

Abstract
Cell cycle activation has been associated with varying types of neurological disorders including brain injury. Cyclin D1 is a critical modulator of cell cycle activation and upregulation of Cyclin D1 in neurons contributes to the pathology associated with traumatic brain injury (TBI). Mitochondrial mass is a critical factor to maintain the mitochondrial function, and it can be regulated by different signaling cascades and transcription factors including NRF1. However, the underlying mechanism of how TBI leads to impairment of mitochondrial mass following TBI remains obscure. Our results indicate that augmentation of CyclinD1 attenuates mitochondrial mass formation following TBI. To elucidate the molecular mechanism, we found that Cyclin D1 interacts with a transcription factor NRF1 in the nucleus and prevents NRF1’s interaction with p300 in the pericontusional cortex following TBI. As a result, the acetylation level of NRF1 was decreased, and its transcriptional activity was attenuated. This event leads to a loss of mitochondrial mass in the pericontusional cortex following TBI. Intranasal delivery of Cyclin D1 RNAi immediately after TBI rescues transcriptional activation of NRF1 and recovers mitochondrial mass after TBI.
Keywords: TBI, Mitochondria, Cyclin D1, NRF1
1. Introduction
Neurons are post-mitotic cells have permanently entered G0 phase and were incapable of entering the cell cycle (Frade and Ovejero-Benito, 2015). However, recent studies have shown that matured differentiated neurons can enter into the cell-cycle reentry phase which ultimately results in cell death rather than proliferation (Herrup and Yang, 2007; Kabadi et al., 2012; Kranenburg et al., 1996). The sequential activation of Ser/Thr kinases called the cyclin-dependent kinases (CDK), and their positive regulators (Kabadi and Faden, 2014; Malumbres, 2014) such as activation of Cyclin D1 are essential to maintaining the different stages of cell cycle. Dr. Faden and his associates have convincingly shown that cell cycle activation is responsible for cell death in apoptosis in the post-mitotic cells like neuron (Kabadi and Faden, 2014). In addition, activation of the cell cycle in microglial cells results in the release of pro-inflammatory and neurotoxic molecules which contributes significantly to the pathology of TBI (Byrnes and Faden, 2007; Byrnes et al., 2007; Di Giovanni et al., 2005; Hilton et al., 2008; Wu et al., 2011). However, its role in mitochondrial function is mostly unknown.
Mitochondrial dysfunction has been implicated in the TBI pathology (Fischer et al., 2016; Hill et al., 2017; Singh et al., 2006) and the maintenance of the mitochondrial mass has been suggested to function as a critical factor to maintain the mitochondrial function and production of ATP inside cells (Wang et al., 2017b). The proper intracellular distribution of mitochondria is assumed to be critical for normal physiology of neuronal cells (Cheng et al., 2012; Li et al., 2004). The changes in mitochondrial mass are correlated with the development and morphological plasticity of spines (Cheng et al., 2010; Li et al., 2004). Mitochondrial mass, by itself, represents the net balance between rates of biogenesis and degradation (Dominy and Puigserver, 2013) and mitochondrial mass can be regulated by mitochondrial DNA (mtDNA) content which is known to be synthesized inside the nucleus through activation of several transcription factors (Dominy and Puigserver, 2013; Jornayvaz and Shulman, 2010). PGC-1α is a co-transcriptional regulation factor that induces mitochondrial mass by activating different transcription factors, including NRF1, which promotes the expression of mitochondrial transcription factor A or TFAM (Dominy and Puigserver, 2013; Jornayvaz and Shulman, 2010; Scarpulla, 2011; Ventura-Clapier et al., 2008). NRF1 is an essential contributor to the sequence of events leading to the increase in transcription of key mitochondrial enzymes, and it has been shown to regulate TFAM, which drives transcription and replication of mtDNA (Jezierska-Drutel et al., 2013; Scarpulla, 2008). Previously it was shown that NRF1 could interact and acetylated by an acetyltransferase p300/CBP and acetylation of NRF1 enhances its transcriptional activation by augmenting its DNA binding (Izumi et al., 2003).
Here we have shown that mitochondrial mass was impaired following TBI due to the loss of mtDNA content. As a part of the mechanism, we found that activation of the cell cycle, notably, cyclin D1 directly contributes to the deficiency in mtDNA content by manipulating the transcriptional efficiency of NRF1.
