CD90). MCSP-1 was used as a melanoma tumor surface marker to assess the purity of the tumor by flow cytometry once enough cells were grown. All cell lines were tested for mycoplasma and fingerprinted before use. Melanoma cell lines were maintained in RPMI1640 with 4 mmol/L L-glutamine, 1 mmol/L nonessential amino acids, 10 mmol/L sodium pyruvate, and 50 U/mL penicillin, 50 mg/mL streptomycin, and 10% FBS (Tissue Culture Biologicals).
All melanoma cell lines numbered from 2042 to 2800 were derived from melanoma patient tumors at the MD Anderson Cancer Center TIL Laboratory between 2006 and 2014. Other cell lines were purchased commercially or obtained from trusted collaborators during the same time period. All cell lines were authenticated by DNA fingerprinting, tested for mycoplasma using a PCR-based test prior to use in the described experiments, and used within 2 to 3 weeks of thawing cryopreserved samples.
Peripheral blood mononuclear cells (PBMC) samples were obtained from healthy donors expressing HLA-A*0201 or HLA-A*2402. Lymphoblastoid cell lines (LCLs) used as feeder cells were cultured in RPMI1640 containing 10% FBS, 50 U/mL penicillin, and 50 mg/mL streptomycin. CTL media for T-cell culture contained 10% FBS, 2 mmol/L L-glutamine, β-mercaptoethanol, penicillin (50 U/mL), and streptomycin (50 mg/mL). Primary neonatal epidermal melanocytes were a kind gift from Dr. Peter P. Lee (City of Hope, Duarte, CA) and were cultured in medium 254 with HMGS-2 supplement (Gibco).
MDA gene expression analysis
Four different techniques were used to analyze the transcriptional expression of MDAs. For RT-PCR analysis of melanoma and primary melanocytes, total cellular RNA was extracted using a guanidine-isothiocyanate/cesium chloride procedure. High-capacity cDNA reverse transcription was used to make cDNA from 1 μg of RNA, which was then amplified by 30 cycles of PCR with primers specific for SLC45A2, MART-1, PMEL, or tyrosinase (Supplementary Table S1). Resulting PCR products were run on a 2% agarose gel and visualized by Gel Red. qRT-PCR was also performed with MDA-specific primers using a Power SYBR Green PCR Master Mix (Applied Biosystems; Life Technologies). In these experiments, MDA expression values were normalized relative to GAPDH expression.
RNA-seq analysis was performed on RNA derived from 66 MD Anderson–derived melanoma cell lines and four melanocyte lines using the following protocol/parameters: whole transcriptome sequencing was performed using the Illumina TruSeq Stranded Total RNA Kit with Ribo-Zero Gold with approximately 2 × 108 paired-end reads for each tumor RNA sample (The Broad Institute and Avera Institute for Human Genetics). All transcript expression values were normalized to transcripts per million (TPM) and compared with RNA-seq datasets from TCGA and GTex portal normal tissue expression database (17, 21). Thresholds for determining MDA positivity in melanoma cell lines were 5 TPM for MART-1 and PMEL and 1 TPM for tyrosinase and SLC45A2. RNA isolated from lentivirally transduced melanocytes was hybridized onto an Affymetrix Human Genome U133A 2.0 Array and analyzed by Expression Analyses as described previously (22).
Lentiviral transduction
SLC45A2-expressing Mel888 melanoma cells (HLA-A*0101/A*2402) were transduced to overexpress HLA-A*0201 or HLA-A*2402 using lentiviral gene transfer vectors as described previously (23, 24). The human phosphoglycerate kinase (hPGK) promoter was used to drive HLA gene expression, and flow cytometry with HLA allotype-specific antibodies was used to confirm tumor cell surface expression. Primary neonatal epidermal melanocytes were transduced with lentiviral vectors that used a CMV promoter to drive expression of either wild-type (WT) BRAF or mutated BRAF(V600E), along with eGFP expression driven by a downstream IRES element, allowing for flow cytometric sorting of transduced cells followed by selection of equivalently transduced lines based on GFP expression. Three days following transduction, GFP-positive melanocytes were sorted and RNA was prepared for Affymetrix microarray gene expression analysis as described previously (22).
Isolation and expansion of SLC45A2-specific CD8 T cells
Tumor antigen–specific CTLs were generated as described previously (25, 26). HLA-A*0201 or HLA-A*2402–positive PBMCs were stimulated by autologous dendritic cells (DC) pulsed with the appropriate antigenic peptide. For induction of DCs, adherent PBMCs were cultured with GM-CSF (800 U/mL) and IL4 (500 U/mL) in AIM-V medium (Invitrogen Life Technologies) for 6 days and then matured using a 1-day incubation in IL1β (2 ng/mL), IL6 (1,000 U/mL), TNFα (10 ng/mL), and PGE2(1,000 ng/mL). Mature DCs were pulsed with peptide (40 μg/mL) at 2 × 106 cells/mL in 1% human serum albumin/PBS in the presence of l β2-microglubulin (3 μg/m) for 4 hours at room temperature and irradiated (5,000 rads). After washing, DCs were mixed with PBMCs (DC:PBMC = 1:35) and plated in 1 mL media in 48-well plate. IL21 (30 ng/mL) was added initially and again after 3 to 4 days of culture. IL2 (10 U/mL) and IL7 (5 ng/mL) were added one day after starting the second DC stimulation to expand Ag-activated T cells.
Six days after the secondary stimulation, cultured cells were stained with SLC45A2382-390peptide/HLA-A*0201 or SLC45A2393-402 peptide/HLA-A*2402–PE–conjugated custom tetramers (Fred Hutchinson Cancer Research Center, Seattle, WA) for 20 minutes, washed, and then stained with allophycocyanin (APC)-conjugated CD8 antibody for 15 minutes. After washing, cells were analyzed by flow cytometry (LSRFortessa X-20 Analyzer). CD8+/tetramer+ cells were sorted by ARIA II, and the sorted SLC45A2-specific CD8+ T cells were expanded using the Rapid Expansion Protocol (REP) with PBMC and LCL feeder cells, as described previously (25).
Phenotyping of expanded, antigen-specific T cells
The T-cell receptor (TCR) Vβ repertoire of expanded CD8 T cells was assessed using the IOTest Beta Mark TCR Vβ Repertoire Kit. This assay utilizes 24 TCR Vβ–specific antibodies, collectively covering approximately 70% of the normal human TCR Vβ repertoire, conjugated with FITC, phycoerythrin (PE), or both fluorophores. Prior to performing flow analysis of the TCR-Vβ repertoire, staining with anti-CD8 APC was performed to enable CTL-specific gating. Expanded T cells were also analyzed by flow cytometry for markers CD45RA, CCR7, CD62L, and CD28 to assess effector/central memory phenotype.
