

Abstract
Despite the ability of some gastrointestinal hormones to reliably reduce meal size when administered prior to a meal, it is not understood why the repeated administration or genetic knockout of these hormones appear largely ineffective in reducing food intake and body weight. Here, we review evidence that the ability of GI peptides such as cholecystokinin (CCK) to elicit satiation is a consequence of prior learning. Evidence includes first, that the ability of some of these signals to modify food intake depends upon past experience and is malleable with new experience. Additionally, the ability of CCK and other gut signals to reduce food intake may not be hard-wired; i.e., any so-called “satiation” signal that reduces food intake in a single-meal situation may not continue to do so over repeated trials. The individual will respond to the signal only so long as it provides reliable information about caloric content. If a particular signal becomes unreliable, the individual will rely on other signals to end meals. Thus, gut peptides/hormones have important metabolic effects such as mediating absorption, digestion, and many aspects of the distribution of ingested nutrients throughout the body; and, if they have been reliably associated with natural stimuli that mediate satiation, they also inform behavior.
The behavioral act of eating impacts, and is impacted by, most physiological systems in the body. However, in this report, we confine discussion to interactions of the brain with the gastrointestinal (GI) system as they relate to food intake and consequently to body weight, with a particular focus on the role of GI peptide hormones in the process of satiation. There is continuous crosstalk between the brain and the GI tract, and the flow of information is critical for the proper functioning of digestion, absorption and disposition of ingested nutrients. Additionally, the communication informs the brain of all relevant metabolic activities (see reviews in [1–3]). The flow of information is bidirectional, and it utilizes both nerves and hormones. For much of the GI tract, the vagus nerves serve as the conduit to the brain, with both afferent and efferent vagal fibers innervating every metabolic organ from the esophagus to the stomach to the intestines, as well as innervating the liver, exocrine and endocrine pancreas and other organs.
An important question concerns whether the flow of information along the vagus, in addition to coordinating all aspects of the digestive process, is critical to the regulation of food intake and body weight. Although the literature on this is mixed, the general conclusion can be made that when the vagus is severed below the diaphragm (thus sparing branches to the heart), there is little overall effect on food intake in otherwise normal individuals. The only proviso is that the food source must be in a form that can pass through the now-denervated pyloric sphincter (or the sphincter’s muscular wall must be surgically altered to allow easy passage of chyme from the stomach to the duodenum) [4]. Body weight is often reduced somewhat following vagotomy, but the cause is mainly malabsorption as opposed to a primary effect on food intake [4, 5].
The brain and GI tract also communicate via hormones. While the hypothalamic-pituitary axis secretes numerous hormones that influence metabolism throughout the entire body (e.g., ACTH, growth hormone, TRH), these hormones are not considered to have a major impact on daily food intake except in extreme disease states. Conversely, hormones from the GI tract, particularly peptides that are secreted in response to ingested nutrients, can have profound effects on food intake. This is the topic under consideration.
The pervasive view is that GI peptides are major contributors to the perception of fullness or satiation. This concept began with the pioneering experiment by Gibbs, Smith and colleagues in 1973 [6]. Specific GI peptides or their synthetic analogues had recently become available for research at that time. Gibbs et al. administered an analogue of cholecystokinin (CCK) or a control solution (saline, the vehicle for the administered CCK) to mildly food-deprived rats prior to giving them access to food. Compared with food intake following an intraperitoneal (ip) injection of vehicle, ip CCK caused a significant reduction of meal size. The phenomenon was dose-dependent, with more CCK causing a greater reduction of intake. This one publication provided a fundamental paradigm that has been utilized in one form or another in over a thousand papers investigating the effect on meal size of dozens of GI and related metabolic hormones or peptides.
The generally accepted model is depicted in Figure 1, and consists of a negative feedback design influencing individual meals. As depicted in the upper left rectangle of the figure, the behavioral phenomenon is clear; the individual starts eating, food enters the GI tract and is processed, and the consequent metabolic effects are sensed. As the sensory signal grows in strength during the meal, the individual eventually stops eating at a point at which satiation is considered to have occurred. It is at this point that humans state they are full. Less clear is what causes satiation. Several possibilities have been considered over the years including the volume of food consumed, the degree of gastric distension, the number of calories consumed, the types and amounts of individual macronutrients consumed, the duration of time since the onset of eating, and many more. At another level, satiation has been hypothesized to occur because of an increase of specific nutrients in the blood (e.g., glucose or fatty acids or metabolites) or of their activity at target organs such as the brain. This area of research has been recently reviewed [7].
Figure 1.