2. Materials and Methods
2.1. TBI procedure:
The Committee on Animal Use for Research and Education at the University of Pittsburgh approved all animal studies, in compliance with National Institutes of Health guidelines. The procedure was done based on our previously published protocol (Farook et al., 2013; Kapoor et al., 2013; Sen et al., 2017). Briefly, 8– 12-week old adult male C57BL/6 (Jackson Laboratory) mice were anesthetized with xylazine (8 mg/kg)/ketamine (60 mg/kg) and subjected to a sham injury or controlled cortical impact. Mice were placed in a stereotaxic frame (Ambient Instruments) and a 3.5 mm craniotomy was made in the right parietal bone midway between bregma and lambda with the medial edge lateral to the midline, leaving the dura intact. Mice were impacted at 4.5 m/s with a 20 ms dwell time and 1.2 mm depression using a 3-mm-diameter convex tip, mimicking a moderate TBI. Sham-operated mice underwent the identical surgical procedures but were not impacted. The incision was closed with 3M Vetbond tissue adhesive and mice were allowed to recover in the heat pad. Body temperature was maintained at 37°C using a small animal temperature controller throughout all procedures (Kopf Instruments).
2.2. Western Blot and Co-immunoprecipitation:
Whole tissue lysates were prepared from 3 mm coronal sections centered upon the site of impact. A 1 mm micro punch was collected from the pericontusional cortex or the corresponding pericontusional hemisphere as described previously (Farook et al., 2013; Kapoor et al., 2013; Mir et al., 2014; Sen et al., 2017). Tissue was placed in RIPA buffer (containing protease and phosphatase inhibitor), sonicated, and centrifuged for 5 min at 12,000 × g at 4°C. Fifty micrograms of protein were resolved on a 4–20% SDS-polyacrylamide gel and transferred onto a nitrocellulose membrane. Blots were incubated overnight at 4°C in primary antibody Cyclin Dl, (Cell Signalling, 1:500 dilution), total NRF1 (Santa Cruz Biotech., 1:500 dilution), p300 (Sigma-Aldrich, 1:500 dilution), total PGClα (Santa Cruz Biotech., 1:500 dilution), Nuclear NRF1 (Abeam, abl75932), nuclear PGClα (Abeam ab54481), TFAM (Santa Cruz Biotech., 1:500 dilution), CoxII (Santa Cruz Biotech., 1:500 dilution), and Actin (Sigma-Aldrich, 1:5000 dilution) followed by a 2 h incubation with a Licor IRDye secondary antibody at room temperature. Blots were visualized using a Fi-Cor Odyssey near-infrared imaging system, and densitometry analysis was performed using Quantity One software (Bio-Rad) (Farook et al., 2013; Kapoor et al., 2013; Mir et al., 2014). The intensity of each band was determined by ImageJ software, and the changes in the experimental band were represented as the fold change as described previously (Sen et al., 2017; Sen and Sen, 2016).
Protein-protein interactions and protein acetylation were measured by co-immunoprecipitation assay per our method (Farook et al., 2013; Kapoor et al., 2013; Mir et al., 2014; Sen et al., 2017). Briefly, treated or untreated cells were homogenized in lysis buffer containing 50 mm Tris, pH 7.4, 150mM NaCl, 0.5% (v/v) tween-20, 50mM Tris (pH 7.5), 1mM EDTA with protease and phosphatase inhibitor by passing through 26- gauge syringe needle and centrifuged at 12,000 × g for 5 min. 400ug of the total protein for each sample were incubated overnight with either CyclinD1 antibody (1:100), NRF1(1:100) or anti-IgG antibody for overnight. 30μ1 of protein G agarose was added, and SDS-PAGE resolved co-immunoprecipitates and analyzed by western blotting with either the NRF1 antibody, p300 antibody or acetyl-lysine antibody.