T-cell functional assays
Tumor cell recognition and antitumor killing by expanded SLC45A2-specific CD8+ T cells was assessed using a standard chromium-51 (51Cr) release assay. Target cells were labeled with 100 μCi of 51Cr for 2 hours, and after washing, labeled targets were plated in triplicate wells at 2,000 target cells per well. Effector cells were incubated with targets as various effector-to-target (E:T) ratios for 4 hours, at which time 30 μL of supernatant was collected from each well and 51Cr release was measured with a gamma radiation counter. The percentage of specific lysis was calculated, correcting for background 51Cr release and relative to a maximum release as measured by Triton X-100–lysed target cells. Antigen-specific cytokine release by SLC45A2, Mart-1, and PMEL-specific CD8 T cells was assessed using an IFNγ ELISA assay. Expanded effector T lymphocytes (1e5 cells) were incubated in culture with T2 cells (5e4 cells) pulsed with titrated concentrations of SLC45A2382-390, MART-127-35, or PMEL154-162 peptide. After 48 hours of coincubation, cell supernatants were collected and IFNγ production quantitated by ELISA.
Xenograft model
To investigate the potential therapeutic effect of SLC45A2-specific CTLs in melanoma-bearing mice, nude mice were inoculated subcutaneously in the right flank with 1 × 107 Mel526 cells/mouse. The mice were randomized into three groups: (i) IL2 only; (ii) SLC45A2-specific CTLs + IL2; or (iii) MART-1–specific CTLs + IL2. CTLs (1 × 106) were injected intravenously at day 7, 14, 21, 28, and 35 following tumor inoculation, followed by intraperitoneal injections of IL2 (5,000 units) twice daily for 3 days. Tumor volumes were determined along three orthogonal axes (A, B, and C) and calculated as tumor volume = ABC/2. Tumor volume was measured every 3 days.
MAPK inhibitor experiments
Melanoma cell lines expressing mutant BRAF(V600E) (Mel526 and A375), or wild-type BRAF (MeWo) were treated with the BRAF(V600E)-specific inhibitor dabrafenib (50 nmol/L), the MEK inhibitor trametinib (50 nmol/L, GlaxoSmithKline), or both inhibitors for 48 hours. Inhibitor-treated melanoma cells were then analyzed by qRT-PCR for MDA expression and 51Cr release to assess susceptibility to antigen-specific T-cell killing as described above. Untreated melanoma cells were used as controls.
Statistical analysis
Data analysis was performed using GraphPad Prism version 6.0e. Normally distributed data were analyzed using parametric tests (ANOVA or unpaired t test). Statistical test differences were considered significant if P values were <0.05.
Results
Melanomas present shared peptide antigens derived from SLC45A2
To characterize the immunopeptidome landscape of melanoma, a panel of 55 melanoma patient–derived cell lines (MD Anderson Cancer Center, Houston, TX) was analyzed using HLA class I immunoprecipitation and acid elution, followed by tandem MS. Peptide fragmentation spectra were matched against a SwissProt peptide database representing the complete human proteome (Fig. 1A). The qualities of individual peptide matches were assessed using multiple orthogonal parameters, including Mascot ion score, MS1 mass differential (delta mass), and predicted peptide binding to the patient’s HLA allotypes, as determined by high-resolution genetic sequencing. To determine suitability as tumor-associated antigens, genes encoding eluted peptides were assessed for normal tissue expression using the GTex Portal RNAseq database and for tumor expression using TCGA RNA-seq database (17, 21). Finally, potential tumor-associated antigens of interest were validated in a targeted MS experiment using synthetic peptides for comparison (Fig. 1B and C).
Figure 1.
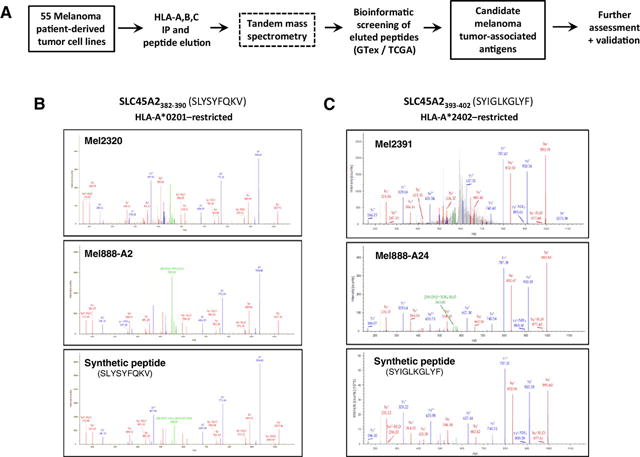
Mass spectrometric identification of SLC45A2-derived peptides from melanoma cell lines. A, Experimental strategy to identify melanoma tumor-specific peptides from melanoma cell lines. B and C, Mass spectra of HLA-A*0201- and HLA-A*2402–restricted tumor cell–derived and synthetic SLC45A2 peptides. B, Peptide eluted from HLA-A*0201+ melanoma cells (top) and HLA-A*0201–transduced Mel888 melanoma cells (middle) showed a corresponding MS fragmentation pattern for the synthetic peptide SLYSYFQKV (bottom). C, Peptide eluted from HLA-A*2402+ melanoma cells (top) and HLA-A*2402–transduced Mel888 melanoma cells (middle) showed a corresponding fragmentation pattern for the synthetic peptide SYIGLKGLYF (bottom).
The 55 melanoma cell line analysis revealed shared expression of a number of high-confidence peptides derived from the highly tissue-restricted melanocyte membrane-associated transporter protein SLC45A2 (Supplementary Table S2). Two HLA-A*0201–restricted peptides were eluted from multiple melanoma cell lines (SLYSYFQKV from 6 cell lines and RLLGTEFQV from 8 lines). In addition, we also eluted two HLA-A*2402–restricted peptides (SYIGLKGLYF from 3 lines and VWFLSPILGF from 2 lines), and one peptide each showing predicted restriction to HLA-A*0301 or HLA-A*3002. To confirm natural processing and presentation of these peptide epitopes, SLC45A2+Mel888 melanoma cells were transduced to express either HLA-A*0201 or HLA-A*2402, as described previously (Supplementary Fig. S1; ref. 23). Both HLA-A*0201–restricted SLC45A2 peptides could only be detected on Mel888 if transduced to express HLA-A*0201 (Fig. 1B; Supplementary Table S3). One of the two HLA-A*2402–restricted peptides (SYIGLKGLYF) was detectable on parental Mel888 cells (which naturally express HLA-A*2402), but showed an increased abundance upon A*2402 overexpression (∼3-fold, Supplementary Fig. S1), which correlated well with a similar increase in fragment ion spectra intensities and Mascot ion score (Supplementary Table S3), thus providing increased confidence in peptide identification. These studies showed that SLC45A2-derived peptides were naturally processed and presented on melanoma cells and could be readily detected by MS in a significant fraction of cases (16/55 cell lines analyzed, 29%). Because HLA-A*0201 and A*2402 are collectively expressed by >60% of melanoma patients, the identified peptide epitopes have the potential to constitute widely shared melanoma target antigens.