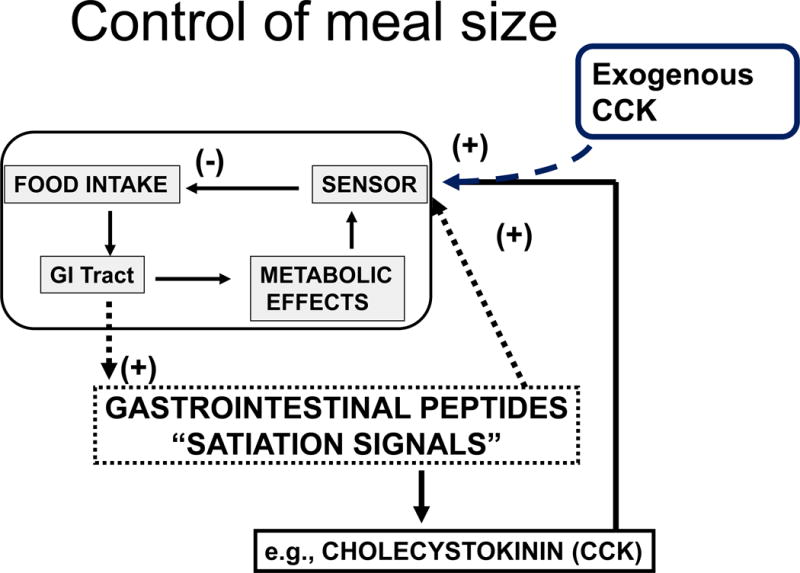
Control of meal size. As depicted in the upper left rectangle, meal size is thought to be controlled by a homeostatic negative-feedback loop. During a meal, food enters the gastrointestinal (GI) tract initiating multiple metabolic effects including digestion and absorption of nutrients. Some (as yet unknown) consequences of the metabolic activities is sensed and the signal is conveyed to the brain and elicits satiation or fullness. The current hypothesis is that the increase of GI peptides/hormones secreted as the food is being processed provides the signal that is sensed and elicits satiation; and the most studied of these hormones is CCK. When any of these hormones such as CCK is administered exogenously as a meal begins, it causes eating to stop prematurely, as depicted in the upper right.
When food is consumed, and enters the GI tract, its nutritional content is evaluated by sensory cells (called enteroendocrine cells) lining the lumen. These cells secrete numerous hormones (including CCK) and paracrines that signal to other cells throughout the GI tract, as well as to the liver and the pancreas, to secrete enzymes into the lumen appropriate for digesting the meal. The digested nutrients are subsequently absorbed, entering the blood and reaching tissues throughout the body. This entire process is customized to the specific food being eaten and is highly coordinated by the enteric nervous system, which is intrinsic within the wall of the GI tract. Ongoing feedback is also exchanged with the central nervous system via the vagus nerves. See reviews in [7] and [8].
The model depicted in Figure 1 implies that satiation occurs as one or more of these secreted GI hormones acts on receptors that send messages to the brain concerning how much food has been eaten, and that this is subsequently translated into feeling full and stopping intake. The poster child as it were for this process has been CCK, because it was the first peptide so-described to reduce the size of a meal and has since been used extensively in research. As depicted in the upper right of the figure, when exogenous CCK (or a different ‘satiation’ compound) is administered at the start of the meal, it is thought to create a false signal that more calories have been consumed than have actually been consumed, and the consequence is that satiation occurs prematurely and results in a reduced meal size.
The GI tract secretes numerous compounds before, during and after meals, many of which have been found to influence food intake when administered exogenously, and many reviews are available [7, 9, 10]. In order to make some general points related to satiation, we focus here on a few of these including CCK, glucagon-like peptide-1 (GLP-1), peptide YY (PYY), amylin and apolipoprotein A-IV (apo AIV) as well as ghrelin, a peptide hormone secreted from the stomach that increases meal size when administered before a meal [11]. There are several important criteria that must be met for a compound to be considered as an endogenous satiation factor [12, 13]. 1) The proposed factor should be elevated prandially, 2) exogenous administration of the factor should reduce meal size, 3) the factor’s onset of action should be rapid, with short duration, 4) any reduction of food intake caused by the factor must not be secondary to illness or malaise, 5) the reduced meal size should occur with physiologically relevant concentrations of the factor, and 6) inhibiting the activity of the factor (e.g., via receptor antagonism) should attenuate the action of the exogenously-administered factor and also increase meal size in the absence of the factor.
