

Abstract
Rhabdoid tumor is a rare, highly aggressive malignancy that primarily affects infants and young children. These tumors typically arise in the brain and kidney, although extrarenal, non–central nervous system tumors in almost all soft-tissue sites have been described. SMARCB1 is a member of the SWI/SNF chromatin-remodeling complex and functions as a tumor suppressor in the vast majority of rhabdoid tumors. Patients with germline mutations or deletions affecting SMARCB1 are predisposed to the development of rhabdoid tumors, as well as the genetic disorder schwannomatosis. The current hypothesis is that rhabdoid tumors are driven by epigenetic dysregulation, as opposed to the alteration of a specific biologic pathway. The strategies for novel therapeutic approaches based on what is currently known about rhabdoid tumor biology are presented.
Keywords: rhabdoid, SMARCB1, SWI/SNF
I. INTRODUCTION
Rhabdoid tumor (RT) is a rare and highly malignant tumor that arises predominantly in the brain (referred to as atypical teratoid/RT [AT/RT]), kidney (RT of the kidney [RTK]), or soft tissues (extrarenal RT, malignant RT [MRT]). Frequent sites for extrarenal RTs include the skin, liver, and lung, although tumors in almost all soft tissues, including the orbit, thymus, uterus, bladder, and neck, have been reported. The peak incidence is between 1 and 4 years of age, although classic RTs in adults have been described.
Children, typically in their first year of life, may also present with more than one primary RT, consistent with a genetic predisposition to cancer. These infants typically have a central nervous system (CNS) AT/RT and RTK, or an AT/RT and a lung or liver tumor. Bilateral RTKs occur but are much less frequent than bilateral Wilms tumors of the kidneys, accounting for 2% of RTK cases.1 Brain and spine imaging studies should, therefore, always be performed in a newly diagnosed patient with a renal or extrarenal RT. Similarly, imaging studies to rule out a renal tumor are indicated for a patient with AT/RT.
Epidemiologic studies of RT have been limited by the fact that this is a rare disease. Heck et al.2 performed the first population-based epidemiologic analysis of RT as part of an Air Pollution and Childhood Cancer Study in the state of California. They reported an association of RT with low birthweight, preterm birth, and late-term delivery. Of interest, twin pregnancies were associated with RT, which also was noted by Nicolaides et al.3 and Bourdeaut et al.4 Nicolaides et al.3 and Cecen et al.5 each reported a single case of RT in a patient born after in vitro fertilization, and we are aware of 3 children conceived by in vitro fertilization who developed AT/RTs in the first year of life (unpublished data). Although some studies suggest a small increased risk for cancer with the use of assisted reproductive technologies,6 this remains controversial.7
Histologically, RTs contain characteristic filamentous cytoplasmic inclusions, large nucleoli, and abundant eosinophilic cytoplasm. A variety of neural, epithelial, mesenchymal, or ependymal patterns may also be present, making the histologic appearance quite variable and clinical diagnosis difficult.8 CNS AT/RTs typically comprise rhabdoid cells and areas of primitive neuroepithelial tissue resembling a primitive neuroectodermal tumor (PNET), as well as mesenchymal and/or epithelial elements.9 In the past, this complex histologic pattern routinely led to misclassification of AT/RTs, most often as medulloblastoma/PNET.9,10 Some AT/RTs display only the PNET component, and the diagnosis relies on molecular genetic analysis. It is assumed that the cell of origin for RTs is a primitive stem cell with the capacity for divergent differentiation, possibly derived from the neural crest.8
II. GENETICS OF RT
The majority of RTs arise as a consequence of homozygous inactivation of the SMARCB1/INI1/hSNF5/BAF47 gene.11,12 Loss of expression of the protein permitted the development of an immunohistochemistry (IHC) assay13 that is based on the loss of nuclear expression of the protein in tumor cells, with retained expression in normal cells (Fig. 1). This IHC assay can be used in the vast majority of cases to help make a clinical diagnosis of RT. Loss of expression of SMARCB1 is also observed in other tumors with inactivation of the locus, including epithelioid sarcoma, cribriform neuroepithelial tumor, chordoma, and medullary renal cell carcinoma.14–20 Therefore, while the loss of SMARCB1 expression by IHC is highly sensitive, it is not specific. Correlation with other histologic and immunophenotypic findings, the patient’s age, and the location of the tumor is required to make a clinical diagnosis. RTs may also arise in the setting of a previously benign tumor in both the brain21,22 and peripheral nervous system23,24 following acquisition of a SMARCB1 mutation and/or deletion. The loss of expression of SMARCB1 by IHC clearly distinguishes the rhabdoid areas from the other (less malignant) components of the tumor.
FIG. 1:

Histology of a metastatic abdominal malignant rhabdoid tumor. A: Hematoxylin and eosin staining (magnification ×60) demonstrates the presence of rhabdoid cells. B: Loss of SMARCB1 is seen by immunohistochemistry (magnification ×40). (Reprinted with permission from Dr. Bruce Pawel, Department nof Pathology & Laboratory Medicine, The Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania.)
Versteege et al.11 first reported somatic mutations of the SMARCB1/hSNF5 gene in renal and extrarenal RT, followed shortly thereafter by a report by Biegel et al.12 of germline and somatic mutations in INI1/hSNF5 in patients with RTs of the brain, kidney, and soft tissues.12 SMARCB1 (SWI/SNF related, matrix-associated, actin-dependent regulator of chromatin, subfamily B, member 1) is now the recommended nomenclature to replace human sucrose nonfermenting gene number 5 (hSNF5), integrase interactor 1 (INI1), or 47-Kd Brg1/Brm-associated factor (BAF47).
SMARCB1 is a member of the human SWI/SNF complex.25,26 The SWI/SNF complex acts in an adenosine triphosphate–dependent manner to remodel chromatin and thus leads to the activation and repression of gene transcription. Whole-exome sequencing studies of primary RTs27 have shown that biallelic mutations or copy number alterations of SMARCB1 seem to be both necessary and sufficient to cause cancer; there were no other consistent coding sequence or copy number changes identified. Stabilization of an epigenetically altered genome is thought to contribute to tumorigenesis, but the specific genes that contribute to transformation are not yet known.
Among patients newly diagnosed with RT, 25–30% have a germline alteration of SMARCB1 that predisposed them to cancer.4,28 In our patient cohort the median age at diagnosis for patients with germline SMARCB1 alterations was 6 months (range, 1 day to 5 years) compared with a median age at diagnosis of 1.5 years (range, 1 day to 32 years) for patients with sporadic tumors.
Virtually all of the complete SMARCB1 deletions or larger 22q11.2 germline deletions that include SMARCB1 are de novo. The majority of germline SMARCB1 mutations in patients with RT are also de novo. Interestingly, the germline SMARCB1 deletions more frequently affect the paternal allele, whereas there seems to be a small bias for mutations to be present on the maternal allele, especially when they are inherited.28 Because of the ascertainment bias for a child with a RT, inherited germline mutations often are passed down from an unaffected parent. Because there is reduced penetrance of RT associated with a germline alteration of SMARCB1, the long-term risk for cancer in carriers of mutations or deletions of SMARCB1 is not yet known. Gonadal mosaicism also has been observed in several families28,29; therefore, parents need to be counseled appropriately about their recurrence risks and options for prenatal testing. Schwannomatosis (OMIM 162091) is characterized by the presence of multiple nerve tumors, which are histologically benign but may cause serious morbidity. Patients may also have meningiomas, and, in some cases, the schwannomas may transform into malignant sarcomas, requiring surgical intervention and chemotherapy. Approximately 50–60% of families with schwannomatosis have germline mutations in SMARCB1.30,31 We and others have described families with germline mutations or intragenic deletions or duplications in SMARCB1 in which the adult carriers of the mutations had fibromas or schwannomas and their affected children had RTs. Because the schwannomas may not develop until the third or fourth decade of life, individuals who have a bgermline SMARCB1 alteration must, therefore, be counseled about their own risk for both benign and malignant tumors, in addition to the cancer risks for their offspring. There does seem to be some genotype–phenotype correlation for the types of mutations that occur in schwannomatosis versus RT, as described below.
The spectrum of germline mutations, deletions, and duplications from 70 patients with RT is shown in Fig. 2. Approximately 20% of the germline alterations are deletions in chromosome band 22q11.2 that include all of SMARCB1, whereas 25% of the patients have a partial deletion or duplication involving 1–5 exons of the gene. The remaining patients have a variety of truncating mutations caused by single base point mutations or insertions/deletions leading to a frameshift. Splicesite mutations are the least common type of mutations observed in children who first present with a RT. By contrast, splicesite mutations and point mutations in exons 1 and 9 are more frequent in families with schwannomatosis.30,31
FIG. 2:
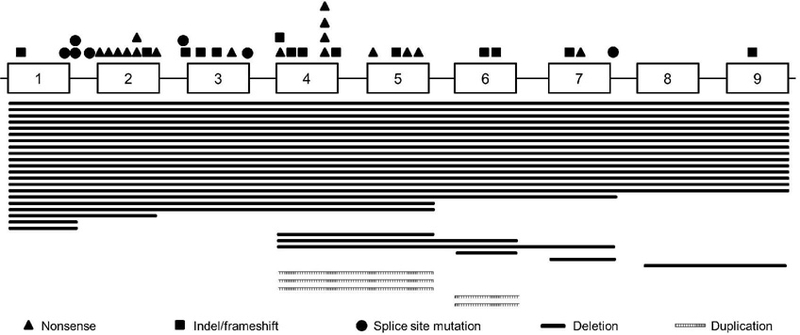
Distribution of germline mutations, deletions, and duplications in SMARCB1 from 70 patients with rhabdoid tumor. Exons, introns, and genetic alterations are not drawn to scale. Stacked symbols are used to identify mutations recurring at the same nucleotide position. Indel, insertion/deletion.