SLC45A2 is expressed by a majority of human melanomas
To assess the overall prevalence of SLC45A2 expression in human melanoma, mRNA was isolated from a panel of 24 melanoma tumor cell lines and subjected to RT-PCR analysis. SLC45A2 was expressed by the majority (18/24, 75%) of melanoma lines (Fig. 2A and B), and in most cases was coexpressed with other tumor-associated melanocyte differentiation antigens MART1, PMEL, and tyrosinase, consistent with its reported role as a target gene for melanogenesis-associated transcription factor (MITF; ref. 14). A separate RNA-seq transcriptome analysis performed on a larger panel of 66 melanoma cell lines showed similar results, demonstrating SLC45A2 expression in 48 of 66 (72.7%) of cell lines analyzed (Fig. 2C). We also examined a panel of 8 tumor cell lines derived from nonmelanoma cancers, including colon, breast, prostate, lung, and different hematologic malignancies, for SLC45A2, but no expression could be detected, results that were consistent with those of the Cancer Cell Line Encyclopedia database (Supplementary Figs. S2 and S3; ref. 27). The status of SLC45A2 as a melanoma-specific tumor-associated antigen was further confirmed by RNA-seq data from TCGA, showing that SLC45A2 RNA is expressed by approximately 80% of cutaneous melanomas and 100% of uveal melanomas tested in this study, but in no other tumor types, with the exception of a small subset of prostate and liver cancers (Supplementary Fig. S4; ref. 17). RNA-seq data from the GTex Portal showed that SLC45A2 transcript expression was absent in nearly all normal tissues analyzed (21). SLC45A2 transcript expression in normal heart was very low or absent (mean 0.07 TPM), whereas the mean transcript expression in normal brain was comparable with that of other MDAs (0.27 TPM; Table 1; Supplementary Fig. S4). The highest normal tissue expression was detected in testis and skin, but at transcript concentrations 1% to 2% of melanoma (Supplementary Fig. S4). In contrast to PMEL, MART1, and TYR, mean SLC45A2 transcript expression among primary melanocytes was 0.8% to 1.4% of these other MDAs, as quantitated by RNA-seq (Table 1).
Figure 2.
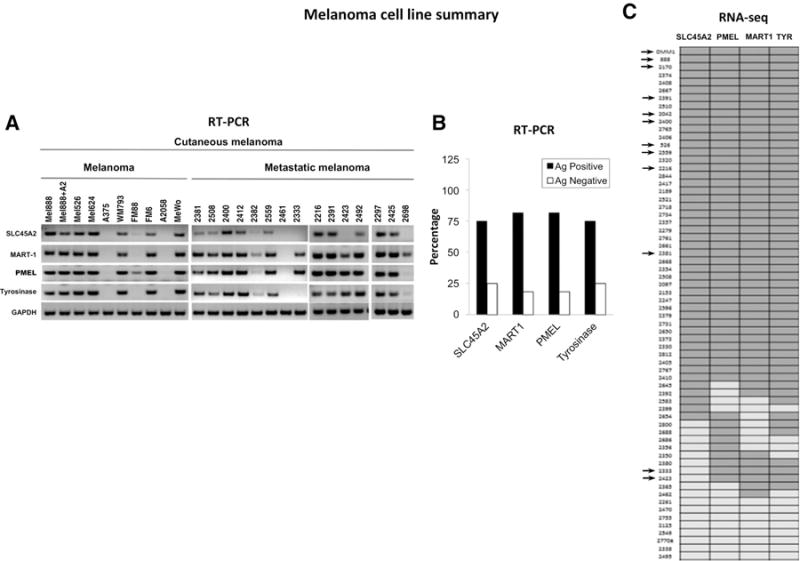
SLC45A2 is a melanocyte differentiation antigen expressed by a majority of melanomas. A, RT-PCR analysis showed that SLC45A2 mRNA was detectable in most melanoma cells and was typically coexpressed along with MDAs MART-1, PMEL, and tyrosinase. B, Summary of results showing MDA expression in 24 melanoma cell lines, as determined by RT-PCR. C, Expression map of MDAs SLC45A2, PMEL, MART-1, and tyrosinase in 66 melanoma cell lines, as determined by RNA-seq analysis. Dark gray, antigen-positive cell lines; light gray, antigen-negative lines; arrows, cell lines that were also analyzed for MDA expression by RT-PCR and tested for susceptibility to MDA-specific CTL killing.
Table 1.
Melanocyte differentiation antigen expression in melanomas and normal tissues
| Tissue expression (RNA-seq, mean TPM) | Samples (n) | PMEL | MART1 | TYR | SLC45A2 | |
|---|---|---|---|---|---|---|
| Gtex normal tissue | Heart | 133 | 0.81 | 0.29 | 0.05 | 0.07 |
| Brain | 357 | 0.15 | 0.21 | 0.09 | 0.27 | |
| Skin | 167 | 42.9 | 11.1 | 8.63 | 0.55 | |
| Cell lines | Melanomas (SLC45A2+) | 48 | 2,361 | 273 | 392 | 99 |
| Primary melanocytes | 4 | 5,354 | 2,804 | 2,268 | 39 |
SLC45A2-specific CD8+ T cells can be readily expanded from normal donors
We next generated CD8+ T cells with specificity for the SLC45A2-derived peptides SLYSYFQKV and SYIGLKGLYF, restricted to HLA-A*0201 and A*2402, respectively. To isolate and expand low frequency CD8+ T cells from peripheral blood, donor PBMCs were stimulated twice with SLC45A2 peptide–pulsed DCs in the presence of IL21, as described previously (Fig. 3A; refs. 26, 28). Using this method, we successfully generated CD8+ T cells specific for the HLA-A*0201–restricted SLYSYFQKV peptide from 4 of 5 healthy donors. Following DC stimulation, SLC45A2 tetramer+CD8+ T cells were typically detected at a frequency of 2% to 4% of lymphocyte-gated cells and 5% to 10% of CD8+ T cells (Fig. 3B, top; Supplementary Fig. S5A). SLC45A2 tetramer+ CD8+ T cells were sorted from single wells and expanded for 2 weeks using the previously described rapid expansion protocol (REP; ref. 29). Following rapid expansion, the frequency of SLC45A2 tetramer+cells was typically >90% of the lymphocyte-gated population. Using the same methodology, SLC45A2-specific CD8+ T cells recognizing the SYIGLKGLYF peptide were also successfully isolated and expanded from two of two healthy HLA-A*2402+ donors (Fig. 3C). TCR Vβ analysis demonstrated that a single Vβ was utilized in most expanded T-cell cultures: Vβ3, Vβ8, Vβ18, and Vβ21.3 in the case of these donors (Fig. 3B and C; Supplementary Fig. S5A). Phenotypic analysis of the expanded SLC45A2-specific CD8 T cells showed low to absent expression of CD45RA, CCR7, and CD62L, but elevated expression of CD28, suggesting an effector memory–like phenotype (Supplementary Fig. S5B).
Figure 3.