2.3. Immunohistochemistry:
Deeply anesthetized mice were perfused with phosphate buffer saline (0.1 M PBS, pH 7.4), followed by fixation with 4% paraformaldehyde in PBS. Brains were post-fixed for 1h in 4% paraformaldehyde, followed by cryoprotection with 30% sucrose in PBS until brains permeated. Serial coronal sections of 20μm were prepared using a cryostat microtome (Leica) and mounted directly onto glass slides before allowing them for drying for 1h at room temperature. For immunofluorescence analysis sections were incubated 10 min at room temperature with 0.1% Triton X-100 in PBS containing, followed by overnight incubation at 4°C with the primary antibody against CyclinD1 (1:100), NRF1 (1:100), PGClα (1:100) or p300 (1:100 dilution). Sections were then washed with PBS and incubated with the appropriate Alexa Fluor-tagged secondary antibody for 2h at room temperature in the dark. Sections were washed and mounted with sufficient drops of ProLong Gold Antifade Mountant with DAPI solution (Molecular Probes). Imaging was performed with the help of Olympus fluorescent microscope (IX83) and Nikon’s C1 Digital Eclipse Modular Confocal Microscope Systems. The omission of primary antibody served as a negative control (Farook et al., 2013; Kapoor et al., 2013; Mir et al., 2014; Sen et al., 2017).
2.4. Intranasal delivery of Cyclin D1-RNAi:
CyclinD1-RNAi (Santa Cruz Biotechnology) were administered to 8–12 weeks C57BL/6J mice through intranasal route using in vivo jetPEI (PolyPlus) transfection reagent as described previously with modifications (Bitko and Barik, 2008; Rodriguez et al., 2017). The RNAi-JetPEI complex was prepared according to the manufacturer’s protocol with modifications (Aigner, 2006; Rodriguez et al., 2017). Briefly, either the cyclinD1 RNAi or control RNAi along with JetPEI were separately diluted into half the injection volume in a 10% sterile glucose solution where the final glucose concentration would have to be 5%. This formulation corresponds to nitrogen and phosphate (N/P) ratio of 7. Both the solutions were mixed by slight vortexing, and the JetPEI-RNAi mixture was incubated 15 min at room temperature. Intranasal administration of the Jet-PEI complex was performed 30 min after either sham or TBI surgery with the pipette tip to each nostril of the mouse. A 5ul of the jetPEI-RNAi complex was slowly administered to the nostrils maintaining a 2–3 sec interval up to l0u1 total/nostril of a mouse.After 5–10 s another 10u1 of the solution was administered to the other nostril following the similar way for a total of 20ul/mouse and 10ug of siRNA/mouse. Mice were under observation for the entire solution disappears through the nasal cavity and till their consciousness. After 24 hours all the mice were sacrificed, and brain samples were collected for further experiments.
2.5. Chromatin immunoprecipitation (ChIP) assay:
For chromatin immunoprecipitation (ChIP) assays, we used a chromatin immunoprecipitation assay kit purchased from Millipore and followed the instructions from the supplier. ChIP assay was performed as described previously (Mir et al., 2014; Sen et al., 2017). Briefly, after sonication, tissue lysates containing soluble chromatin were incubated overnight with an anti-NRF1 antibody or with normal rabbit IgG. DNA-protein immunocomplexes were precipitated with protein A-agarose beads, washed, and eluted. The eluates were used as templates in PCR using the primers 5’-TTTGCTGTTGGGCA −3’ and 5’-CGGCGGCTTACCCCA −3’. The expected DNA fragment that was amplified is in the TFAM promoter region, which encompassed the NRF1 binding site.
2.6. Analysis of mitochondrial DNA (mtDNA):
mtDNA was isolated from the using a Mitochondrial DNA Isolation Kit (Biovison). Briefly, tissues were incubated with extraction buffer and homogenized using Dounce homogenizer. The homogenates were separated into the cytosol (supernatant) and mitochondrial fractions (pellet) by differential centrifugation following manufacturer’s protocol. The mitochondrial pellets were lysed overnight using mitochondrial lysis buffer. mtDNA will be isolated by ethanol precipitation. An aliquot of homogenates was reserved for protein quantification, and mtDNA content was normalized to the protein concentration (Cheng et al., 2012; Zhang et al., 2014). Total nuclear DNA was isolated from the nuclear fraction using QIAamp DNA Mini Kit (Qiagen), according to the manufacturer’s protocol. The purified mitochondrial DNA was quantified by quantitative PCR with SYBR Green master mix (Quanta Biosystems) as described previously (Gonzalez-Hunt et al., 2016; Rooney et al., 2015). Mitochondrial DNA content was represented by primers towards two mtDNA- encoded genes, mitochondrial cyclooxygenase II (CoxII), and NADH dehydrogenase subunit 1 (ND1, Realtimesprimer.com) normalized to a nuclear intron of β-globin. The primer sequences were as follows: Cox2, 5’-GCCGACTAAATCAAGCAACA-3’ (forward) and 5’-CAATGGGCATAAAGCTATGG-3’ (reverse); and β-globin, 5 ‘-GAAGCGATTCTAGGGAGCAG-3’ (forward) and 5’-GGAGCAGC GATTCTGAGTAGA-3’ (reverse). The relative mtDNA to nuclear DNA copy number ratio was determined using the comparative DDCT method (Ballista-Hemandez et al., 2017; Gonzalez-Hunt et al., 2016; Lien et al., 2017), in which NDl/β-globin and Cox2/β-globin ratios were calculated.