The 3 most frequently detected germline specific mutations in SMARCB1 are c.118C>T in exon 2, c.157C>T in exon 2, and c.472C>T in exon 4.4,12,28 With the exception of the exon 9 frameshift mutations (described below), the same mutations predispose carriers to AT/RT, renal RT, and, to a lesser extent, extrarenal RT. The majority of extrarenal RTs are sporadic and arise as a consequence of homozygous loss of SMARCB1 caused by deletions, unbalanced 22q11.2 translocations, or monosomy 22. The most frequent second hit in patients with a germline mutation is a large 22q deletion or monosomy 22, or a copy number neutral loss of heterozygosity (CN-LOH) generating event that unmasks the mutation or deletion on the remaining allele.
The distribution of SMARCB1-and chromosome 22–inactivating mutations, deletions, and CN-LOH in 200 sporadic AT/RTs, renal RTs, and extrarenal RTs is shown in Table 1. In the majority of tumors (43%) there is a mutation in one allele, and the second copy of the gene is lost as a result of a structural deletion in 22q11.2, monosomy 22, or an acquired CN-LOH event. Compound heterozygous mutations are infrequent in these patients (4%). Partial deletions and duplications are detected in approximately 15% of tumors. Homozygous deletions of exons 1– 9 of SMARCB1 are present in approximately 40% of RTs overall, although there is an unequal distribution with respect to anatomic location. Approximately 25% of AT/RTs, 40% of renal RTs, and 70% of extrarenal RTs have homozygous deletions of the entire locus.
TABLE 1:
Acquired SMARCB1 Alterations in 200 Sporadic Rhabdoid Tumors
| Allele 1 Alteration |
Allele 2 Alteration | Total | |||
|---|---|---|---|---|---|
| Mutation | Partial Gene Deletion / Duplication |
Whole Gene Deletion |
CN-LOH | ||
| Mutation | 8 (4%) | 1 (0.50%) | 58 (29%) | 27 (13.5%) | 94 (47%) |
| Partial gene deletion / duplication |
— | 5 (2.5%) | 14 (7%) | 11 (5.3%) | 30 (15 %) |
| Whole gene deletion |
— | — | 76 (38%) | — | 76 (38%) |
| Total | 8 (4%) | 6 (3%) | 148 (74%) | 38 (19%) | 200 (100%) |
CN-LOH, copy number neutral loss of heterozygosity.
The mutations in sporadic RTs include single base pair point mutations and insertion/deletion or frameshift mutations that are predicted to introduce a novel stop codon (Fig. 3). The majority of mutations result in nonsense-mediated decay, although this has not formally been proven in most cases The highest frequency of coding sequence mutations among the sporadic RTs occurs in exon 9 (Fig. 3). Two single base deletions in codons 381 (c.1143delG) and 382 (c.1145delC) are somatic in origin and are associated exclusively with AT/RT.12 Mutations in exon 2 and exons 4–7 are frequently observed in RTK and AT/RT. Four specific mutations— c.118C T, c.157C>T, c.472C>T, and c.601C>T in exons 2, 2, 4, and 5, respectively—are highly recurrent, although they do not seem to be specific to the brain or kidney.4,12,28 Mutations in exons 1 and 3 are rare, and a mutation in exon 8 has been documented in only one RT in our cohort (unpublished data) in a patient with schwannomatosis.32 Splice site mutations are rare in primary RTs, and missense mutations have not yet been reported.4,28
FIG. 3:

Somatic point mutations (A) and frameshift mutations (B) identified in 200 sporadic rhabdoid tumors. Somatic point mutations and frameshift mutations were identified in a total of 46 and 53 patients, respectively.
The 2 most common mutations in AT/RT are single base deletions in exon 9: c.1143delG and c.1145delC (Fig. 3B). Interestingly, neither of these frameshift mutations has been detected as a predisposing mutation in blood from patients with RT or schwannomatosis. These 2 frameshift mutations are not predicted to be subject to nonsense-mediated decay, and theoretically this would result in the addition of 100 amino acids to the protein. Similar to other RTs with coding sequence alterations, there is no expression of the protein by IHC in AT/RTs with these 2 exon 9 deletions.13 It is possible that this mutation functions as a dominant-negative mutation during early development, which is an area for future research.
Although SMARCB1 is the predominant gene altered in RTs, approximately 2–3% of tumors with rhabdoid histology retain expression of the SMARCB1 protein on IHC and do not display inactivating mutations in the gene. A small number of families and patients with RT with germline or somatic mutations of SMARCA4, which is the primary ATPase in the SWI/SNF complex, have now been reported.33,34 A variety of solid tumors in children and adults, such as medulloblastoma, the most common malignant brain tumor in children, have mutations in SMARCA4. To date, however, the only other tumor type to demonstrate biallelic inactivation of SMARCA4, consistent with a cancer-predisposing germline mutation and second somatic alteration, is small cell carcinoma of the ovary, hypercalcemic type (SCCOHT).35–37 Based on the relatively early age at presentation, and the presence of rhabdoid-appearing cells on histology, it has been proposed that SCCOHT represents another type of extrarenal RT.38 The germline mutations in SCCOHT included both missense and truncating mutations, typically with loss of the wild-type allele as the second inactivating event in the tumor.
Interestingly, mutations in a variety of SWI/SNF proteins, including ARID1A, ARID1B, SMARCA2, SMARCA4, SMARCE1 and SMARCB1, have recently been reported in patients with genomic disorders such as Coffin-Siris syndrome (CSS)39–43 or Nicolaides Barrister syndrome43–45 who do not seem to be at increased risk for cancer, as well as in patients with unexplained intellectual disability or autism.46,47 Most patients with CSS and SMARCB1 alterations have heterozygous missense mutations, which are so far distinct from the typical nonsense mutations that occur in patients with RT or the splice site mutations that often occur in familial schwannomatosis. We studied 1 patient with CSS and a missense mutation in exon 9 who developed multiple schwannomas but not RT.48 As whole-genome sequencing moves into the area of prenatal testing, the prediction of whether such mutations will result in a genomic disorder or increased risk for malignancy will become extremely challenging.
III. HISTORICAL TREATMENT AND OUTCOMES
Patients with AT/RT have, until recently, been treated according to institutional preference or nonspecific infant brain tumor protocols, combining surgery, possible radiation therapy, and chemotherapy. In general, drugs have included some combination of platinum agents, epipodophyllotoxins, oxazaphosphorines, vinca alkaloids, methotrexate, and anthracycline, with or without intrathecal directed medications (methotrexate, hydrocortisone, cytarabine, mafosfamide) and/or high-dose chemotherapy with stem cell rescue.49–56 While the optimal “standard” therapy remains debated, the prognosis has improved from early reports to nearly 50% overall for AT/RT, but for RTK it remains unchanged at approximately 20–25%.1,57,58 Efforts by the Children’s Oncology Group (COG) to improve the cure rate of AT/RT (protocol ACNS0333 [www.clinicaltrials.gov identifier NCT00653068]) use a combination of surgery; 2 cycles of induction chemotherapy (cisplatin, cyclophosphamide, etoposide, vincristine, methotrexate); consolidation therapy with 3 cycles of high-dose chemotherapy with stem cell rescue (thiotepa, carboplatin); and age- and stage-directed radiation therapy. By contrast, the Dana Farber Consortium AT/RT study (www.clinicaltrials.gov identifier NCT00084838) uses a more protracted approach to combination therapy with surgery, age- and stage-directed radiation, and chemotherapy lasting approximately 1 year in duration, in part based on historic rhabdomyosarcoma group therapy, including vincristine, dactinomycin, cyclophosphamide, cisplatin, doxorubicin, temozolomide, and intrathecal methotrexate, cytarabine, and hydrocortisone.51 In Europe the registry study (Eu-Rhab) for all RT (AT/RT, RTK, MRT) recommends using combination therapy including surgery, radiotherapy, and chemotherapy (vincristine, dactinomycin, cyclophosphamide, doxorubicin, ifosfamide, carboplatin, etoposide), and intrathecal methotrexate and permissive use of high-dose chemotherapy with stem cell rescue (carboplatin, thiotepa).59
Before study AREN0321 (www.clinicaltrials.gov identifier NCT00335556), in the United States, RTKs specifically were historically treated along-side Wilms’ tumors in National Wilms’ Tumor Study trials with regimens used for the treatment of Wilms’ tumors, including vincristine, dactinomycin, and doxorubicin, with or without cyclophosphamide. The outcomes attained with these regimens were poor.1,57 NWTS-5 adopted a different treatment strategy consisting of carboplatin and etoposide alternating with cyclophosphamide (regimen RTK). Preliminary analysis of patients treated with regimen RTK revealed no clear improvement compared with previous studies, leading to study closure. Subsequent case reports demonstrated that ifosfamide-carboplatin-etoposide or ifosfamide-etoposide chemotherapy alternating with vincristine-doxorubicin-cyclophosphamide can be efficacious against RTK,52,60,61 providing the rationale for study AREN0321 regimen UH-1 (vincristine-doxorubicin-cyclophosphamide alternating with cyclophosphamide, carboplatin, etoposide), as well as the similar Eu-Rhab registry regimen. In study AREN0321, patients with stage 4 measurable RTK or MRT were initially eligible for vincristine/irinotecan window therapy, but none of the 3 patients with RT who were enrolled in the window responded, leading to closure of such window therapy for patients with RT. Preliminary analysis of regimen UH-1 does not, unfortunately, demonstrate clear improved outcome compared with historical data; however, further analyses are in process. While some authors discuss a potential role for even higher doses of alkylator therapy and/or high-dose chemotherapy for RTK and MRT, analogous to approaches drafted for AT/RT,62,63 no formal trial has demonstrated a therapeutic advantage in the treatment of non-CNS RT, and any further intensification of therapy is challenged by the fact that current regimens already maintain a toxicity-related mortality of approximately 5%, as well as significant morbidity.64
In sum, while the prognosis for select patients—particularly those with localized RT associated with an older age and lower stage disease—has improved some,1,65,66 the overall outcomes of RT remain poor despite maximized therapy intensity, mandating the discovery and integration of targeted novel therapy, which is likely to emerge from a deeper understanding of RT biology and additional preclinical investigation
IV. RT BIOLOGY AND TARGETED THERAPY
Current preclinical investigations aiming to expand therapy options and improve the survival of infants and children with RT have focused largely on the specific interrogation of SMARCB1-related biology and potential therapeutic targets (Table 2), as well as nonspecific preclinical efforts conducted through the Pediatric Preclinical Testing Program (PPTP), using 2 RT cell lines and xenografts for testing of new agents emerging from the pharmaceutical industry.