As more and more hormones were found to influence food intake, our group found it useful to summarize what was known in a model that has been highly influential [14–16]. The model partitioned signals originating in metabolic organs (GI tract, endocrine pancreas, adipose tissue) into those that influence meal size (satiation signals) and those that monitor body fat (and consequently body weight; adiposity signals). Satiation signals reach the brain either directly via the blood (e.g., amylin and ghrelin) or else neuronally via the vagus nerves (e.g., CCK, GLP-1, apo AIV) [17]. All of the satiation signals converge in the nucleus of the solitary tract (NTS) in the hindbrain, and from there, this information is relayed to other, more-anterior, brain areas. Adiposity signals, on the other hand, circulate to the brain and are actively transported into the hypothalamus (e.g., insulin, leptin) [18]. This is a homeostatic-based model that describes how meal-related signals interact with adiposity-related signals to maintain a relatively constant level of body fat/weight.
Although it was not explicitly stated, an implication of the model is that the impact of these peripheral signals is hard-wired. While other influences on food intake (such as emotions, stress, the social situation, learning, etc.) were acknowledged, their impact was considered to be superimposed onto the hypothalamic homeostatic control system for body weight maintenance.
The broad application of molecular genetic techniques to research on food intake and body weight regulation in recent years has caused this model to be reconsidered. As it became possible and even commonplace to knock out specific genes, the function of many genes encoding for purported satiation factors or their related actions could be determined. Ghrelin, the gastric hormone that increases meal size, was among the first, and the results were surprising. Given that exogenous ghrelin potently stimulates food intake in animals and humans, the a priori hypothesis would be that animals lacking ghrelin would be hypophagic and underweight. In fact, both food intake and body weight of ghrelin-knockout mice were the same as in wild-type controls [19, 20]. A quote from one of those reports is telling. “In contrast to predictions made from the pharmacology of ghrelin, ghrelin-null mice are not anorexic dwarfs; their size, growth rate, food intake, body composition, reproduction, gross behavior, and tissue pathology are indistinguishable from wild-type littermates” [19].
Analogous results have been found for purported satiation factors. Glucagon-like peptide-1 (GLP-1) is secreted from the intestines and is an important incretin hormone, facilitating insulin secretion. Its exogenous administration also reduces meal size [21]. To selectively eliminate glucagon-like peptide (GLP)-1 activity, it is necessary to knock out its receptor (GLP-1r) since the gene for GLP-1 makes several other biologically active peptides. Again, contrary to the hypothesis that mice lacking GLP-1 activity would eat more and have accelerated body weight gain, GLP-1r knockout mice have normal food intake and body weight [22, 23]. Our lab found that mice lacking the gene for CCK have identical daily food intake, body weight and body fat as controls, although the diurnal food intake pattern shifted slightly [24]. Similar results have been made for mice lacking apo AIV [25] or amylin [26]; i.e., the respective knockout mice have normal food intake and body weight. Although there is one report of the opposite effect [27], most reports of genetically knocking out PYY function report no profound changes of food intake or body weight [28, 29]. Schonhoff et al. state that “Despite the anorectic effects of exogenous peptide YY3-36 following intraperitoneal administration, mice lacking peptide YY showed normal growth, food intake, energy expenditure, and responsiveness to peptide YY3-36” (p 4189) [28].
The point is that whereas the exogenous administration of each of these ‘satiation’ peptides reduces intake of single meals, genetically removing its activity has little or no effect on food take or body weight. It is important to note, however, that the metabolic effects of these peptides related to the handling of nutrients are greatly impacted in these genetic knockout models such that only feeding behavior is unaffected. For example, GLP-1r knockout mice have glucose intolerance but normal satiation [22] and ghrelin knockout mice retain the altered fat deposition action of ghrelin despite having no change of food intake or body weight [20]. Thus, while evidence based on pharmacological manipulation of GI peptides and their metabolic action is often consistent with evidence based on mutagenesis, discrepancies occur when considering food intake [30]. With regard to satiation, it can therefore be concluded that any GI peptide that is sufficient to influence meal size when administered exogenously is not necessary for maintenance of normal food intake and body weight. This begs the question as to whether manipulating meal size in this manner is simply a pharmacological phenomenon.
The increasing use of bariatric surgery to treat obesity over the last two decades has lent support to the idea that endogenous GI peptides can have a significant role in the control of food intake and body weight. The reason is that a number of different procedures in which the plumbing and the intercellular communication network of the GI tract is altered, including Roux-en-Y gastric bypass (RYGB) and vertical sleeve gastrectomy (VSG), result in significant weight loss that is often accompanied by a transient hypophagia. Although the underlying mechanism(s) remain unknown, a universal finding is that those surgical procedures that result in significant weight loss also result in large changes in the secretion and circulating levels of GI peptides. Specifically, there is a reduction in the food intake-stimulating peptide, ghrelin, and an increase in peptides that reduce food intake, including PYY, GLP-1, apo AIV and CCK. That said, when these bariatric procedures are applied to animals genetically lacking one or another specific GI peptide or its action, they exhibit the same amount of hypophagia and weight loss as occurs in wildtype controls. As examples, GLP-1r knockout mice have the same reduction of food intake and loss of body weight following RYGB as wildtype control mice [31], and mice lacking ghrelin have identical weight loss and hypophagia following VSG as wildtype controls [32]. Thus, the beneficial effects of bariatric surgery do not seem to be due to the profound changes of GI-secreted satiation peptides, since the effect is the same whether the respective peptides are there or not. Collectively, the data from genetic knockout animals as well as from bariatric surgery imply that the role of gut peptides in the control of food intake needs reconsideration.