2.7. Determination of mitochondrial mass:
For mitochondrial mass measurements freshly prepared mitochondrial fraction from coronal tissues sections were made by differential centrifugation following previous publication (Sen et al., 2007). The isolated mitochondria were loaded with MitoTracker Green (Molecular Probes) at a final concentration of 500 nM and incubated for 20 min. The fluorescence intensity will be measured at ex. 485 and em. 535 nm (Cheng et al., 2012) using BioTek’s Fluorescence Microplate Readers. The values will be normalized by protein concentrations.
2.8. Citrate Synthase Activity:
Citrate synthase activity was determined in homogenates prepared from pericontusional cortex using a citrate synthase assay kit (K318, Biovision)(Liu et al., 2015; Wang et al., 2017a) Total protein was determined in triplicate by the method of Bradford and the protein concentration of all samples was equalized. Citrate synthase activity was determined in triplicate based on the formation of 2-nitro-5- thiobenzoic acid at a wavelength of 412 nm at 25°C on a spectrophotometer. In each well, 8 μl of sample was added to a reaction medium containing 178 μl of assay buffer, 2 μl of 30 mmol/L acetyl coenzyme A, and 10 mmol/L 2-nitro-5-thiobenzoic acid. The baseline solution absorbance was recorded, reactions were initiated by the addition of 10 μl of oxaloacetic acid, and the change in absorbance measured every 15 seconds for 2 minutes.
2.8. Statistical Analysis:
All data are presented as the mean ± SEM (standard error of the mean). The effects of treatments were analyzed using a one-way ANOVA. Results are expressed as mean ± SEM (n = 5–7). A p-value <0.05 was considered as statistically significant. Male C57BL/6 mice were used for all experiments.
3. Results:
3.1. TBI leads to a decrease in mitochondrial mass
Mice were subjected to TBI, and pericontusional cortex was isolated after 24h of TBI. For the measurement of mitochondrial mass, mitochondrial fractions from pericontusional cortex were isolated and incubated with MitoTracker Green. We found that the green fluorescent intensity was decreased significantly after 24h of TBI compared to a sham control to more than 35%. This data indicates that TBI may affect mitochondrial mass (Fig. 1A).
Figure 1: TBI causes a decrease in mitochondrial mass.
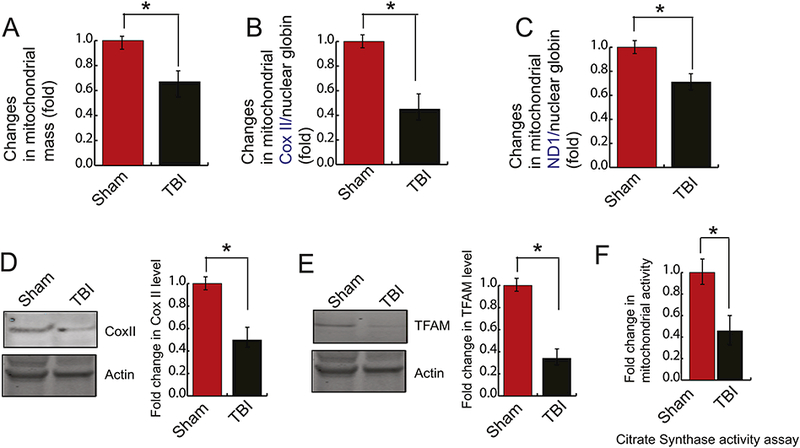
(A) Fold changes in mitochondrial mass obtained from staining from MitoTracker Green was monitored in both sham and TBI samples by a spectrofluorometer. It was shown that TBI causes a decrease in mitochondrial mass (n=3–5; one-way ANOVA, p<0.005). (B-C) Mitochondrial DNA quantification by Real-time PCR was represented by mitochondrial COXII (B) and mitochondrial ND1 (C) normalized to nuclear ß-globin isolated from nuclear DNA. It was shown that COXII and ND1 was decreased significantly after TBI. (n=3–5; one-way ANOVA, p<0.005) (D-E) Western blot and quantitative analysis of the expression of TFAM (D) and COXII (E) level in both sham and TBI samples. The band intensity of TFAM (D) and COXII (E) was monitored by Image J. The expression level of TFAM and COXII was decreased significantly after TBI. (n=3–5; one-way ANOVA, *p<0.005).