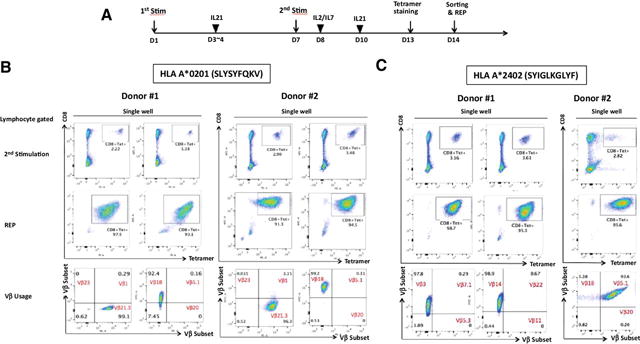
Generation of SLC45A2 antigen-specific CTLs from normal donor peripheral blood. A, Schematic showing the experimental schedule for generating SLC45A2-specific CD8 T cells from normal donor PBMCs. B and C, Induction of SLC45A2-specfic CD8 T cells from PBMCs. HLA-A*0201- (B) or A*2402-restricted healthy donors (C) were stimulated with autologous SLYSYFQKV or SYIGLKGLYF peptide-pulsed DCs, respectively. SLC45A2 tetramer-positive CD8+ T cells were sorted by ARIA sorter after two stimulations (top), and the sorted SLC45A2-tetramer+ CD8+ T cells were expanded according to the REP. Expanded cells (middle) were subsequently tested for antitumor activity. TCR repertoire analysis of expanded SLC45A2-specific CTLs was also performed using Vβ antibodies corresponding to 24 different specificities (bottom).
SLC45A2-specific T cells kill a majority of HLA-matched melanomas
Assessment of melanoma cell recognition and killing by SLC45A2-specific CD8+ T cells was performed using a standard 51Cr release assay. Although expanded A*0201-restricted (SLYSYFQKV peptide-reactive) T cells did not recognize Mel888 (A*0201−/SLC45A2+) or A375 (A*0201+/SLC45A2−) melanoma cells, they demonstrated robust killing of Mel526 (A*0201+/SLC45A2+) and Mel888 cells transduced to express HLA-A*0201 (Fig. 4A). SLC45A2-specific T-cell recognition was also tested against 12 additional allogeneic A*0201+ cutaneous melanoma cell lines, demonstrating specific killing of all HLA-A*0201+ SLC45A2+ targets, and correlating with tumor cell SLC45A2 transcript expression (Figs. 4A and 2A). Similarly, A*2402-restricted (SYIGLKGLYF-specific) T cells lysed four HLA-A*2402–positive cell lines expressing SLC45A2, but did not recognize Mel526 (A*2402−/SLC45A2+) control cells (Fig. 4B).
Figure 4.
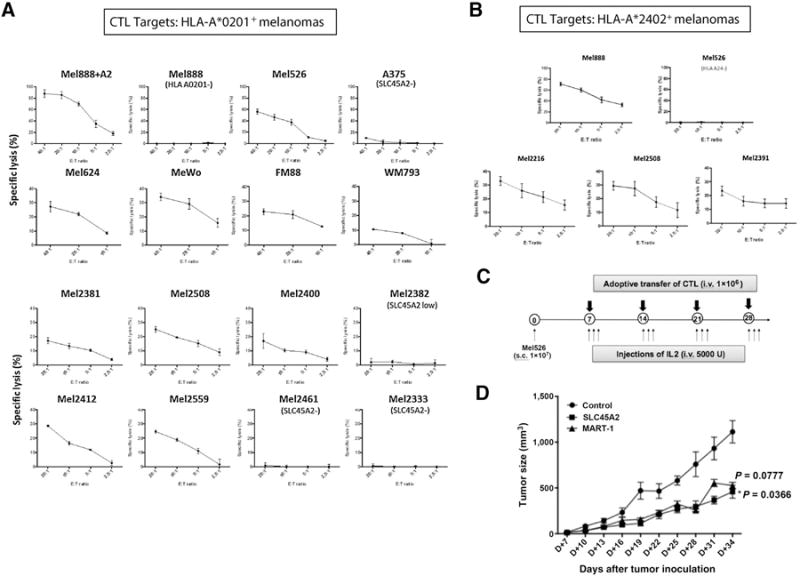
SLC45A2-specific CTLs kill a majority of HLA-matched cutaneous melanoma cell lines. A, Expanded HLA-A*0201–restricted SLC45A2-specific CD8+ T cells were cocultured with a panel of 15 HLA-A*0201–positive cutaneous melanoma target cell lines in a standard 51Cr release assay to measure cytotoxic activity at different effector-to-target (E:T) cell ratios. B, Expanded HLA-A*2402–restricted SLC45A2-specific CTLs were cocultured with a panel of 4 HLA-A*2402+ cutaneous melanoma target cell lines in a standard 51Cr release assay. All melanoma cells expressed SLC45A2 unless otherwise indicated. C, Schematic showing the experimental timeline of adoptive T-cell transfer using a melanoma xenograft model. D, Tumor growth curves showing the therapeutic effect of adoptively transferred HLA-A*0201–restricted SLC45A2-specific or MART1-specific CTLs against human melanoma Mel526 xenografts. Nude mice were inoculated subcutaneously with 1 × 107 Mel526 cells. Seven days following tumor challenge, SLC45A2- or MART-1–specific CTLs (1 × 106) were injected intravenously once per week for 4 weeks and tumor growth was monitored. P values indicate the comparison between MART1 and SLC45A2 T cell treatments (P = 0.078) and between control and SLC45A2 T cell treatments (P = 0.037). All results show one representative experiment of at least three performed.
The ability of SLC45A2-specific T cells to induce antitumor effects in vivo was also tested in a xenogeneic mouse model (Fig. 4C). Infusion of A*0201-restricted SLC45A2-specific CTLs induced a significant delay in tumor growth in Mel526 tumor–bearing mice compared with that of control mice (P < 0.05), and was comparable with MART1-specific CTL treatment (P < 0.05, Fig. 4D).
The activity of SLC45A2-specific T cells against uveal and mucosal melanoma cell lines was also evaluated (30). Four of five uveal melanoma cell lines showed robust SLC45A2 transcript expression (Supplementary Fig. S6A). Of the two A*0201+ cell lines, OMM1 cells were lysed efficiently by SLYSYFQKV-specific T cells, but UPMD1 cells demonstrating reduced SLC45A2 expression were not (Supplementary Fig. S6B). Only one of the five uveal cell lines expressed HLA-A*2402 (UPMD2), and these cells were targets for killing by SYIGLKGLYF-specific T cells (Supplementary Fig. S6C). Similarly, both mucosal melanoma cell lines analyzed showed SLC45A2 transcript expression (Supplementary Fig. S6D), and the one A*0201+ cell line (Mel2170) was also robustly killed by SLC45A2-specific T cells (Supplementary Fig. S6E). These results show that the SLC45A2382-390 and SLC45A2393-402 peptide epitopes are naturally processed and presented by HLA-A*0201 and HLA-A*2402 on a majority of cutaneous, uveal, and mucosal melanomas, rendering them susceptible to lysis by SLC45A2 antigen-specific CTLs.