We present a different conception to help understand the role of GI peptides in the normal control of food intake. Meals tend to be regular phenomena, often occurring at the same time or in the same situations from day to day [33–35]. The predictability of when meals will occur is an optimal circumstance for learned associations to be made; i.e., a reliable and persistent association between two events is necessary for classical conditioning to occur. As Pavlov first taught us, stimuli associated with the presentation of food can be conditioned to elicit salivary as well as gastric secretions. We now know that numerous hormones are also secreted in anticipation of predictable meals (e.g., insulin, ghrelin, GLP-1) [36, 37], presumably to prepare the GI tract for the pending caloric load; i.e., making conditioned responses prior to beginning to eat enables far-more efficient processing of the food [33, 34, 38]. In fact, individuals who fail to secrete anticipatory insulin prior to a meal are glucose intolerant and appear acutely diabetic [39–42]. Learning is also important for determining which foods are safe and which are not [43, 44]. Because both when to eat and what to eat are greatly influenced by conditioning, it is therefore reasonable to contemplate whether or not conditioning might also account for the impact of GI secretions on satiation.
As seen in the top of Figure 2, conditioned salivation occurs after stimuli such as a bell or the sound of a metronome have reliably been associated with food on the tongue. The presence of the bell at the same time that the food (unconditioned stimulus, US) elicits salivation (unconditioned response, UR) is the key to the process, enabling the bell to become a conditioned stimulus (CS) and elicit salivation (the conditioned response, CR) in the absence of the food. In the lower section of Figure 2, the specific consequence of processing the nutrients in a meal that actually causes satiation is not known. Nonetheless, whatever the US that unconditionally elicits the UR (satiation) is, it is always temporally associated with increased levels of ‘satiation’ hormones, setting the stage for each of the hormones to develop the ability to elicit conditioned satiation on their own. Consequently, administering one of these peptides exogenously at the start of the meal would be expected to elicit premature satiation.
Figure 2.
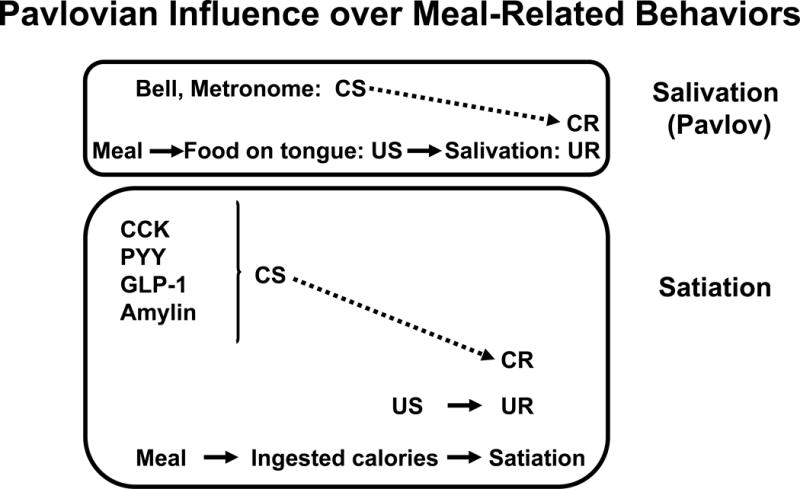
Pavlovian Influence over Meal-Related Behaviors. Top panel: Food on the tongue, as occurs during meals, unconditionally elicits salivation. Food on the tongue is the unconditioned stimulus (US) and salivation is the unconditioned response (UR). When the UR to US reflex is reliably associated with another stimulus, one that does not normally elicit salivation such as a bell, the bell can develop the ability to elicit salivation without the necessity of food on the tongue; i.e., the bell becomes a conditioned stimulus (CS) and the salivation it elicits is a conditioned response (CR). Bottom panel: Analogous to the association between the bell and salivation in the top panel, gastrointestinal hormones such as CCK, glucagon-like peptide-1 (GLP-1), peptide YY (PYY), amylin and apolipoprotein A-IV, that are secreted during meals can become associated with meal-elicited satiation. Thus, these hormones can be considered as CSs that elicit premature satiation (CR).