Since the content of mtDNA is a critical determinant of mitochondrial mass (de la Monte et al., 2000; Lanza and Nair, 2010; Lee and Wei, 2005; May-Panloup et al., 2005), we monitored the mtDNA content and the level of mitochondrial genes e.g. ND1 and CoxII using real-time-PCR using mitochondrial DNA as a template. We found that the level of mtDNA content was decreased significantly after TBI which was evidenced by a decrease in levels of ND1 and COXII genes (Fig. 1B and 1C). To further confirm, we have monitored the protein level of CoxII using the pericontusional cortex and found that the expression of COXII was decreased significantly following TBI (Fig. 1D). Since little is known to regulate the expression of mitochondrial genes, we monitored the expression of TFAM in the mitochondrial fraction of TBI. We found that the protein level of TFAM was decreased following TBI (Fig. 1E). This data suggests that TBI affects the mtDNA content inside the brain.
To see whether the loss of mtDNA affects the mitochondrial function following TBI, we monitored the mitochondrial function by citrate synthase assay. Citrate Synthase is a key enzyme that is present in all living organisms. It catalyzes the conversion of acetyl-CoA and oxaloacetate into citrate and serves as a marker for intact mitochondria and it is considered as a biochemical determination of mitochondrial mass (Lopez-Lluch et al., 2006; Yin et al., 2008). We found that, following TBI, mitochondria function was decreased more than 70% compared to sham mice after 24h (Fig. 1F).
3.2. TBI leads to inactivation of NRF1 by decreasing its acetylation via p300
Since, the nuclear transcription factors such as PGC1α and NRF1 are known to regulate the synthesis of mitochondrial genes (Fernandez-Marcos and Auwerx, 2011; Jornayvaz and Shulman, 2010; Scarpulla, 2011), we monitored the level of these transcription factors using pericontusional cortex isolated after 24h of TBI. Interestingly, we did not find a significant change in the expression level of either nuclear or total level of PGC1α or NRF1 (Fig. 2A). To further confirm, we have carried out confocal microscopic analysis of both PGC1α and NRF1 in the pericontusional cortex. Similar to our western blot data, we found that the total expression level (supplementary Fig 1A and Fig 1B) or nuclear level (Fig 2B and Fig 2C) of both PGClα and NRF1 remains unaltered compared to the sham condition. Previously it was shown that the activation of PGClα depends on its acetylation status (Gurd, 2011; Jeninga et al., 2010). To test whether TBI has any difference in acetylation level of PGClα, we performed the acetylation assay of PGClα. We found that the acetylation status of PGClα remained unaltered following TBI (Fig. 2D). To further confirm, we monitored the interaction between PGClα and acetyltransferase, p300 and found that there is a change in their level of interaction (Fig. 2E). This data leads us to test whether transcriptional activity of NRF1 contributes to the alteration in mtDNA following TBI.
Figure 2: TBI does not affect the expression level of PGC1α and NRF1; however, decreases acetylation level of NRF1.