SLC45A2-specific T cells demonstrate low cytotoxicity against primary melanocytes
Serious autoimmune consequences have resulted from CTL targeting of the MDAs MART-1 and PMEL, mostly due to on-target destruction of normal melanocytes (3, 6, 31). Therefore, we next assessed the cytotoxic activity of SLYSYFQKV-specific CD8 T cells against normal primary A*0201-expressing melanocytes compared with T cells specific for other known A*0201-restricted MDA peptides. Normal donor PBMC-derived CTLs were generated that demonstrated strong recognition of the well-characterized A*0201-restricted PMEL154-162 and MART-127-35 peptide targets (Supplementary Fig. S7; Supplementary Table S4). RT-PCR analysis of three HLA-A*0201+ human epidermal primary melanocyte lines revealed that SLC45A2 transcript expression was appreciably lower than that of MDAs MART-1, PMEL, and tyrosinase (Fig. 5A). When melanocyte lines were used as targets for SLC45A2-, PMEL-, or MART-1-specific CTLs, all melanocyte lines were efficiently killed by PMEL and MART-1 CTLs, but not by SLC45A2-specific T cells (Fig. 5B). The normal melanocytes could present A*0201-restricted peptides, as shown by high HLA-A*0201 surface expression (Supplementary Fig. S8A), and pulsing the melanocytes with the SLYSYFQKV peptide rendered them targets for SLC45A2-specific CTL recognition (Supplementary Fig. S8B). Differential killing of melanocytes and melanoma cells was also observed in SLC45A2-specific T cells derived from three additional PBMC donors (Supplementary Fig. S9).
Figure 5.
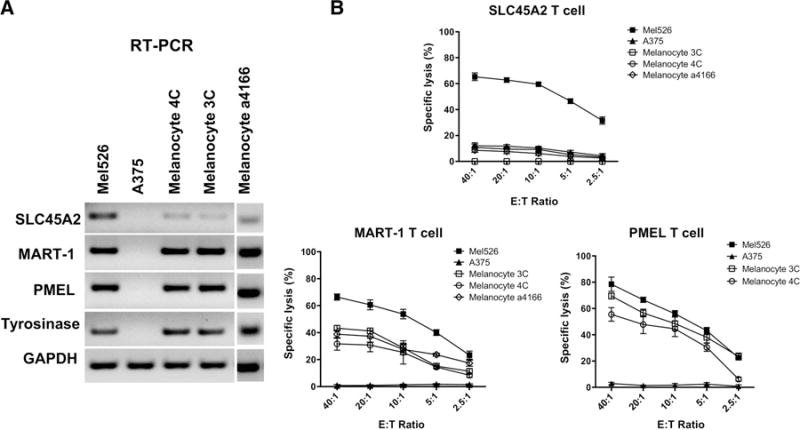
SLC45A2-specific CTLs demonstrate reduced lysis of primary melanocytes compared with other MDA-specific CTLs. A, Relative gene expression of MDAs SLC45A2, MART-1, PMEL, and tyrosinase as assessed using RT-PCR in three different primary melanocyte lines, in addition to SLC45A2+ and SLC45A2− control melanoma cell lines, Mel526 and A375, respectively. B, Cytolytic activity of SLC45A2-specific, MART1-specific, or PMEL-specific CTLs when cocultured with three HLA-A*0201+ primary melanocyte lines (3C, 4C, and a4166), Mel526, or A375, using the indicated E:T ratios in a standard 51Cr release assay.
To confirm antigen expression differences among our melanoma cell lines and primary melanocytes, we performed a more quantitative RNA-seq–based assessment of MDA transcript expression (Table 1). RNA-seq analysis of 48 SLC45A2-positive melanoma cell lines uncovered highly significant differences in expression levels among the different MDAs. The highest expression by a large margin was PMEL (mean 2,361 TPM), followed by tyrosinase (392 TPM), MART-1 (273 TPM), and SLC45A2 (99 TPM). RNA-seq data from the melanoma TCGA showed a highly similar decreasing trend in MDA expression, but with a higher overall observed expression for PMEL and MART-1 in both cutaneous and uveal melanoma (Table 1; ref. 17). To better understand the differential melanocyte killing by PMEL-, MART1-, and SLC45A2-specific CTLs, we also performed RNA-seq analysis on four primary human melanocyte cell lines. These experiments revealed that mean RNA transcript expression of well-known MDAs PMEL (5,354 TPM), MART-1 (2,804 TPM), and tyrosinase (2,268 TPM) were significantly higher (3- to 9-fold) in cultured primary melanocytes compared with melanoma cells. In contrast, mean SLC45A2 transcript expression in primary melanocytes was only 39 TPM, less than half of that expressed by melanoma cells (Table 1). These results show that SLC45A2 has a more favorable melanoma-to-melanocyte overexpression ratio and significantly lower overall expression compared with PMEL, MART-1, and tyrosinase, likely contributing to the ability of SLC45A2-specific CTLs to effectively kill melanoma cells without destroying normal primary melanocytes.
SLC45A2 expression and melanoma cell killing is enhanced by MAPK inhibitors
Similar to other MDAs, SLC45A2 is regulated by MITF, which is suppressed in melanoma by oncogenic BRAF through ERK-mediated phosphorylation and degradation (32, 33). To investigate whether MAP kinase pathway activation through mutated BRAF(V600E) could modulate SLC45A2 expression, we transduced primary melanocytes to express GFP, wild-type (WT) BRAF, or mutant BRAF(V600E) (22). Gene expression microarray analysis showed that BRAF(V600E) expression led to a significant downregulation of MDAs PMEL, MART-1, TYRP1, TYR, and SLC45A2, ranging from approximately 17% to 33% of the expression in control GFP-transduced cells (Fig. 6A). Thus, we next investigated whether clinically relevant BRAF and MEK inhibitors could upregulate the expression of SLC45A2 in melanoma cells, as has been demonstrated for other MDAs (34). Melanoma cells were treated with sublethal doses of the BRAF(V600E)-specific inhibitor dabrafenib, MEK inhibitor trametinib, or both inhibitors, for 48 hours, after which mRNA was isolated and expression of SLC45A2 and MART-1 analyzed by qRT-PCR. A significant increase in both SLC45A2 and MART-1 gene expression was observed in BRAF(V600E)-expressing Mel526 cells after treatment with both inhibitors, alone or in combination (Fig. 6B). As expected based upon MDA upregulation, significantly enhanced killing of Mel526 cells by both SLC45A2-specific and MART-1-specific CTLs was observed following inhibitor treatment, while MDA-negative A375 cells were not lysed under any treatment condition (Fig. 6C). Collectively, these data indicate that MAPK pathway inhibitors can effectively upregulate the expression of SLC45A2 in BRAF(V600E)-mutated melanomas, leading to enhanced tumor cell recognition and killing by SLC45A2-specific CD8+ T cells.
Figure 6.