TABLE 2:
Molecular Targets and Potential Inhibitors of Rhabdoid Tumors
| Epigenetic Target |
Mechanism of Action |
Agent | Pediatric Development Comments |
|---|---|---|---|
| EZH2 | Histone methylation | E7458, EPZ-6438, GSK2816126 |
Agents pending phase I investigation |
| DNMT | DNA methylation | Decitabine, 5-Azacitadine SGI-110 |
Decitabine: phase I single and combination studies complete |
| HDAC | Histone deacetylation | Vorinostat, Valproic acid, Romidepsin, Panobinostat, Quisinostat, Others |
Vorinostat: phase I single and combination studies complete/ongoing |
| CDK4/cyclinD | G1 cell cycle arrest | Palbociclib, LEE011, P276-00, LY2835219 |
LEE-011: pediatric phase I/II (rhabdoid) |
| Aurora-A-kinase | Antimitotic | Alisertib, TAS-119, ENMD-2076, AMG900 |
Alisertib: pediatric phase II (rhabdoid) |
CDK4, cyclin-dependent kinase 4; DNMT, DNA methyltransferase; EZH2, enhancer of zeste 2 polycomb repressor complex 2; HDAC, histone deacetylase.
SMARCB1 plays a critical role in epigenetic regulation, cell cycle progression, and signaling crosstalk, all of which provide fertile ground for preclinical and clinical investigation. SMARCB1 functions as a classic tumor suppressor and is the primary gene responsible for malignant RT pathophysiology. While homozygous inactivation of Smarcb1 in mice exhibits embryonic lethality, 20% of heterozygous Smarcb1 mice that are normal at birth ultimately develop sarcomas at a median age of 1 year, following a second hit to the Smarcb1 locus. All mice with conditional biallelic inactivation of Smarcb1 develop cancer, with a median onset of 11 weeks, making this one of the most aggressive cancer predisposition genotype–phenotype correlations yet described.67,68 As mentioned, RTs are generally diploid and genomically stable, lacking additional recurrent gene amplifications or deletions beyond Smarcb1 loss. The SWI/SNF complex, perturbed in the setting of Smarcb1 loss, acts in an adenosine triphosphate–dependent manner to remodel chromatin, regulating gene transcription and DNA repair. Considering the lack of cooperating mutations and aggressive neoplasia, RT is perhaps the quintessential tumor driven by epigenetic dysregulation.
A. Epigenetic Targeting
The evolving field of epigenetics has provided access to targeted therapy aiming to alter methylation and acetylation patterns within cancer cells.69
Somewhat speculative at this point, the loss of SMARCB1 is postulated to result in a global failure to release the repressive H3K27 trimethylation mark present on bivalently modified histones, mediated by the polycomb complex 2, resulting in widespread epigenetic modifications and leading to arrested development and abnormal proliferation, potentially via histone methylation processes.70 The polycomb group family of proteins represses transcription by mediating histone 3 lysine-27 trimethylation. Two members of the polycomb complex 2, CBX6 and EZH2—the latter a histone methyltransferase—are upregulated in RT (Fig. 4). ZNF217, an organizer of repressive histones, is also significantly upregulated in RT and is capable of demethylating H3K4me3 and methylating H3K27 through interaction with EZH2.70,71 EZH2 inhibitors are now in clinical development (Table 2), and one report by investigators associated with the company Epizyme documents in vitro and in vivo activity against RT, albeit delayed,72 potentially limiting efficacy in more rapidly dividing and morbidly aggressive RT. EZH2 inhibition also has been shown to sensitize RT cells to the effects of radiation.73
FIG. 4:

Regulation of gene expression by SWI/SNF and Polycomb repressive complex 2 (PRC2) complexes. During lineage-specific differentiation, the SWI/SNF complex, which includes SMARCB1, interacts with transcription factors, histone acetyltransferases, and transcriptional regulators to activate expression of target genes. Acetylation of histone H3K27 is present at transcriptionally active genes. Opposing the SWI/SNF complex is the PRC2 complex, which contains EZH2. PRC2 interacts with DNA methyltransferases and histone deacetylases to silence gene expression. The transcriptionally inactive genes are marked by methylation at histone H3K27. In rhabdoid tumors the loss of SMARCB1 expression prohibits the normal functions of the SWI/SNF complex, resulting in altered gene expression.
In addition to EZH2 histone methyltransferase targeting, preclinical investigations have demonstrated anti-RT effects with histone deacetylase inhibitors (HDACi) and demethylating agents.74–79 HDACi sensitization of RTs to radiation therapy and sensitization to anthracycline-based therapy also has been demonstrated in RT cell lines.74,75,80
Importantly, the strategy for future treatment of patients with RT should include the addition of new agents concurrent with, or before, standard chemotherapy. DNA damage response pathways, apoptosis signaling components, DNA repair components, and drug transporters each include genes subject to epigenetic control in cancer and relevant to chemotherapy disease resistance.81–83 For example, multidrug (doxorubicin and cisplatin)–resistant human MCF-7 breast adenocarcinoma cells demonstrate loss of global DNA methylation, loss of histone H4 lysine 20 trimethylation, increased phosphorylation of histone H2 serine 10, and diminished expression of Suv4–20h2 histone methyltransferase compared with parental MCF-7 cells.84 Subsequent investigations have demonstrated that DNA methyltransferase inhibition with 5-azacytidine reduces MDR1 promoter methylation in MCF-7 cells, with changes in chromatin structure.85 MLL1, a histone methyltransferase specific for H3K4 that is transcriptionally activated through interaction with SMARCB1, as previously discussed, has been shown to be required for MDR1 promoter methylation and chemoresistance.86 Chemotherapeutic drugs can upregulate MDR1 with associated H3 acetylation and induction of methylated H32K4 within the MDR1 locus.87 Similar to MDR1, ABCG2 gene expression is dependent on DNA methylation.88 EZH2, upregulated in RT, is essential for chemotherapy resistance in cisplatin resistant cell lines, likely through H3K27 methylation.89 Removal of H3K27 methylation resensitizes drug-resistant ovarian carcinoma cells to cisplatin by in-creasing DNA-platinum adduct formation resulting from increased access of cisplatin to target DNA sequences.90 Further, DNA methyltransferase inhibition enhances chemosensitivity to cisplatin.91 Last, microRNAs themselves, which are suppressed in RT, can mediate drug sensitivity.92 Specifically, suppression of mir451 imparts doxorubicin resistance in MCF-7 cells.93 Thus, it is possible that DNA or histone methylation inhibitors may sensitize cells to the effects of standard chemotherapy via a reversal of resistance mechanisms.
The COG completed a trial of decitabine in combination with doxorubicin and cyclophosphamide in children, which is of potential interest in RT, though concerns regarding adequate pharmacodynamic demethylation and toxicity have thus far limited advancement.94 In pediatric-focused trials, decitabine/vorinostat chemotherapy combination therapies have advanced with anthracycline-based combination therapy in relapsed leukemia (www.clinicaltrials.gov identifier NCT01483690) and with alkylator therapy for brain tumors.95
B. CDK4/CDK6/CyclinD/RB
Reports to date have demonstrated that SMARCB1 loss can promote cell cycle progression resulting from upregulation of targets of the p16INK4a-Rb-E2F pathway, primarily including cyclin D1 (upregulated in primary RTs) as well as several cyclin-dependent kinases (CDKs).70,96 Rb family loss has been shown to increase rhabdoid tumorigenesis,97 and reintroduction of SMARCB1 into RT cell lines leads to G1 arrest and decreased cyclin D1 transcription,98 whereas ablation of CyclinD1 abrogates malignant RT evolution in mouse models.99 Tumor development in Smarcb1-deficient mice is greatly accelerated in the absence of functional p53 protein.100 These findings suggest a cooperative effect between SMARCB1 and the pRB, CyclinD1, and Tp53 pathways.