While the possibility that the reduction of meal size elicited by the exogenous administration of, for example, CCK may seem unlikely, it can be tested empirically. If classical conditioning accounts for the ability of a peptide such as CCK to reduce meal size when exogenously administered, then the phenomenon should follow the laws of learning. Consistent with such a possibility, it is known that arbitrary exogenous stimuli repeatedly associated with meals can become conditioned either to increase [45, 46] or to reduce meal size [47, 48] as well as can orosensory stimuli associated with eating [49]. It is therefore not too much of a stretch to presume that endogenous hormones can do the same (see [34, 50]).
More to the point, if the anorexic action of one or more digestion-related compounds is actually a conditioned as opposed to a hard-wired unconditioned response, it should be subject to the process of extinction. In a clever experiment, Goodison and Siegel [51] assessed this possibility. They had two distinctive environments (A and B) in which rats were placed to eat a meal each day. One group of rats always received a control injection of saline before eating their meal on the days they ate in Environment A, and an injection of CCK before their meals on other days when they ate in Environment B. Hence, Environment A became associated with a control injection and Environment B became associated with CCK. A second group of rats also received CCK or saline each day, but with the opposite contingencies; i.e., Environment A was always associated with CCK and Environment B was always associated with saline. Animals had one trial per day and received an equal number of trials in Environment A and in Environment B. Following the initial injection of CCK on Trial 1, all rats in both groups ate significantly less food than when they received saline (in the alternate environment), demonstrating CCK’s satiating effect. However, whereas the meal size in response to saline remained relatively constant for each group over trials, the ability of CCK to reduce meal size waned over trials. After eight trials with CCK, its administration no longer reduced food intake, and the rats ate the same amount as if they had been administered saline. Thus, the rats appeared to undergo extinction (or ‘tolerance’ as the authors called it) in the CCK-associated environment.
That demonstration aside, the power of the experimental design allowed an important further characterization. On a subsequent trial, the rats were given CCK, but in the environment previously only associated with getting saline. Their food intake was reduced similarly to their intake on the first CCK trial. In other words, whereas the satiating effect of CCK had extinguished in one environment, it was normal in a second environment not previously associated with CCK. Further, when the rats were administered saline, but in the CCK-associated environment, they actually ate significantly more food than on other saline days. These data strongly imply that CCK’s satiating effect can be brought under stimulus control based on prior associations; i.e., that it is due to conditioning. The basic findings of the experiment have been replicated by others [52].
The important point is that whether or not CCK, or any other GI hormone secreted during the process of digestion, is able to elicit a reduction of food intake (i.e., satiation) depends upon its history of association with other stimuli present during meals. In typical laboratory experiments, the food is constant from day to day and from meal to meal, such that the same cocktail of GI hormones is secreted during every meal and easily becomes associated with eating and with how many calories have been ingested. We have written elsewhere that it is to the individual’s advantage to be able to gauge how many calories have been eaten as soon as possible once a meal begins, so as to elicit satiation when appropriate and consequently not overeat [34, 38, 53]. Responding to stimuli such as CCK and other GI hormones that it has learned to associate with calories consumed is a convenient way to do this, but only so long as the signal is reliable. Once the signal (CCK in the example) becomes dissociated from calories consumed and hence is no longer reliable, the individual ignores it but continues to respond to other satiation peptides.
In conclusion, we have presented important implications with regard to GI peptides and satiation. First, the ability of some of these signals to modify food intake depends upon past experience and is malleable with new experience. Second, the ability of CCK and other gut signals to reduce food intake may not be hard-wired; i.e., any so-called “satiation” signal that reduces food intake in a single-meal situation may not continue to do so over repeated trials. The individual will respond to the signal only so long as it provides reliable information about caloric content. If a particular signal becomes unreliable, the individual will utilize other signals to end meals. Thus, gut peptides/hormones have important metabolic effects such as mediating absorption, digestion and the disposal of ingested nutrients into tissues; and, if they have been reliably associated with an unconditioned stimulus mediating satiation, they also inform behavior.
Highlights.
Individual GI peptides are sufficient but not necessary to elicit satiation.
Anorectic effects of CCK undergo extinction when dissociated from consumed calories.
Anorectic effects of GI peptides persist if they provide accurate caloric information.
We conclude that GI peptides are classically conditioned to mediate satiation.
Acknowledgments
This work was funded by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK): DK017844, DK092779, DK095440, DK059803 and DK007673 and by the Australian Research Council: DE160100088 and DP170100063.