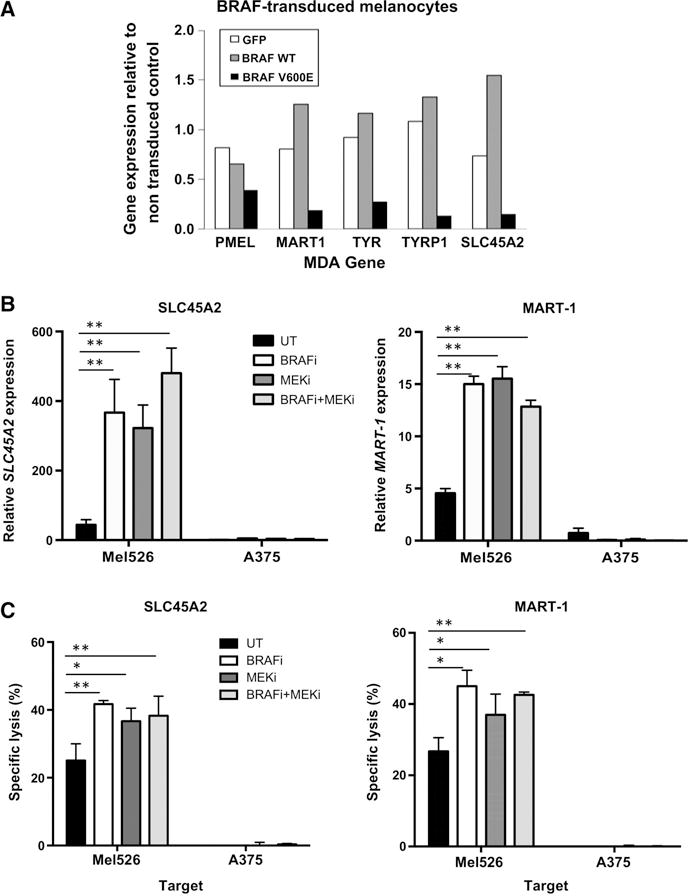
Control of SLC45A2 expression by the MAPK pathway and enhanced melanoma cell CTL killing after MAPK inhibitor treatment. A, Melanocyte differentiation antigen expression in primary melanocytes transduced to express GFP, wild-type BRAF, or mutant BRAF(V600E), as assessed by gene expression microarray analysis. Shown is relative expression of SLC45A2, MART-1, PMEL, tyrosinase-related protein and tyrosinase compared with nontransduced cells. B, BRAF(V600E)+melanoma cell lines Mel526 or A375 were treated with the BRAF(V600E)-specific inhibitor dabrafenib (50 nmol/L), the MEK inhibitor trametinib (50 nmol/L), or both inhibitors. After 48 hours, mRNA expression of SLC45A2 and MART-1 was analyzed by quantitative RT-PCR. Untreated melanoma cells were used as controls. C, SLC45A2-specific T-cell–mediated cytotoxic killing of melanoma cell lines Mel526 or A375 following 48 hours treatment with BRAFi, MEKi, or both inhibitors. Cytotoxic activity of SLC45A2-specific CTLs in a standard 51Cr release assay (E:T ratio = 20:1) against drug-treated targets is shown in comparison with untreated targets. *, P < 0.05; **, P < 0.01.
Discussion
Eliciting potent T-cell–mediated antitumor immunity without concurrent autoimmunity has proven to be one of the greatest ongoing challenges for tumor immunologists. The specific targeting of tumor-associated neoantigens may largely preclude this issue; however, the unique mutational profile within each patient’s tumor necessitates a personalized approach that is neither practical nor currently feasible for the vast majority of cancer patients (35–39). A number of immune-based strategies have shown effectiveness in metastatic melanoma, particularly immune checkpoint inhibitors that can induce long-term disease control (40–42). However, even this promising modality eventually fails in more than 60% of patients, and one third experience serious autoimmune toxicities (41, 43). Adoptive TIL therapy has also proven effective in melanoma, often leading to long-term durable complete responses after only a single infusion (8, 44). However, TIL therapy regimens typically utilize lymphodepletion conditioning, followed by high-dose IL2 administration, which can lead to serious toxicities (45, 46). Thus, employing T cells with defined specificity against shared tumor-associated antigens holds great promise. Melanoma cells express a number of nonmutated genes specific to the melanocyte lineage that can be targeted by T cells, most commonly the MDAs MART-1, PMEL, and tyrosinase, which are collectively expressed under the control of MITF (47). The use of endogenous T cells targeting MDAs has been successful at mediating tumor regressions and in some cases durable complete responses (28, 29, 48). Alternative approaches employing recombinant viral vectors expressing high-affinity MDA-specific TCR genes to genetically modify peripheral blood lymphocytes have also proved successful at eradicating large tumor burdens. However, this treatment was also accompanied by a significant increase in the incidence of serious dose-limiting toxicities, including uveitis, vestibular toxicity, and hearing impairment, likely as the result of T-cell–mediated destruction of melanocytes in normal tissues (6).
The aforementioned studies have led to the idea that MDA-specific T cells may be incapable of inducing antitumor immunity without concurrent autoimmunity through targeting normal melanocytes (7). However, in this study, we provide evidence that different MDAs demonstrate distinctly differential expression profiles in normal and tumor tissues and thus could differ in their propensity to induce autoimmune side effects. SLC45A2 in cutaneous melanoma distinguishes itself from other MDAs by expressing significantly fewer transcripts: 2.5% to 5% that of PMEL, and 17% to 33% that of MART-1 and tyrosinase. Nonetheless, several SLC45A2-derived, HLA class I–restricted peptides could be detected in an MS-based immunopeptidome survey of 55 melanoma cell lines. Antigen-specific CTLs were raised against two SLC45A2-derived peptides from multiple donors, demonstrating their immunogenicity. SLC45A2-reactive CTLs could lyse most HLA-matched allogeneic cutaneous, uveal, and mucosal melanoma cell lines tested. Perhaps the key distinguishing feature of SLC45A2 was its significantly reduced expression in normal melanocytes. Whereas PMEL transcript levels were comparable between melanocytes and melanomas, MART-1 and tyrosinase transcript expression in primary melanocytes exceeded mean transcript numbers in melanoma cell lines and TCGA melanoma tumors by 3- to 10-fold. In contrast, SLC45A2 transcript expression in normal melanocytes was 1% to 2% that of PMEL, MART-1, or tyrosinase. These differential MDA gene expression profiles are consistent with our data showing that HLA-A*0201+primary melanocytes were readily lysed by A*0201-restricted PMEL- and MART1-specific CTLs but not by SLC45A2-specific CTLs, despite the ability of all 3 CTL lines to robustly kill melanoma tumor cells.
Most MDAs function at different stages of the melanin synthesis pathway in normal melanocytes, which may provide further clues to understanding differential targeting by different MDA-specific CTLs. SLC45A2 is a solute transporter found within the membranes of melanosomes and functions by elevating the pH of these organelles using a proton gradient (11). Under conditions of elevated pH, copper binds to tyrosinase, resulting in its activation and promoting early melanin synthesis; following this process, melanosomal pH is restored to neutral. In contrast, MART-1 and PMEL are melanosome matrix proteins responsible for late-stage melanin synthesis (49). Thus, it is possible that SLC45A2 may be preferentially expressed during the early stages of melanocyte development. Although further studies are required to confirm this, it is conceivable that differences in timing of MDA gene expression during melanocyte differentiation may lead to the reduced expression of SLC45A2 in mature melanocytes compared with other more abundantly expressed MDAs. However, differences in transcript expression cannot fully explain the preferential killing of melanoma cells over melanocytes by SLC45A2-specific CTLs. No SLC45A2 peptides were detected by MS analysis of primary melanocytes, in spite of high HLA-A*0201 surface expression. Furthermore, these refractory melanocytes quickly became robust targets for CTL killing after being pulsed with low concentrations of SLC45A2 peptide. Collectively, these data suggest that SLC45A2 peptides show significantly reduced abundance on normal melanocytes, possibly due to differential processing of antigenic peptides by the proteasome or other antigen-processing components, or differential posttranslational modification of this peptide between melanocytes and melanoma cells.