Flavopiridol, a nonspecific CDK inhibitor, has inhibited RT cell growth with synergy demonstrated with tamoxifen in tumor models.101 The CDK4/6 inhibitor LEE011 is currently in phase I/II investigation in pediatric patients with perturbed RB/CyclinD1/CDK4/6 pathway signaling, with specific focus on RT and neuroblastoma (www.clinicaltrials.gov identifier NCT01747876). Other CDK4/6 inhibitors are in active development (Table 2).
C. Aurora-A-Kinase
Aurora-A-kinase is expressed at high levels in RT and is repressible with SMARCB1 reintroduction into RT cells via transcriptional downregulation. In addition, small interfering RNA targeting of Aurora A induces RT cell death in vitro,102 and additional data from PPTP testing of the aurora-A-kinase inhibitor MLN8237 (Alisertib) demonstrated in vivo activity in RT xenografts.103 Such data prompted the COG Rhabdoid Tumor Working Group to endorse the phase II trial of Alisertib in pediatric solid tumors via the inclusion of an RT stratum (ADVL0921; www.clinicaltrials.gov identifier NCT01154816). Unfortunately, none of 4 patients demonstrated an objective response to further accruals before study closure. Nonetheless, an institutional study (www.clinicaltrials.gov identifier NCT02114229) continues to investigate Alisertib in patients with RT, either as a single agent for recurrent/refractory disease or as part of combination therapy with chemotherapy, surgery, and radiation to treat AT/RT. While trial NCT02114229 is not designed to test concurrent Alisertib and radiation exposures, preclinical data suggest a potential role for Alisertib as a radiation sensitizer in the treatment of RT.104
D. Additional Potential Targets
SMARCB1 loss leads to increased expression of GLI1, noted in RT primary tumors, supporting a role in the biology of the sonic hedgehog pathway and suggesting that downstream inhibition of the pathway is worth further preclinical and possibly clinical testing.59 Microarray experiments have further suggested interferon therapy or downmodulation of PLK1,105 as well as osteopontin and endostatin,70 as worthy of further consideration. Additional genomic studies are underway as part of the National Cancer Institute–sponsored “Therapeutically Applicable Research to Generate Effective Treatments” (TARGET) initiative, inclusive of RTs derived from the COG biobank (AREN10B2).
E. PPTP Investigation in RT
The PPTP uses several RT cell lines (BT-12 and CHLA-266) and xenografts (BT29, KT16, KT14, KT12) in the study of new agents. Interestingly, these studies started with the validation of traditional chemotherapeutic agents such as vincristine, cyclophosphamide, and cisplatin. While widely used, vincristine failed to show an effect in BT29 and yielded growth delay in KT14 and KT12 only.106,107 Cyclophosphamide and cisplatin therapy each resulted in a partial response and a complete response in KT16, respectively (a xenograft that trended toward increased sensitivity with other agents as well), and growth delay in BT29 and KT14. Additional activity was noted for AZD2171 (vascular endothelial growth factor [VEGF] receptor inhibitor), ispinesib (antimitotic), SU11248 (VEGF receptor inhibitor), rapamycin (mammalian target of rapamycin inhibitor), SVV001 (oncolytic), PR-104 (alkylator), GSK923295A (centromere protein E inhibitor), MLN8237 (aurora-A-kinase inhibitor), cabozantinib (VEGF receptor/c-Met inhibitor), and RG7112 (MDM2 inhibitor).103,108–116 Interesting additional negative results included in vitro assessment of the HDACi vorinostat (SAHA), showing high half-maximal inhibitory concentration (>2 µM) values, as well as limited activity with the HDACi JNJ-26481585 (quisinostat),117,118 limited growth delay in vivo with topotecan,119 and lack of in vitro or in vivo activity with CDK1/2/5/9 inhibitor SCH727965 (Dinaciclib).120 While correlations of such preclinical testing with clinical activity in patients remains unproven, these data suggest several classes of drugs worth consideration of further clinical investigation, including VEGF multi–tyrosine kinase inhibitors as well as novel antimitotic therapies.
V. CONCLUSIONS AND FUTURE DIRECTIONS
RT remains a biologically fascinating, quintessential model of epigenetically controlled aggressive neo-The efforts of registry studies, cooperative group biological and clinical trials, and independent investigator– driven exploration of rhabdoid genomics; exploration of SMARCB1-driven biology, targeting epigenetic determinants of disease; and collaboration with the pharmaceutical industry to advance further preclinical and clinical testing are all imperative to advancing this important cause. Importantly, not only will such advances benefit patients and families affected by rhabdoid and related tumors, the results of such investigations are likely to be generalizable to a wide array of SMARCB1-dependent cancers and the epigenetic control of neoplasia in general.
ACKNOWLEDGMENTS
The authors thank Dr. Bruce Pawel for the rhabdoid tumor images in Fig. 1. The authors’ work is sup-ported by the National Institutes of Health (grant no. CA46274, to J.A.B.) and by the Children’s Brain Tumor Research Foundation (to J.A.B.). J.J.R. is sup-ported by a National Institutes of Health/National Institute of General Medical Sciences Medical Genetics Research Training Grant (T32-GM008638).
ABBREVIATIONS:
- AT/RT
atypical teratoid/rhabdoid tumor
- CNS
central nervous system
- COG
Children’s Oncology Group
- CSS
Coffin-Siris syndrome
- CN-LOH
copy number neutral loss of heterozygosity
- CDK
cyclin-dependent kinase
- HDACi
histone deacetylase inhibitor
- IHC
immunohistochemistry
- MRT
malignant rhabdoid tumor
- PPTP
Pediatric Preclinical Testing Program
- PNET
primitive neuroectodermal tumor
- RT
rhabdoid tumor
- RTK
rhabdoid tumor of the kidney
- SCCOHT
small cell carcinoma of the ovary, hypercalcemic type
- SMARCB1
SWI/SNF related, matrix-associated, actin-dependent regulator of chromatin, subfamily B, member 1
- VEGF
vascular endothelial growth factor
REFERENCES
- 1.Tomlinson GE, Breslow NE, Dome J, Guthrie KA, Norkool P, Li S, Thomas PR, Perlman E, Beckwith JB, D’Angio GJ, Green DM. Rhabdoid tumor of the kidney in the National Wilms’ Tumor Study: age at diagnosis as a prognostic factor. J Clin Oncol 2005;23(30):7641–5. [DOI] [PubMed] [Google Scholar]
- 2.Heck JE, Wu J, Lombardi C, Qiu J, Meyers TJ, Wilhelm M, Cockburn M, Ritz B. Childhood cancer and traffic-related air pollution exposure in pregnancy and early life. Environ Health Perspect 2013;121(11–12):1385–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicolaides T, Tihan T, Horn B, Biegel J, Prados M, Banerjee A. High-dose chemotherapy and autologous stem cell rescue for atypical teratoid/rhabdoid tumor of the central nervous system. J Neurooncol 2010;98(1):117–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bourdeaut F, Lequin D, Brugieres L, Reynaud S, Dufour C, Doz F, Andre N, Stephan JL, Perel Y, Oberlin O, Orbach D, Bergeron C, Rialland X, Freneaux P, Ranchere D, Figarella-Branger D, Audry G, Puget S, Evans DG, Pinas JC, Capra V, Mosseri V, Coupier I, Gauthier-Villars M, Pierron G, Delattre O. Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res 2011;17(1):31–8. [DOI] [PubMed] [Google Scholar]
- 5.Cecen E, Gunes D, Uysal KM, Yuceer N, Ozer E. Atypical teratoid/rhabdoid tumor in an infant conceived by in vitro fertilization. Childs Nerv Syst 2010;26(2):263–6. [DOI] [PubMed] [Google Scholar]
- 6.Kallen B, Finnstrom O, Lindam A, Nilsson E, Nygren KG, Olausson PO. Cancer risk in children and young adults conceived by in vitro fertilization. Pediatrics 2010;126(2):270–6. [DOI] [PubMed] [Google Scholar]
- 7.Lerner-Geva L, Toren A, Chetrit A, Modan B, Mandel M, Rechavi G, Dor J. The risk for cancer among children of women who underwent in vitro fertilization. Cancer 2000;88(12):2845–7. [DOI] [PubMed] [Google Scholar]
- 8.Parham DM, Weeks DA, Beckwith JB. The clinicopathologic spectrum of putative extrarenal rhabdoid tumors. An analysis of 42 cases studied with immunohis-tochemistry or electron microscopy. Am J Surg Pathol 1994;18(10):1010–29. [DOI] [PubMed] [Google Scholar]