References
- 1.Berthoud HR, Morrison C. The brain, appetite, and obesity. Annu Rev Psychol. 2008;59:55–92. doi: 10.1146/annurev.psych.59.103006.093551. [DOI] [PubMed] [Google Scholar]
- 2.Begg DP, Woods SC. Interactions between the central nervous system and pancreatic islet secretions: a historical perspective. Adv Physiol Educ. 2013 Mar;37:53–60. doi: 10.1152/advan.00167.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sclafani A. Gut-brain nutrient signaling. Appetition vs. satiation. Appetite. 2013 Dec;71:454–8. doi: 10.1016/j.appet.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kral JG, Paez W, Wolfe BM. Vagal nerve function in obesity: therapeutic implications. World J Surg. 2009 Oct;33:1995–2006. doi: 10.1007/s00268-009-0138-8. [DOI] [PubMed] [Google Scholar]
- 5.Kral JG. Overview of surgical techniques for treating obesity. Am J Clin Nutr. 1992 Feb;55:552S–555S. doi: 10.1093/ajcn/55.2.552s. [DOI] [PubMed] [Google Scholar]
- 6.Gibbs J, Young RC, Smith GP. Cholecystokinin decreases food intake in rats. J Comp Physiol Psychol. 1973 Sep;84:488–95. doi: 10.1037/h0034870. [DOI] [PubMed] [Google Scholar]
- 7.Moran TH, Ladenheim EE. Physiologic and Neural Controls of Eating. Gastroenterol Clin North Am. 2016 Dec;45:581–599. doi: 10.1016/j.gtc.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clemmensen C, Muller TD, Woods SC, Berthoud HR, Seeley RJ, Tschop MH. Gut-Brain Cross-Talk in Metabolic Control. Cell. 2017 Feb 23;168:758–774. doi: 10.1016/j.cell.2017.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strader AD, Woods SC. Gastrointestinal hormones and food intake. Gastroenterology. 2005 Jan;128:175–91. doi: 10.1053/j.gastro.2004.10.043. [DOI] [PubMed] [Google Scholar]
- 10.Woods SC, D’Alessio DA. Central control of body weight and appetite. J Clin Endocrinol Metab. 2008 Nov;93:S37–50. doi: 10.1210/jc.2008-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller TD, Tschop MH. Ghrelin - a key pleiotropic hormone-regulating systemic energy metabolism. Endocr Dev. 2013;25:91–100. doi: 10.1159/000346590. [DOI] [PubMed] [Google Scholar]
- 12.Smith G, Gibbs J. The development and proof of the cholecystokinin hypothesis of satiety. In: Dourish CT, Cooper SJ, Iversen SD, Iversen LL, editors. Multiple Cholecystokinin Receptors in the CNS. Oxford University Press; Oxford: 1991. [Google Scholar]
- 13.Moran TH. Gut peptides in the control of food intake: 30 years of ideas. Physiol Behav. 2004 Aug;82:175–80. doi: 10.1016/j.physbeh.2004.04.048. [DOI] [PubMed] [Google Scholar]
- 14.Woods SC, Seeley RJ, Porte D, Jr, Schwartz MW. Signals that regulate food intake and energy homeostasis. Science. 1998 May 29;280:1378–83. doi: 10.1126/science.280.5368.1378. [DOI] [PubMed] [Google Scholar]
- 15.Schwartz MW, Woods SC, Porte D, Seeley RJ, Jr, Baskin DG. Central nervous system control of food intake. Nature. 2000 Apr 06;404:661–71. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 16.Woods SC, Seeley RJ. Adiposity signals and the control of energy homeostasis. Nutrition. 2000 Oct;16:894–902. doi: 10.1016/s0899-9007(00)00454-8. [DOI] [PubMed] [Google Scholar]
- 17.May AA, Liu M, Woods SC, Begg DP. CCK increases the transport of insulin into the brain. Physiol Behav. 2016 Oct 15;165:392–7. doi: 10.1016/j.physbeh.2016.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woods SC, May AA, Liu M, Tso P, Begg DP. Using the cerebrospinal fluid to understand ingestive behavior. Physiol Behav. 2017 Sep 01;178:172–178. doi: 10.1016/j.physbeh.2016.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun Y, Ahmed S, Smith RG. Deletion of ghrelin impairs neither growth nor appetite. Mol Cell Biol. 2003 Nov;23:7973–81. doi: 10.1128/MCB.23.22.7973-7981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wortley KE, Anderson KD, Garcia K, Murray JD, Malinova L, Liu R, Moncrieffe M, Thabet K, Cox HJ, Yancopoulos GD, Wiegand SJ, Sleeman MW. Genetic deletion of ghrelin does not decrease food intake but influences metabolic fuel preference. Proc Natl Acad Sci U S A. 2004 May 25;101:8227–32. doi: 10.1073/pnas.0402763101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larsen PJ, Tang-Christensen M, Goke R, Jessop DS, Fink-Jensen A, Moller M, Sheikh SP. Central administration of GLP-1-(7-36) amide inhibits food and water intake in rats. Am J Physiol. 1996 Oct;271:R848–56. doi: 10.1152/ajpregu.1996.271.4.R848. [DOI] [PubMed] [Google Scholar]