The SLC45A2 protein demonstrates a number of attributes that argue in favor of its use as a specific target for melanoma immunotherapy. The gene is expressed by a majority of cutaneous, uveal, and mucosal melanomas, it is not observed in other nonmelanin-producing tumor types, and it shows little or no expression in normal tissues (17, 21). The preferential killing by SLC45A2-specific T cells of melanoma cells over primary melanocytes predicts that this tumor-associated antigen may have a greater therapeutic window compared with other MDAs; however, this has yet to be tested in vivo. We have now initiated clinical trials to test the safety and efficacy of adoptive transfer with endogenous, expanded SLC45A2-specific T cells in metastatic uveal and cutaneous melanoma patients, with or without checkpoint blockade. Beyond these efforts, reports showing MAPK inhibitor–induced upregulation of MDAs and HLA class I antigen presentation suggest that combining SLC45A2-specific T-cell therapy with BRAF or MEK inhibition may lead to therapeutic synergy in future trials (34, 50). In light of past experience with MDA-specific T cells, monitoring for autoimmunity in these patients will be of prime interest, in addition to assessing antitumor efficacy. These crucial clinical parameters will ultimately determine whether SLC45A2 can distinguish itself from other MDAs as an effective target for melanoma immunotherapy.
Supplementary Material
Acknowledgments
We thank Dr. Peter P. Lee for kindly providing two HLA A*0201-positive melanocytes, and thank the UT-MDACC Proteomics and Metabolomics Facility for their invaluable contributions to the MDACC antigen discovery pipeline.
Grant Support
Research supported by a Stand Up To Cancer-Cancer Research Institute Cancer Immunology Translational Research Grant. Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research. Cassian Yee is a Scholar of the Cancer Prevention Research Institute of Texas (CPRIT). This work was supported by the Cancer Prevention Research Institute of Texas (CPRIT) grant RP130397, The National Institutes of Health (NIH) grant 1S10OD012304-01, National Research Foundation of Korea Grants NRF-2013M3A9D3045719 and NRF-2016M3A9B6948342, and by generous philanthropic contributions to The University of Texas MD Anderson Melanoma Moon Shots Program.
Footnotes
Disclosure of Potential Conflicts of Interest
C. Yee is a consultant/advisory board member for Adaptive Biotechnologies and Immatics. No potential conflicts of interest were disclosed by the other authors.
Authors’ Contributions
Conception and design: J. Park, P. Hwu, K.-M. Lee, G. Lizée, C. Yee
Development of methodology: A.H. Talukder, J.S. Khalili, D. Hawke, J. Roszik, G. Lizée, C. Yee
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): A.H. Talukder, S.A. Lim, S.D. Bradley, K.R. Jackson, C. Creasy, B.-F. Pan, C. Bernatchez, D. Hawke, G. Lizée, C. Yee
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): J. Park, S.A. Lim, K.R. Jackson, J.S. Khalili, S.E. Woodman, D. Hawke, P. Hwu, K.-M. Lee, J. Roszik, G. Lizée, C. Yee
Writing, review, and/or revision of the manuscript: J. Park, S.E. Woodman, G. Lizée, C. Yee
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): J. Park, K. Kim, K. Pan, B. Melendez, K.R. Jackson, J.S. Khalili, J. Wang, C. Bernatchez, K.-M. Lee, G. Lizée, C. Yee
Study supervision: P. Hwu, G. Lizée, C. Yee
Note: Supplementary data for this article are available at Cancer Immunology Research Online (http://cancerimmunolres.aacrjournals.org/).
References
- 1.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–4. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yee C, Thompson JA, Roche P, Byrd DR, Lee PP, Piepkorn M, et al. Melanocyte destruction after antigen-specific immunotherapy of melanoma: direct evidence of t cell-mediated vitiligo. J Exp Med. 2000;192:1637–44. doi: 10.1084/jem.192.11.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brichard V, Van Pel A, Wölfel T, Wölfel C, De Plaen E, Lethé B, et al. The tyrosinase gene codes for an antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med. 1993;178:489–95. doi: 10.1084/jem.178.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–80. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–46. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chandran SS, Paria BC, Srivastava AK, Rothermel LD, Stephens DJ, Dudley ME, et al. Persistence of CTL clones targeting melanocyte differentiation antigens was insufficient to mediate significant melanoma regression in humans. Clin Cancer Res. 2015;21:534–43. doi: 10.1158/1078-0432.CCR-14-2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–57. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newton JM, Cohen-Barak O, Hagiwara N, Gardner JM, Davisson MT, King RA, et al. Mutations in the human orthologue of the mouse underwhite gene (uw) underlie a new form of oculocutaneous albinism, OCA4. Am J Hum Genet. 2001;69:981–8. doi: 10.1086/324340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costin GE, Valencia JC, Vieira WD, Lamoreux ML, Hearing VJ. Tyrosinase processing and intracellular trafficking is disrupted in mouse primary melanocytes carrying the underwhite (uw) mutation. A model for oculocutaneous albinism (OCA) type 4. J Cell Sci. 2003;116(Pt 15):3203–12. doi: 10.1242/jcs.00598. [DOI] [PubMed] [Google Scholar]
- 11.Bin BH, Bhin J, Yang SH, Shin M, Nam YJ, Choi DH, et al. Membrane-associated transporter protein (MATP) regulates melanosomal pH and influences tyrosinase activity. PLoS One. 2015;10:e0129273. doi: 10.1371/journal.pone.0129273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fukamachi S, Shimada A, Shima A. Mutations in the gene encoding B, a novel transporter protein, reduce melanin content in medaka. Nat Genet. 2001;28:381–5. doi: 10.1038/ng584. [DOI] [PubMed] [Google Scholar]
- 13.Graf J, Hodgson R, van Daal A. Single nucleotide polymorphisms in the MATP gene are associated with normal human pigmentation variation. Hum Mutat. 2005;25:278–84. doi: 10.1002/humu.20143. [DOI] [PubMed] [Google Scholar]
- 14.Du J, Fisher DE. Identification of Aim-1 as the underwhite mouse mutant and its transcriptional regulation by MITF. J Biol Chem. 2002;277:402–6. doi: 10.1074/jbc.M110229200. [DOI] [PubMed] [Google Scholar]
- 15.Guedj M, Bourillon A, Combadières C, Rodero M, Dieudé P, Descamps V, et al. Variants of the MATP/SLC45A2 gene are protective for melanoma in the French population. Hum Mutat. 2008;29:1154–60. doi: 10.1002/humu.20823. [DOI] [PubMed] [Google Scholar]
- 16.Harada M, Li YF, El-Gamil M, Rosenberg SA, Robbins PF. Use of an in vitro immunoselected tumor line to identify shared melanoma antigens recognized by HLA-A*0201-restricted T cells. Cancer Res. 2001;61:1089–94. [PubMed] [Google Scholar]
- 17.The Cancer Genome Atlas Research Network. Bethesda, MD: NIH; Available from: http://cancergenome.nih.gov/ [Google Scholar]