- 9.Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg 1996;85(1):56–65. [DOI] [PubMed] [Google Scholar]
- 10.Burger PC, Yu IT, Tihan T, Friedman HS, Strother DR, Kepner JL, Duffner PK, Kun LE, Perlman EJ. Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol 1998;22(9):1083– 92. [DOI] [PubMed] [Google Scholar]
- 11.Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998;394(6689):203–6. [DOI] [PubMed] [Google Scholar]
- 12.Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B. Germline and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 1999;59(1):74–9. [PubMed] [Google Scholar]
- 13.Judkins AR, Mauger J, Ht A, Rorke LB, Biegel JA. Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am J Surg Pathol 2004;28(5):644–50. [DOI] [PubMed] [Google Scholar]
- 14.Le Loarer F, Zhang L, Fletcher CD, Ribeiro A, Singer S, Italiano A, Neuville A, Houlier A, Chibon F, Coindre JM, Antonescu CR. Consistent SMARCB1 homozygous deletions in epithelioid sarcoma and in a subset of myoepithelial carcinomas can be reliably detected by FISH in archival material. Genes Chromosomes Cancer 2014;53(6):475–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sullivan LM, Folpe AL, Pawel BR, Judkins AR, Biegel JA. Epithelioid sarcoma is associated with a high percentage of SMARCB1 deletions. Mod Pathol 2013;26(3):385–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hasselblatt M, Oyen F, Gesk S, Kordes U, Wrede B, Bergmann M, Schmid H, Fruhwald MC, Schneppenheim R, Siebert R, Paulus W. Cribriform neuroepithelial tumor (CRINET): a nonrhabdoid ventricular tumor with INI1 loss and relatively favorable prognosis. J Neuropathol Exp Neurol 2009;68(12):1249–55. [DOI] [PubMed] [Google Scholar]
- 17.Arnold MA, Stallings-Archer K, Marlin E, Grondin R, Olshefski R, Biegel JA, Pierson CR. Cribriform neuroepithelial tumor arising in the lateral ventricle. Pediatr Dev Pathol 2013;16(4):301–7. [DOI] [PubMed] [Google Scholar]
- 18.Liu Q, Galli S, Srinivasan R, Linehan WM, Tsokos M, Merino MJ. Renal medullary carcinoma: molecular, immunohistochemistry, and morphologic correlation. Am J Surg Pathol 2013;37(3):368–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calderaro J, Moroch J, Pierron G, Pedeutour F, Grison C, Maille P, Soyeux P, de la Taille A, Couturier J, Vieillefond A, Rousselet MC, Delattre O, Allory Y. SMARCB1/INI1 inactivation in renal medullary carcinoma. Histopathology 2012;61(3):428–35. [DOI] [PubMed] [Google Scholar]
- 20.Mobley BC, McKenney JK, Bangs CD, Callahan K, Yeom KW, Schneppenheim R, Hayden MG, Cherry AM, Gokden M, Edwards MS, Fisher PG, Vogel H. Loss of SMARCB1/INI1 expression in poorly differentiated chordomas. Acta Neuropathol 2010;120(6):745–53. [DOI] [PubMed] [Google Scholar]
- 21.Allen JC, Judkins AR, Rosenblum MK, Biegel JA. Atypical teratoid/rhabdoid tumor evolving from an optic pathway ganglioglioma: case study. Neuro Oncol 2006;8(1):79–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chacko G, Chacko AG, Dunham CP, Judkins AR, Biegel JA, Perry A. Atypical teratoid/rhabdoid tumor arising in the setting of a pleomorphic xanthoastrocytoma. J Neurooncol 2007;84(2):217–22. [DOI] [PubMed] [Google Scholar]
- 23.Carter JM, O’Hara C, Dundas G, Gilchrist D, Collins MS, Eaton K, Judkins AR, Biegel JA, Folpe AL. Epithelioid malignant peripheral nerve sheath tumor arising in a schwannoma, in a patient with “neuroblastomalike” schwannomatosis and a novel germline SMARCB1 mutation. Am J Surg Pathol 2012;36(1):154–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rizzo D, Freneaux P, Brisse H, Louvrier C, Lequin D, Nicolas A, Ranchere D, Verkarre V, Jouvet A, Dufour C, Edan C, Stephan JL, Orbach D, Sarnacki S, Pierron G, Parfait B, Peuchmaur M, Delattre O, Bourdeaut F. SMARCB1 deficiency in tumors from the peripheral nervous system: a link between schwannomas and rhabdoid tumors? Am J Surg Pathol 2012;36(7):964–72. [DOI] [PubMed] [Google Scholar]
- 25.Phelan ML, Sif S, Narlikar GJ, Kingston RE. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol Cell 1999;3(2):247–53. [DOI] [PubMed] [Google Scholar]
- 26.Wang W, Xue Y, Zhou S, Kuo A, Cairns BR, Crabtree GR. Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev 1996;10(17):2117–30. [DOI] [PubMed] [Google Scholar]
- 27.Lee RS, Stewart C, Carter SL, Ambrogio L, Cibulskis K, Sougnez C, Lawrence MS, Auclair D, Mora J, Golub TR, Biegel JA, Getz G, Roberts CW. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 2012;122(8):2983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eaton KW, Tooke LS, Wainwright LM, Judkins AR, Biegel JA. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 2011;56(1):7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sevenet N, Sheridan E, Amram D, Schneider P, Handgretinger R, Delattre O. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet 1999;65(5):1342–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith MJ, Wallace AJ, Bowers NL, Eaton H, Evans DG. SMARCB1 mutations in schwannomatosis and genotype correlations with rhabdoid tumors. Cancer Genet 2014;207(9):373–8. [DOI] [PubMed] [Google Scholar]
- 31.Smith MJ, Wallace AJ, Bowers NL, Rustad CF, Woods CG, Leschziner GD, Ferner RE, Evans DG. Frequency of SMARCB1 mutations in familial and sporadic schwannomatosis. Neurogenetics 2012;13(2):141–5. [DOI] [PubMed] [Google Scholar]
- 32.Hadfield KD, Newman WG, Bowers NL, Wallace A, Bolger C, Colley A, McCann E, Trump D, Prescott T, Evans DG. Molecular characterisation of SMARCB1 and NF2 in familial and sporadic schwannomatosis. J Med Genet 2008;45(6):332–9. [DOI] [PubMed] [Google Scholar]
- 33.Hasselblatt M, Gesk S, Oyen F, Rossi S, Viscardi E, Giangaspero F, Giannini C, Judkins AR, Fruhwald MC, Obser T, Schneppenheim R, Siebert R, Paulus W. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 2011;35(6):933–5. [DOI] [PubMed] [Google Scholar]
- 34.Schneppenheim R, Fruhwald MC, Gesk S, Hasselblatt M, Jeibmann A, Kordes U, Kreuz M, Leuschner I, Martin Subero JI, Obser T, Oyen F, Vater I, Siebert R. Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet 2010;86(2):279–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramos P, Karnezis AN, Craig DW, Sekulic A, Russell ML, Hendricks WP, Corneveaux JJ, Barrett MT, Shumansky K, Yang Y, Shah SP, Prentice LM, Marra MA, Kiefer J, Zismann VL, McEachron TA, Salhia B, Prat J, D’Angelo E, Clarke BA, Pressey JG, Farley JH, Anthony SP, Roden RB, Cunliffe HE, Huntsman DG, Trent JM. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat Genet 2014;46(5):427–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Witkowski L, Carrot-Zhang J, Albrecht S, Fahiminiya S, Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet 2014;46(5):438–43. [DOI] [PubMed] [Google Scholar]
- 37.Jelinic P, Mueller JJ, Olvera N, Dao F, Scott SN, Shah R, Gao J, Schultz N, Gonen M, Soslow RA, Berger MF, Levine DA. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat Genet 2014;46(5):424–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foulkes WD, Clarke BA, Hasselblatt M, Majewski J, Albrecht S, McCluggage WG. No small surprise -small cell carcinoma of the ovary, hypercalcaemic type, is a malignant rhabdoid tumour. J Pathol 2014;233(3):209–14. [DOI] [PubMed] [Google Scholar]
- 39.Santen GW, Aten E, Sun Y, Almomani R, Gilissen C, Nielsen M, Kant SG, Snoeck IN, Peeters EA, Hilhorst-Hofstee Y, Wessels MW, den Hollander NS, Ruivenkamp CA, van Ommen GJ, Breuning MH, den Dunnen JT, van Haeringen A, Kriek M. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin Siris syndrome. Nat Genet 2012;44(4):379–80. [DOI] [PubMed] [Google Scholar]
- 40.Santen GW, Aten E, Vulto-van Silfhout AT, Pottinger C, van Bon BW, van Minderhout IJ, Snowdowne R, van der Lans CA, Boogaard M, Linssen MM, Vijfhuizen L, van der Wielen MJ, Vollebregt MJ, the Coffin-Siris Consortium, Breuning MH, Kriek M, van Haeringen A, den Dunnen JT, Hoischen A, Clayton-Smith J, de Vries BB, Hennekam RC, van Belzen MJ, Almureikhi M, Baban A, Barbosa M, Ben-Omran T, Berry K, Bigoni S, Boute O, Brueton L, van der Burgt I, Canham N, Chandler KE, Chrzanowska K, Collins AL, de Toni T, Dean J, den Hollander NS, Flore LA, Fryer A, Gardham A, Graham JM, Jr, Harrison V, Horn D, Jongmans MC, Josifova D, Kant, Kapoor S, Kingston H, Kini U, Kleefstra T, Krajewska-Walasek M, Kramer N, Maas SM, Maciel P, Mancini GM, Maystadt I, McKee S, Milunsky JM, Nampoothiri S, Newbury-Ecob R, Nikkel SM, Parker MJ, Perez-Jurado LA, Robertson SP, Rooryck C, Shears D, Silengo M, Singh A, Smigiel R, Soares G, Splitt M, Stewart H, Sweeney E, Tassabehji M, Tuysuz B, van Eerde AM, Vincent-Delorme C, Wilson LC, Yesil G. Coffin-Siris syndrome and the BAF complex: genotypephenotype study in 63 patients. Hum Mutat 2013;34(11):1519–28. [DOI] [PubMed] [Google Scholar]