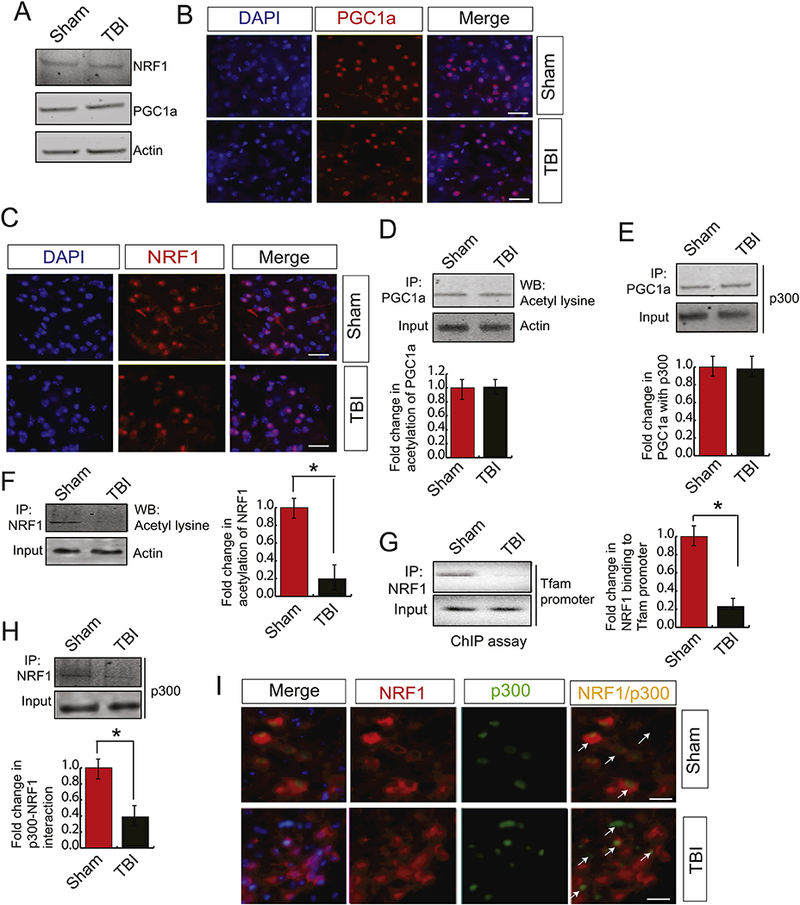
(A) Western blot analysis of NRF1 and PGClα in both sham and TBI samples and it was shown that the expression level of NRF1 remains the same after TBI. (B-C) Confocal microscopic analysis of PGClα (B) and NRF1 (C) in both sham and TBI samples and it shows that there is no change in the nuclear expression level of NRF1 and PGCla following TBI. Scale bar 10μΜ. (D) Immunoprecipitation assay detected acetylation of PGClα in both sham and TBI samples with PGClα antibody followed by western blotting using the acetyl- lysine antibody as described in the methods. The band intensity of acetylated PGClα was monitored by Image J and it shows that there is no change in acetylation level of PGClα. (n=3–5; one-way ANOVA, p<0.005) (E) Immunoprecipitation assay detected the interaction between PGClα and p300 in both sham and TBI samples by PGClα antibody followed by western blotting by p300 antibody as described in the methods. Image J monitored the band intensity of the interaction between PGClα and p300. The data shows that there is no change in acetylation level of PGClα (n=3–5; one-way ANOVA, p<0.005) (F) Acetylation of NRFl in both sham and TBI samples were detected by immunoprecipitation assay by NRF1 antibody followed by western blotting with the acetyl-lysine antibody. The band intensity of acetylated NRF1 was monitored by Image J and it shows that the acetylation level of NRFl was decreased significantly after TBI (n=3–5; one-way ANOVA, *p<0.005) (G) ChIP assay to demonstrate NRF1 binding to TFAM promoter. The band intensity of acetylated PGClα was monitored by Image J and it shows that NRF1 binding to TFAM promoter was decreased significantly after TBI (n=3–5; one-way ANOVA, p<0.005) (H) Immunoprecipitation assay detected the interaction between NRF1 and p300 in both sham and TBI samples by NRF1 antibody followed by western blotting by p300 antibody as described in the methods. Image J monitored the band intensity of the interaction between NRF1 and p300. The data shows that the interaction between NRF1 and p300 was decreased significantly after TBI (n=3–5; one-way ANOVA, *p<0.005). (I) Confocal microscopic analysis of p300 and NRF1 in both sham and TBI samples. The arrowhead shows colocalization and interaction between NRF1 and p300 in sham and TBI samples. The results show that the interaction between NRF1 and p300 was decreased in TBI mice. Scale bar 10μΜ.
Previously it was shown that acetylation of NRF1 is required for its transcription (Izumi et al., 2003). To test whether TBI has any influence on acetylation level of NRF1, we have carried out acetylation assay using the pericontusional cortex after 24h of TBI. Interestingly, we found that the acetylation of NRF1 was decreased significantly compared to sham condition (Fig. 2F). This data indicates that the lack of acetylation of NRF1 may affect its transcriptional activity following TBI. To measure whether the DNA binding affinity of NRF1 is altered in the pericontusional cortex after TBI, we have performed the chromatin immunoprecipitation (ChIP) assay using TFAM promoter. We found that the binding of NRF1 to the TFAM promoter was decreased significantly in TBI mice (Fig. 2G); suggests that the transcriptional activity of NRF1 was attenuated after TBI due to the loss of its acetylation status.