- 18.NetMHC3.4 server. Lyngby, Denmark: Center for Biological Sequence Analysis. Available from: http://www.cbs.dtu.dk/services/NetMHC-3.4/
- 19.Kalkum M, Lyon GJ, Chait BT. Detection of secreted peptides by using hypothesis-driven multistage mass spectrometry. Proc Natl Acad Sci USA. 2003;100:2795–800. doi: 10.1073/pnas.0436605100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yi Z, Luo M, Carroll CA, Weintraub ST, Mandarino LJ. Identification of phosphorylation sites in insulin receptor substrate-1 by hypothesis-driven high-performance liquid chromatography-electrospray ionization tandem mass spectrometry. Anal Chem. 2005;77:5693–9. doi: 10.1021/ac050760y. [DOI] [PubMed] [Google Scholar]
- 21.GTex Portal. Broad Institute. Cambridge, MA: Available from: http://www.gtexportal.org/home/ [Google Scholar]
- 22.Khalili JS, Liu S, Rodríguez-Cruz TG, Whittington M, Wardell S, Liu C, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res. 2012;18:5329–40. doi: 10.1158/1078-0432.CCR-12-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bradley SD, Chen Z, Melendez B, Talukder A, Khalili JS, Rodriguez-Cruz T, et al. BRAFV600E Co-opts a conserved MHC class I internalization pathway to diminish antigen presentation and CD8+ T-cell recognition of melanoma. Cancer Immunol Res. 2015;3:602–9. doi: 10.1158/2326-6066.CIR-15-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodriguez-Cruz TG, Liu S, Khalili JS, Whittington M, Zhang M, Overwijk W, et al. Natural splice variant of MHC class I cytoplasmic tail enhances dendritic cell-induced CD8+ T-cell responses and boosts anti-tumor immunity. PLoS One. 2011;6:e22939. doi: 10.1371/journal.pone.0022939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Bleakley M, Yee C. IL-21 influences the frequency, phenotype, and affinity of the antigen-specific CD8 T cell response. J Immunol. 2005;175:2261–9. doi: 10.4049/jimmunol.175.4.2261. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Yee C. IL-21 mediated Foxp3 suppression leads to enhanced generation of antigen-specific CD8+ cytotoxic T lymphocytes. Blood. 2008;111:229–35. doi: 10.1182/blood-2007-05-089375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cancer Cell Line Encyclopedia (CCLE) Cambridge, MA: Broad Institute; http://software.broadinstitute.org/software/cprg/?q=node/11. [Google Scholar]
- 28.Chapuis AG, Lee SM, Thompson JA, Roberts IM, Margolin KA, Bhatia S, et al. Combined IL-21-primed polyclonal CTL plus CTLA4 blockade controls refractory metastatic melanoma in a patient. J Exp Med. 2016;213:1133–9. doi: 10.1084/jem.20152021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA. 2002;99:16168–73. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Griewank KG, Yu X, Khalili J, Sozen MM, Stempke-Hale K, Bernatchez C, et al. Genetic and molecular characterization of uveal melanoma cell lines. Pigment Cell Melanoma Res. 2012;25:182–7. doi: 10.1111/j.1755-148X.2012.00971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palmer DC, Chan CC, Gattinoni L, Wrzesinski C, Paulos CM, Hinrichs CS, et al. Effective tumor treatment targeting a melanoma/melanocyte-associated antigen triggers severe ocular autoimmunity. Proc Natl Acad Sci USA. 2008;105:8061–6. doi: 10.1073/pnas.0710929105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wellbrock C, Arozarena I. Microphthalmia-associated transcription factor in melanoma development and MAP-kinase pathway targeted therapy. Pigment Cell Melanoma Res. 2015;28:390–406. doi: 10.1111/pcmr.12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kono M, Dunn IS, Durda PJ, Butera D, Rose LB, Haggerty TJ, et al. Role of the mitogen-activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Mol Cancer Res. 2006;4:779–92. doi: 10.1158/1541-7786.MCR-06-0077. [DOI] [PubMed] [Google Scholar]
- 34.Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70:5213–9. doi: 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 35.Heemskerk B, Kvistborg P, Schumacher TN. The cancer antigenome. EMBO J. 2013;32:194–203. doi: 10.1038/emboj.2012.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li F, Chen C, Ju T, Gao J, Yan J, Wang P, et al. Rapid tumor regression in an Asian lung cancer patient following personalized neo-epitope peptide vaccination. Oncoimmunology. 2016;5:e1238539. doi: 10.1080/2162402X.2016.1238539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348:803–8. doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest. 2015;125:3413–21. doi: 10.1172/JCI80008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 40.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–11. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Della Vittoria Scarpati G, Fusciello C, Perri F, Sabbatino F, Ferrone S, Carlomagno C, et al. Ipilimumab in the treatment of metastatic melanoma: management of adverse events. Onco Targets Ther. 2014;7:203–9. doi: 10.2147/OTT.S57335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buque A, Bloy N, Aranda F, Castoldi F, Eggermont A, Cremer I, et al. Trial Watch: Immunomodulatory monoclonal antibodies for oncological indications. Oncoimmunology. 2015;4:e1008814. doi: 10.1080/2162402X.2015.1008814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haanen JB, Thienen H, Blank CU. Toxicity patterns with immunomodulating antibodies and their combinations. Semin Oncol. 2015;42:423–8. doi: 10.1053/j.seminoncol.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 44.Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res. 2012;18:6758–70. doi: 10.1158/1078-0432.CCR-12-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bernatchez C, Radvanyi LG, Hwu P. Advances in the treatment of metastatic melanoma: adoptive T-cell therapy. Semin Oncol. 2012;39:215–26. doi: 10.1053/j.seminoncol.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wrzesinski C, Paulos CM, Kaiser A, Muranski P, Palmer DC, Gattinoni L, et al. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J Immunother. 2010;33:1–7. doi: 10.1097/CJI.0b013e3181b88ffc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheli Y, Ohanna M, Ballotti R, Bertolotto C. Fifteen-year quest for microphthalmia-associated transcription factor target genes. Pigment Cell Melanoma Res. 2010;23:27–40. doi: 10.1111/j.1755-148X.2009.00653.x. [DOI] [PubMed] [Google Scholar]
- 48.Yee C, Lizee G, Schueneman AJ. Endogenous T-cell therapy: clinical experience. Cancer J. 2015;21:492–500. doi: 10.1097/PPO.0000000000000158. [DOI] [PubMed] [Google Scholar]
- 49.Raposo G, Marks MS. Melanosomes–dark organelles enlighten endosomal membrane transport. Nat Rev Mol Cell Biol. 2007;8:786–97. doi: 10.1038/nrm2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bradley SD, Melendez B, Talukder A, Lizée G. Trouble at the core: BRAF(V600E) drives multiple modes of T-cell suppression in melanoma. Oncoimmunology. 2016;5:e1078966. doi: 10.1080/2162402X.2015.1078966. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.