- 41.Tsurusaki Y, Okamoto N, Ohashi H, Mizuno S, Mat-sumoto N, Makita Y, Fukuda M, Isidor B, Perrier J, Aggarwal S, Dalal A, Al-Kindy A, Liebelt J, Mowat D, Nakashima M, Saitsu H, Miyake N, Matsumoto N. Coffin-Siris syndrome is a SWI/SNF complex disorder. Clin Genet 2014;85(6):548–54. [DOI] [PubMed] [Google Scholar]
- 42.Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, Kaname T, Naritomi K, Kawame H, Wakui K, Fukushima Y, Homma T, Kato M, Hiraki Y, Yamagata T, Yano S, Mizuno S, Sakazume S, Ishii T, Nagai T, Shiina M, Ogata K, Ohta T, Niikawa N, Miyatake S, Okada I, Mizuguchi T, Doi H, Saitsu H, Miyake N, Matsumoto N. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet 2012;44(4):376–8. [DOI] [PubMed] [Google Scholar]
- 43.Wieczorek D, Bogershausen N, Beleggia F, Steiner-Haldenstatt S, Pohl E, Li Y, Milz E, Martin M, Thiele H, Altmuller J, Alanay Y, Kayserili H, Klein-Hitpass L, Bohringer S, Wollstein A, Albrecht B, Boduroglu K, Caliebe A, Chrzanowska K, Cogulu O, Cristofoli F, Czeschik JC, Devriendt K, Dotti MT, Elcioglu N, Gener B, Goecke TO, Krajewska-Walasek M, Guillen-Navarro E, Hayek J, Houge G, Kilic E, Simsek-Kiper PO, Lopez-Gonzalez V, Kuechler A, Lyonnet S, Mari F, Marozza A, Mathieu Dramard M, Mikat B, Morin G, Morice-Picard F, Ozkynay F, Rauch A, Renieri A, Tinschert S, Utine GE, Vilain C, Vivarelli R, Zweier C, Nurnberg P, Rahmann S, Vermeesch J, Ludecke HJ, Zeschnigk M, Wollnik A comprehensive molecular study on Coffin-Siris and Nicolaides-Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum Mol Genet 2013;22(25):5121– 35. [DOI] [PubMed] [Google Scholar]
- 44.Wolff D, Endele S, Azzarello-Burri S, Hoyer J, Zweier M, Schanze I, Schmitt B, Rauch A, Reis A, Zweier In-frame deletion and missense mutations of the C-terminal helicase domain of SMARCA2 in three patients with Nicolaides-Baraitser syndrome. Mol Syndromol 2012;2(6):237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Houdt JK, Nowakowska BA, Sousa SB, van Schaik BD, Seuntjens E, Avonce N, Sifrim A, Abdul-Rahman OA, van den Boogaard MJ, Bottani A, Castori M, Cormier-Daire V, Deardorff MA, Filges I, Fryer A, Fryns JP, Gana S, Garavelli L, Gillessen-Kaesbach G, Hall BD, Horn D, Huylebroeck D, Klapecki J, Krajewska-Walasek M, Kuechler A, Lines MA, Maas S, Macdermot KD, Mc-Kee S, Magee A, de Man SA, Moreau Y, Morice-Picard F, Obersztyn E, Pilch J, Rosser E, Shannon N, Stolte-Dijkstra I, Van Dijck P, Vilain C, Vogels A, Wakeling E, Wieczorek D, Wilson L, Zuffardi O, van Kampen AH, Devriendt K, Hennekam R, Vermeesch JR. Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat Genet 2012;44(4):445–9, S1. [DOI] [PubMed] [Google Scholar]
- 46.Hoyer J, Ekici AB, Endele S, Popp B, Zweier C, Wiesener A, Wohlleber E, Dufke A, Rossier E, Petsch C, Zweier M, Gohring I, Zink AM, Rappold G, Schrock E, Wieczorek D, Riess O, Engels H, Rauch A, Reis A. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet 2012;90(3):565– 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kosho T, Okamoto N, Ohashi H, Tsurusaki Y, Imai Y, Hibi-Ko Y, Kawame H, Homma T, Tanabe S, Kato M, Hiraki Y, Yamagata T, Yano S, Sakazume S, Ishii T, Nagai T, Ohta T, Niikawa N, Mizuno S, Kaname T, Naritomi K, Narumi Y, Wakui K, Fukushima Y, Miyatake S, Mizuguchi T, Saitsu H, Miyake N, Matsumoto N. Clinical correlations of mutations affecting six components of the SWI/SNF complex: detailed description of 21 patients and a review of the literature. Am J Med Genet A 2013;161A(6):1221–37. [DOI] [PubMed] [Google Scholar]
- 48.Moertel CL, Biegel JA, Dahlheime TR, Berr SA. Report of a patient with constitutional missense mutation of INI1/SMARCB1, Coffin-Siris phenotype and schwannomatosis. Poster presented at: The Children’s Tumor Foundation NF Conference; 2009. June 3–6; Portland, Oregon. [Google Scholar]
- 49.Hilden JM, Meerbaum S, Burger P, Finlay J, Janss A, Scheithauer BW, Walter AW, Rorke LB, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol 2004;22(14):2877–84. [DOI] [PubMed] [Google Scholar]
- 50.Geyer JR, Sposto R, Jennings M, Boyett JM, Axtell RA, Breiger D, Broxson E, Donahue B, Finlay JL, Goldwein JW, Heier LA, Johnson D, Mazewski C, Miller DC, Packer R, Puccetti D, Radcliffe J, Tao ML, Shiminski-Maher T; Children’s Cancer Group. Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children’s Cancer Group. J Clin Oncol 2005;23(30):7621–31. [DOI] [PubMed] [Google Scholar]
- 51.Chi SN, Zimmerman MA, Yao X, Cohen KJ, Burger P, Biegel JA, Rorke-Adams LB, Fisher MJ, Janss A, Mazewski C, Goldman S, Manley PE, Bowers DC, Bendel A, Rubin J, Turner CD, Marcus KJ, Goumnerova L, Ullrich NJ, Kieran MW. Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol 2009;27(3):385–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fidani P, De Ioris MA, Serra A, De Sio L, Ilari I, Cozza R, Boldrini R, Milano GM, Garre ML, Donfrancesco A. A multimodal strategy based on surgery, radiotherapy, ICE regimen and high dose chemotherapy in atypical teratoid/rhabdoid tumours: a single institution experience. J Neurooncol 2009;92(2):177–83. [DOI] [PubMed] [Google Scholar]
- 53.Zaky W, Dhall G, Ji L, Haley K, Allen J, Atlas M, Bertolone S, Cornelius A, Gardner S, Patel R, Pradhan K, Shen V, Thompson S, Torkildson J, Sposto R, Finlay JL. Intensive induction chemotherapy followed by myeloablative chemotherapy with autologous hematopoietic progenitor cell rescue for young children newly-diagnosed with central nervous system atypical teratoid/rhabdoid tumors: the Head Start III experience. Pediatr Blood Cancer 2014;61(1):95–101. [DOI] [PubMed] [Google Scholar]
- 54.Dufour C, Beaugrand A, Le Deley MC, Bourdeaut F, Andre N, Leblond P, Bertozzi AI, Frappaz D, Rialland X, Fouyssac F, Edan C, Grill J, Quidot M, Varlet P. Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: a multicenter study. Cancer 2012;118(15):3812–21. [DOI] [PubMed] [Google Scholar]
- 55.Lafay-Cousin L, Hawkins C, Carret AS, Johnston D, Zel-cer S, Wilson B, Jabado N, Scheinemann K, Eisenstat D, Fryer C, Fleming A, Mpofu C, Larouche V, Strother D, Bouffet E, Huang A. Central nervous system atypical teratoid rhabdoid tumours: the Canadian Paediatric Brain Tumour Consortium experience. Eur J Cancer 2012;48(3):353–9. [DOI] [PubMed] [Google Scholar]
- 56.Blaney SM, Kocak M, Gajjar A, Chintagumpala M, Merchant T, Kieran M, Pollack IF, Gururangan S, Geyer R, Phillips P, McLendon RE, Packer R, Goldman S, Banerjee A, Heideman R, Boyett JM, Kun L. Pilot study of systemic and intrathecal mafosfamide followed by conformal radiation for infants with intracranial central nervous system tumors: a pediatric brain tumor consortium study (PBTC-001). J Neurooncol 2012;109(3):565–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weeks DA, Beckwith JB, Mierau GW, Luckey DW. Rhabdoid tumor of kidney. A report of 111 cases from the National Wilms’ Tumor Study Pathology Center. Am J Surg Pathol 1989;13(6):439–58. [PubMed] [Google Scholar]
- 58.van den Heuvel-Eibrink MM, van Tinteren H, Rehorst H, Coulombe A, Patte C, de Camargo B, de Kraker J, Leuschner I, Lugtenberg R, Pritchard-Jones K, Sandstedt B, Spreafico F, Graf N, Vujanic GM. Malignant rhabdoid tumours of the kidney (MRTKs), registered on recent SIOP protocols from 1993 to 2005: a report of the SIOP Renal Tumour Study Group. Pediatr Blood Cancer 2011;56(5):733–7. [DOI] [PubMed] [Google Scholar]
- 59.Kerl K, Holsten T, Fruhwald MC. Rhabdoid tumors: clinical approaches and molecular targets for innovative therapy. Pediatr Hematol Oncol 2013;30(7):587–604. [DOI] [PubMed] [Google Scholar]
- 60.Wagner L, Hill DA, Fuller C, Pedrosa M, Bhakta M, Perry A, Dome JS. Treatment of metastatic rhabdoid tumor of the kidney. J Pediatr Hematol Oncol 2002;24(5):385–8. [DOI] [PubMed] [Google Scholar]