- 22.Scrocchi LA, Brown TJ, Brubaker PL, MaClusky N, Auerbach AB, Joyner AL, Drucker DJ. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon-like peptide 1 receptor gene. Nat Med. 1996 Nov;2:1254–8. doi: 10.1038/nm1196-1254. [DOI] [PubMed] [Google Scholar]
- 23.Cook S, MacLusky NJ, Scrocchi L, Shin J, Kim J, Vaccarino F, Asa SL, Drucker DJ. Neuroendocrine function and response to stress in mice with complete disruption of glucagon-like peptide-1 receptor signaling. Endocrinology. 2000 Feb;141:752–62. doi: 10.1210/endo.141.2.7326. [DOI] [PubMed] [Google Scholar]
- 24.Lo CM, Zhang DM, Pearson K, Ma L, Sun W, Sakai RR, Davidson WS, Liu M, Raybould HE, Woods SC, Tso P. Interaction of apolipoprotein AIV with cholecystokinin on the control of food intake. Am J Physiol Regul Integr Comp Physiol. 2007 Oct;293:R1490–4. doi: 10.1152/ajpregu.00329.2007. [DOI] [PubMed] [Google Scholar]
- 25.Weinstock PH, Bisgaier CL, Hayek T, Sehayek E, Aalto-Setala K, Wu L, Sheiffele P, Merkel M, Essenburg AD, Breslow JL. Decreased HDL cholesterol levels but normal lipid absorption, growth, and feeding behavior in apolipoprotein A-IV knockout mice. J Lipid Res. 1997 Sep;38:1782–94. [PubMed] [Google Scholar]
- 26.Mulder S, Gebre-Medhin H, Pekny M, Westermark G, Tornell J, Westermark P, Sundler F, Ahren B, Betsholtz C. Increased insulin secretion and glucose tolerance in mice lacking islet amyloid polypeptide (amylin) Biochem Biophys Res Commun. 1998 Sep 18;250:271–7. doi: 10.1006/bbrc.1998.9308. [DOI] [PubMed] [Google Scholar]
- 27.Batterham RL, Heffron H, Kapoor S, Chivers JE, Chandarana K, Herzog H, Roux CWL, Thomas EL, Bell JD, Withers DJ. Critical role for peptide YY in protein-mediated satiation and body-weight regulation. Cell Metab. 2006 Sep;4:223–33. doi: 10.1016/j.cmet.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 28.Schonhoff S, Baggio L, Ratineau C, Ray SK, Lindner J, Magnuson MA, Drucker DJ, Leiter AB. Energy homeostasis and gastrointestinal endocrine differentiation do not require the anorectic hormone peptide YY. Mol Cell Biol. 2005 May;25:4189–99. doi: 10.1128/MCB.25.10.4189-4199.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wortley KE, Garcia K, Okamoto H, Thabet K, Anderson KD, Shen V, Herman JP, Valenzuela D, Yancopoulos GD, Tschop MH, Murphy A, Sleeman MW. Peptide YY regulates bone turnover in rodents. Gastroenterology. 2007 Nov;133:1534–43. doi: 10.1053/j.gastro.2007.08.024. [DOI] [PubMed] [Google Scholar]
- 30.Seeley RJ, Woods SC, D’Alessio D. Targeted gene disruption in endocrine research--the case of glucagon-like peptide-1 and neuroendocrine function. Endocrinology. 2000 Feb;141:473–5. doi: 10.1210/endo.141.2.7372. [DOI] [PubMed] [Google Scholar]
- 31.Ye J, Hao Z, Mumphrey MB, Townsend RL, Patterson LM, Stylopoulos N, Munzberg H, Morrison CD, Drucker DJ, Berthoud HR. GLP-1 receptor signaling is not required for reduced body weight after RYGB in rodents. Am J Physiol Regul Integr Comp Physiol. 2014 Mar 01;306:R352–62. doi: 10.1152/ajpregu.00491.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chambers AP, Kirchner H, Willency JA, Wilson-Perez HE, Hale JE, Gaylinn BD, Thorner MO, Pfluger PT, Gutierrez JA, Tschop MH, Sandoval DA, Seeley RJ. The effects of vertical sleeve gastrectomy in rodents are ghrelin independent. Gastroenterology. 2013 Jan;144:50–52 e5. doi: 10.1053/j.gastro.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woods SC, Strubbe JH. The psychobiology of meals. Psychon Bull Rev. 1994 Jun;1:141–55. doi: 10.3758/BF03200770. [DOI] [PubMed] [Google Scholar]