- 61.Yamamoto M, Suzuki N, Hatakeyama N, Mizue N, Hori T, Kuroiwa Y, Hareyama M, Oda T, Kudoh T, Nui A, Matsuno T, Hirama T, Yokoyama S, Dome JS, Tsutsumi H. Treatment of stage IV malignant rhabdoid tumor of the kidney (MRTK) with ICE and VDCy: a case report. J Pediatr Hematol Oncol 2006;28(5):286–9. [DOI] [PubMed] [Google Scholar]
- 62.Venkatramani R, Shoureshi P, Malvar J, Zhou S, Mascarenhas L. High dose alkylator therapy for extracranial malignant rhabdoid tumors in children. Pediatr Blood Cancer 2014;61(8):1357–61. [DOI] [PubMed] [Google Scholar]
- 63.Koga Y, Matsuzaki A, Suminoe A, Hatano M, Saito Y, Kinoshita Y, Tajiri T, Taguchi T, Kohashi K, Oda Y, Tsuneyoshi M, Hara T. Long-term survival after autologous peripheral blood stem cell transplantation in two patients with malignant rhabdoid tumor of the kidney. Pediatr Blood Cancer 2009;52(7):888–90. [DOI] [PubMed] [Google Scholar]
- 64.Daw NC, Anderson JR, Kalapurakal JA, Hoffer FA, Geller JI, Perlman EJ, Ehrlich PF, Mullen EA, Warwick A, Grundy PE, Dome JS. On behalf of the AREN0321 Study Committee. Treatment of stage II-IV diffuse anaplastic Wilms tumor: results from the Children’s Oncology Group AREN0321 Study [abstract]. Presented at the International Society of Pediatric Oncology (SIOP); 2014. October 22–25; Toronto, Canada. [Google Scholar]
- 65.Reinhard H, Reinert J, Beier R, Furtwangler R, Alkasser M, Rutkowski S, Fruhwald M, Koscielniak E, Leuschner I, Kaatsch P, Graf N. Rhabdoid tumors in children: prognostic factors in 70 patients diagnosed in Germany. Oncol Rep 2008;19(3):819–23. [PubMed] [Google Scholar]
- 66.Tekautz TM, Fuller CE, Blaney S, Fouladi M, Broniscer A, Merchant TE, Krasin M, Dalton J, Hale G, Kun LE, Wallace D, Gilbertson RJ, Gajjar A. Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 2005;23(7):1491–9. [DOI] [PubMed] [Google Scholar]
- 67.Roberts CW, Galusha SA, McMenamin ME, Fletcher CD, Orkin SH. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc Natl Acad Sci U S A 2000;97(25):13796–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roberts CW, Leroux MM, Fleming MD, Orkin SH. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell 2002;2(5):415–25. [DOI] [PubMed] [Google Scholar]
- 69.Lawlor ER, Thiele CJ. Epigenetic changes in pediatric solid tumors: promising new targets. Clin Cancer Res 2012;18(10):2768–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gadd S, Sredni ST, Huang CC, Perlman EJ; Renal Tumor Committee of the Children’s Oncology Group. Rhabdoid tumor: gene expression clues to pathogenesis and potential therapeutic targets. Lab Invest 2010;90(5):724–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Banck MS, Li S, Nishio H, Wang C, Beutler AS, Walsh MJ. The ZNF217 oncogene is a candidate organizer of repressive histone modifiers. Epigenetics 2009;4(2):100– 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM, Kuntz KW, Keilhack H. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 2013;110(19):7922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alimova I, Birks DK, Harris PS, Knipstein JA, Venkataraman S, Marquez VE, Foreman NK, Vibhakar R. Inhibition of EZH2 suppresses selfrenewal and induces radiation sensitivity in atypical rhabdoid teratoid tumor cells. Neuro Oncol 2013;15(2):149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kerl K, Ries D, Unland R, Borchert C, Moreno N, Hassel-blatt M, Jurgens H, Kool M, Gorlich D, Eveslage M, Jung M, Meisterernst M, Fruhwald M. The histone deacetylase inhibitor SAHA acts in synergism with fenretinide and doxorubicin to control growth of rhabdoid tumor cells. BMC Cancer 2013;13:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Knipstein JA, Birks DK, Donson AM, Alimova I, Fore-man NK, Vibhakar R. Histone deacetylase inhibition decreases proliferation and potentiates the effect of ionizing radiation in atypical teratoid/rhabdoid tumor cells. Neuro Oncol 2012;14(2):175–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sredni ST, Halpern AL, Hamm CA, Bonaldo Mde F, Tomita T. Histone deacetylases expression in atypical teratoid rhabdoid tumors. Childs Nerv Syst 2013;29(1):5–9. [DOI] [PubMed] [Google Scholar]
- 77.Watanabe M, Adachi S, Matsubara H, Imai T, Yui Y, Mizushima Y, Hiraumi Y, Watanabe K, Kamitsuji Y, Toyokuni SY, Hosoi H, Sugimoto T, Toguchida J, Nakahata T. Induction of autophagy in malignant rhabdoid tumor cells by the histone deacetylase inhibitor FK228 through AIF translocation. Int J Cancer 2009;124(1):55– 67. [DOI] [PubMed] [Google Scholar]
- 78.Graham C, Tucker C, Creech J, Favours E, Billups CA, Liu T, Fouladi M, Freeman BB 3rd, Stewart CF, Houghton PJ. Evaluation of the antitumor efficacy, pharmacokinetics, and pharmacodynamics of the histone deacetylase inhibitor depsipeptide in childhood cancer models in vivo. Clin Cancer Res 2006;12(1):223–34. [DOI] [PubMed] [Google Scholar]
- 79.Shim KW, Xi G, Farnell BM, Kim DS, Tsurubuchi T, Tomita T, Mayanil CS. Epigenetic modification after inhibition of IGF-1R signaling in human central nervous system atypical teratoid rhabdoid tumor (AT/RT). Childs Nerv Syst 2013;29(8):1245–51. [DOI] [PubMed] [Google Scholar]
- 80.Thiemann M, Oertel S, Ehemann V, Weichert W, Stenzinger A, Bischof M, Weber KJ, Perez RL, Haberkorn U, Kulozik AE, Debus J, Huber PE, Battmann C. In vivo efficacy of the histone deacetylase inhibitor suberoylanilide hydroxamic acid in combination with radiotherapy in a malignant rhabdoid tumor mouse model. Radiat Oncol 2012;7:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol 2010;28(10):1069–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dario LS, Rosa MA, Mariela E, Roberto G, Caterina C. Chromatin remodeling agents for cancer therapy. Rev Recent Clin Trials 2008;3(3):192–203. [DOI] [PubMed] [Google Scholar]
- 83.Perez-Plasencia C, Duenas-Gonzalez A. Can the state of cancer chemotherapy resistance be reverted by epigenetic therapy? Mol Cancer 2006;5:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chekhun VF, Lukyanova NY, Kovalchuk O, Tryndyak VP, Pogribny IP. Epigenetic profiling of multidrug-resistant human MCF-7 breast adenocarcinoma cells reveals novel hyper-and hypomethylated targets. Mol Cancer Ther 2007;6(3):1089–98. [DOI] [PubMed] [Google Scholar]
- 85.David GL, Yegnasubramanian S, Kumar A, Marchi VL, De Marzo AM, Lin X, Nelson WG. MDR1 promoter hypermethylation in MCF-7 human breast cancer cells: changes in chromatin structure induced by treatment with 5-Aza-cytidine. Cancer Biol Ther 2004;3(6):540–8. [DOI] [PubMed] [Google Scholar]
- 86.Huo H, Magro PG, Pietsch EC, Patel BB, Scotto KW. Histone methyltransferase MLL1 regulates MDR1 transcription and chemoresistance. Cancer Res 2010;70(21):8726–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Baker EK, Johnstone RW, Zalcberg JR, El-Osta A. Epigenetic changes to the MDR1 locus in response to chemotherapeutic drugs. Oncogene 2005;24(54):8061– 75. [DOI] [PubMed] [Google Scholar]
- 88.To KK, Zhan Z, Bates SE. Aberrant promoter methylation of the ABCG2 gene in renal carcinoma. Mol Cell Biol 2006;26(22):8572–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hu S, Yu L, Li Z, Shen Y, Wang J, Cai J, Xiao L, Wang Z. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Biol Ther 2010;10(8):788–95. [DOI] [PubMed] [Google Scholar]
- 90.Abbosh PH, Montgomery JS, Starkey JA, Novotny M, Zuhowski EG, Egorin MJ, Moseman AP, Golas A, Brannon KM, Balch C, Huang TH, Nephew KP. Dominant-negative histone H3 lysine 27 mutant derepresses silenced tumor suppressor genes and reverses the drug-resistant phenotype in cancer cells. Cancer Res 2006;66(11):5582–91. [DOI] [PubMed] [Google Scholar]
- 91.Suzuki M, Shinohara F, Nishimura K, Echigo S, Rikiishi H. Epigenetic regulation of chemosensitivity to 5-fluorouracil and cisplatin by zebularine in oral squamous cell carcinoma. Int J Oncol 2007;31(6):1449–56. [PubMed] [Google Scholar]
- 92.Ma J, Dong C, Ji C. MicroRNA and drug resistance. Cancer Gene Ther 2010;17(8):523–31. [DOI] [PubMed] [Google Scholar]
- 93.Kovalchuk O, Filkowski J, Meservy J, Ilnytskyy Y, Tryn-dyak VP, Chekhun VF, Pogribny IP. Involvement of microRNA-451 in resistance of the MCF-7 breast cancer cells to chemotherapeutic drug doxorubicin. Mol Cancer Ther 2008;7(7):2152–9. [DOI] [PubMed] [Google Scholar]
- 94.George RE, Lahti JM, Adamson PC, Zhu K, Finkelstein D, Ingle AM, Reid JM, Krailo M, Neuberg D, Blaney SM, Diller L. Phase I study of decitabine with doxorubicin and cyclophosphamide in children with neuroblastoma and other solid tumors: a Children’s Oncology Group study. Pediatr Blood Cancer 2010;55(4):629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hummel TR, Wagner L, Ahern C, Fouladi M, Reid JM, McGovern RM, Ames MM, Gilbertson RJ, Horton T, Ingle AM, Weigel B, Blaney SM. A pediatric phase 1 trial of vorinostat and temozolomide in relapsed or refractory primary brain or spinal cord tumors: a Children’s Oncology Group phase 1 consortium study. Pediatr Blood Cancer 2013;60(9):1452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fujisawa H, Misaki K, Takabatake Y, Hasegawa M, Yamashita J. Cyclin D1 is overexpressed in atypical teratoid/rhabdoid tumor with hSNF5/INI1 gene inactivation. J Neurooncol 2005;73(2):117–24. [DOI] [PubMed] [Google Scholar]
- 97.Chai J, Lu X, Godfrey V, Fletcher C, Roberts CW, Van Dyke T, Weissman BE. Tumorspecific cooperation of retinoblastoma protein family and Snf5 inactivation. Cancer Res 2007;67(7):3002–9. [DOI] [PubMed] [Google Scholar]
- 98.Zhang ZK, Davies KP, Allen J, Zhu L, Pestell RG, Zagzag D, Kalpana GV. Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Mol Cell Biol 2002;22(16):5975–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tsikitis M, Zhang Z, Edelman W, Zagzag D, Kalpana GV. Genetic ablation of Cyclin D1 abrogates genesis of rhabdoid tumors resulting from Ini1 loss. Proc Natl Acad Sci U S A 2005;102(34):12129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Isakoff MS, Sansam CG, Tamayo P, Subramanian A, Evans JA, Fillmore CM, Wang X, Biegel JA, Pomeroy SL, Mesirov JP, Roberts CW. Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc Natl Acad Sci U S A 2005;102(49):17745–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cimica V, Smith ME, Zhang Z, Mathur D, Mani S, Kalpana GV. Potent inhibition of rhabdoid tumor cells by combination of flavopiridol and 4OH-tamoxifen. BMC Cancer 2010;10:364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee S, Cimica V, Ramachandra N, Zagzag D, Kalpana GV. Aurora A is a repressed effector target of the chromatin remodeling protein INI1/hSNF5 required for rhabdoid tumor cell survival. Cancer Res 2011;71(9):3225– 35. [DOI] [PubMed] [Google Scholar]
- 103.Maris JM, Morton CL, Gorlick R, Kolb EA, Lock R, Carol H, Keir ST, Reynolds CP, Kang MH, Wu J, Smith MA, Houghton PJ. Initial testing of the aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP). Pediatr Blood Cancer 2010;55(1):26– 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Venkataraman S, Alimova I, Tello T, Harris PS, Knipstein JA, Donson AM, Foreman NK, Liu AK, Vibhakar R. Targeting Aurora kinase A enhances radiation sensitivity of atypical teratoid rhabdoid tumor cells. J Neurooncol 2012;107(3):517–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Morozov A, Lee SJ, Zhang ZK, Cimica V, Zagzag D, Kalpana GV. INI1 induces interferon signaling and spindle checkpoint in rhabdoid tumors. Clin Cancer Res 2007;13(16):4721–30. [DOI] [PubMed] [Google Scholar]
- 106.Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, Gorlick R, Kolb EA, Zhang W, Lock R, Carol H, Tajbakhsh M, Reynolds CP, Maris JM, Courtright J, Keir ST, Friedman HS, Stopford C, Zeidner J, Wu J, Liu T, Billups CA, Khan J, Ansher S, Zhang J, Smith MA. The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer 2007;49(7):928–40. [DOI] [PubMed] [Google Scholar]
- 107.Tajbakhsh M, Houghton PJ, Morton CL, Kolb EA, Gorlick R, Maris JM, Keir ST, Wu J, Reynolds CP, Smith MA, Lock RB. Initial testing of cisplatin by the pediatric preclinical testing program. Pediatr Blood Cancer 2008;50(5):992–1000. [DOI] [PubMed] [Google Scholar]
- 108.Maris JM, Courtright J, Houghton PJ, Morton CL, Gorlick R, Kolb EA, Lock R, Tajbakhsh M, Reynolds CP, Keir ST, Wu J, Smith MA. Initial testing of the VEGFR inhibitor AZD2171 by the pediatric preclinical testing program. Pediatr Blood Cancer 2008;50(3):581–7. [DOI] [PubMed] [Google Scholar]
- 109.Carol H, Lock R, Houghton PJ, Morton CL, Kolb EA, Gorlick R, Reynolds CP, Maris JM, Keir ST, Billups CA, Smith MA. Initial testing (stage 1) of the kinesin spindle protein inhibitor ispinesib by the pediatric preclinical testing program. Pediatr Blood Cancer 2009;53(7):1255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Maris JM, Courtright J, Houghton PJ, Morton CL, Kolb EA, Lock R, Tajbakhsh M, Reynolds CP, Keir ST, Wu J, Smith MA. Initial testing (stage 1) of sunitinib by the pediatric preclinical testing program. Pediatr Blood Cancer 2008;51(1):42–8. [DOI] [PubMed] [Google Scholar]
- 111.Houghton PJ, Morton CL, Kolb EA, Gorlick R, Lock R, Carol H, Reynolds CP, Maris JM, Keir ST, Billups CA, Smith MA. Initial testing (stage 1) of the mTOR inhibitor rapamycin by the pediatric preclinical testing program. Pediatr Blood Cancer 2008;50(4):799–805. [DOI] [PubMed] [Google Scholar]
- 112.Houghton PJ, Lock R, Carol H, Morton CL, Phelps D, Gorlick R, Kolb EA, Keir ST, Reynolds CP, Kang MH, Maris JM, Wozniak AW, Gu Y, Wilson WR, Smith MA. Initial testing of the hypoxiaactivated prodrug PR-104 by the pediatric preclinical testing program. Pediatr Blood Cancer 2011;57(3):443–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lock RB, Carol H, Morton CL, Keir ST, Reynolds CP, Kang MH, Maris JM, Wozniak AW, Gorlick R, Kolb EA, Houghton PJ, Smith MA. Initial testing of the CENPE inhibitor GSK923295A by the pediatric preclinical testing program. Pediatr Blood Cancer 2012;58(6):916–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Carol H, Reynolds CP, Kang MH, Keir ST, Maris JM, Gorlick R, Kolb EA, Billups CA, Geier B, Kurmasheva RT, Houghton PJ, Smith MA, Lock RB. Initial testing of the MDM2 inhibitor RG7112 by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer 2013;60(4):633– 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Smith MA, Kang MH, Reynolds CP, Gorlick R, Kolb EA, Maris JM, Keir ST, Billups CA, Kurmasheva RT, Houghton PJ. Pediatric Preclinical Testing Program (PPTP) stage 1 evaluation of cabozantinib. Poster session at the American Association of Cancer Research 104th Annual Meeting; 2013. April 6–10; Washington, DC. [Google Scholar]
- 116.Morton C, Houghton PJ, Maris JM, Gorlick R, Kolb EA, Kang MH, Reynolds CP, Smith MA. Pediatric Preclinical Testing Program (PPTP) evaluation of the oncolytic picornavirus, NTX-010 (SVV-001). Poster session at the 20th European Organization for Research and Treatment of Cancer (EORTC), National Cancer Institute (NCI), and American Association for Cancer Research (AACR) Symposium on Molecular Targets and Cancer Therapeutics; 2008. October 21–24; Geneva, Switzerland. [Google Scholar]
- 117.Keshelava N, Houghton PJ, Morton CL, Lock RB, Carol H, Keir ST, Maris JM, Reynolds CP, Gorlick R, Kolb EA, Wu J, Smith MA. Initial testing (stage 1) of vorinostat (SAHA) by the pediatric preclinical testing program. Pediatr Blood Cancer 2009;53(3):505–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Carol H, Gorlick R, Kolb EA, Morton CL, Manesh DM, Initial testing (stage 1) of the histone deacetylase inhibitor, quisinostat (JNJ-26481585), by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer 2014;61(2):245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carol H, Houghton PJ, Morton CL, Kolb EA, Gorlick R, Reynolds CP, Kang MH, Maris JM, Keir ST, Watkins A, Smith MA, Lock RB. Initial testing of topotecan by the pediatric preclinical testing program. Pediatr Blood Cancer 2010;54(5):707–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gorlick R, Kolb EA, Houghton PJ, Morton CL, Neale G, Keir ST, Carol H, Lock R, Phelps D, Kang MH, Reynolds CP, Maris JM, Billups C, Smith MA. Initial testing (stage 1)of the cyclin dependent kinase inhibitor SCH 727965 (dinaciclib) by the pediatric preclinical testing program. Pediatr Blood Cancer 2012;59(7):1266–74. [DOI] [PMC free article] [PubMed] [Google Scholar]