- 34.Woods SC. The control of food intake: behavioral versus molecular perspectives. Cell Metab. 2009 Jun;9:489–98. doi: 10.1016/j.cmet.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woods SC, Ramsay DS. Food intake, metabolism and homeostasis. Physiol Behav. 2011 Jul 25;104:4–7. doi: 10.1016/j.physbeh.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drazen DL, Vahl TP, Seeley RJ, D’Alessio DA, Woods SC. Effects of a fixed meal pattern on ghrelin secretion: evidence for a learned response independent of nutrient status. Endocrinology. 2006 Jan;147:23–30. doi: 10.1210/en.2005-0973. [DOI] [PubMed] [Google Scholar]
- 37.Teff K. Nutritional implications of the cephalic-phase reflexes: endocrine responses. Appetite. 2000 Apr;34:206–13. doi: 10.1006/appe.1999.0282. [DOI] [PubMed] [Google Scholar]
- 38.Woods SC. The eating paradox: how we tolerate food. Psychol Rev. 1991 Oct;98:488–505. doi: 10.1037/0033-295x.98.4.488. [DOI] [PubMed] [Google Scholar]
- 39.Woods SC. The house economist and the eating paradox. Appetite. 2002 Apr;38:161–5. doi: 10.1006/appe.2001.0468. [DOI] [PubMed] [Google Scholar]
- 40.Steffens AB. Influence of the oral cavity on insulin release in the rat. Am J Physiol. 1976 May;230:1411–5. doi: 10.1152/ajplegacy.1976.230.5.1411. [DOI] [PubMed] [Google Scholar]
- 41.Berthoud HR, Bereiter DA, Trimble ER, Siegel EG, Jeanrenaud B. Cephalic phase, reflex insulin secretion. Neuroanatomical and physiological characterization. Diabetologia. 1981 Mar;20(Suppl):393–401. doi: 10.1007/BF00254508. [DOI] [PubMed] [Google Scholar]
- 42.Lorentzen M, Madsbad S, Kehlet H, Tronier B. Effect of sham-feeding on glucose tolerance and insulin secretion. Acta Endocrinol (Copenh) 1987 May;115:84–6. doi: 10.1530/acta.0.1150084. [DOI] [PubMed] [Google Scholar]
- 43.Rozin P, Kalat JW. Specific hungers and poison avoidance as adaptive specializations of learning. Psychol Rev. 1971 Nov;78:459–86. doi: 10.1037/h0031878. [DOI] [PubMed] [Google Scholar]
- 44.Garcia J, Kimeldorf DJ, Koelling RA. Conditioned aversion to saccharin resulting from exposure to gamma radiation. Science. 1955 Jul 22;122:157–8. [PubMed] [Google Scholar]
- 45.Sclafani A. Post-ingestive positive controls of ingestive behavior. Appetite. 2001 Feb;36:79–83. doi: 10.1006/appe.2000.0370. [DOI] [PubMed] [Google Scholar]
- 46.Touzani K, Bodnar RJ, Sclafani A. Neuropharmacology of learned flavor preferences. Pharmacol Biochem Behav. 2010 Nov;97:55–62. doi: 10.1016/j.pbb.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benoit SC, Davis JF, Davidson TL. Learned and cognitive controls of food intake. Brain Res. 2010 Sep 02;1350:71–6. doi: 10.1016/j.brainres.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davidson TL, Altizer AM, Benoit SC, Walls EK, Powley TL. Encoding and selective activation of “metabolic memories” in the rat. Behav Neurosci. 1997 Oct;111:1014–30. doi: 10.1037//0735-7044.111.5.1014. [DOI] [PubMed] [Google Scholar]
- 49.Davis JD, Smith GP. The conditioned satiating effect of orosensory stimuli. Physiol Behav. 2009 Jun 22;97:293–303. doi: 10.1016/j.physbeh.2009.03.028. [DOI] [PubMed] [Google Scholar]
- 50.Woods SC, Ramsay DS. Pavlovian influences over food and drug intake. Behav Brain Res. 2000 Jun 01;110:175–82. doi: 10.1016/s0166-4328(99)00194-1. [DOI] [PubMed] [Google Scholar]
- 51.Goodison T, Siegel S. Learning and tolerance to the intake suppressive effect of cholecystokinin in rats. Behav Neurosci. 1995 Feb;109:62–70. [PubMed] [Google Scholar]
- 52.Duncan EA, Davita G, Woods SC. Changes in the satiating effect of cholecystokinin over repeated trials. Physiol Behav. 2005 Jul 21;85:387–93. doi: 10.1016/j.physbeh.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 53.Ramsay DS, Woods SC. Physiological Regulation: How It Really Works. Cell Metab. 2016 Sep 13;24:361–4. doi: 10.1016/j.cmet.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]