We were interested in understanding the underlying mechanism responsible for a decrease in acetylation of NRF1. Previously it was shown that NRF1 interacts with an acetyltransferase p300 which in turn acetylates NRF1. We monitored the interaction between NRF1 and p300 in the pericontusional cortex after TBI through the immunoprecipitation (IP) assay, and we found that the binding between NRF1 and p300 was decreased significantly compared to sham control (Fig. 2H). To further confirm, we have done the analysis using confocal microscopy, and we found that the interaction between NRF1 and p300 was decreased in the pericontusional cortex compared to sham control (Fig. 2I).
3.3. TBI leads to an augmentation in Cyclin D1 and the interaction between Cyclin D1 and NRF1 prevents acetylation of NRF1.
This data raised the question what the possible mechanism for a decrease in interaction between p300 and NRF1 is. Previously it was shown that TBI causes cell cycle activation and the level of cyclin D1 (CD1) was increased significantly after TBI (Kabadi et al., 2012). We monitored the activation of CD1 in mice following TBI using CCI method. Consistent with the previous finding, we found that within 6h of TBI, the CD1 level was increased and it was further augmented at 12h and 24h of TBI. Then the CD1 level remains unaltered up to 3 days of TBI (Fig. 3A). To further confirm, we have carried out the confocal microscopy for the cyclin D1 level in both TBI and sham conditions. We found that TBI leads to an increase in Cyclin D1 level compared to sham condition (Fig. 3B). However, the western blot data cannot distinguish whether neuronal cells and glial cells contribute to the increase in CD1 following TBI. To confirm whether glial cells can also express an increased level of CD1, we stained glial cells with anti-Ibal antibody along with anti-CDl antibody. Neuronal cells were stained with NeuN. Interestingly, we found that CD1 expression was increased in the nucleus of Iba1-positive cells along with NeuN positive cells; indicates that glial cells also express higher level of CD1 like neuronal cells following TBI (Fig. 3C).
Figure 3: TBI causes an increase in Cyclin D1 which interacts with NRF1.
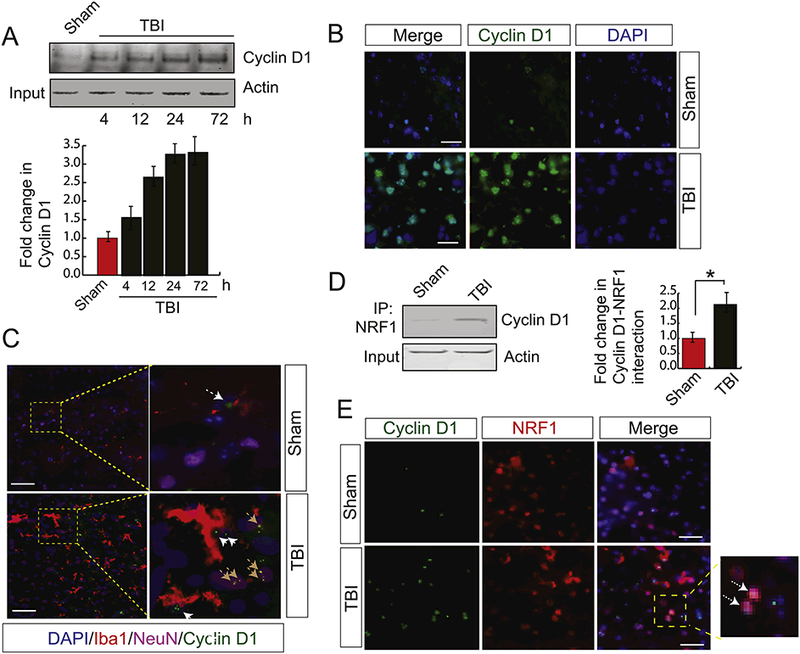
(A) Western blot analysis of cyclin D1 in both sham and TBI samples. The band intensity of cyclin D1 was monitored by Image J and the data shows that the expression level of CD1 was increased in time dependent manner (n=3–5; one-way ANOVA, *p<0.005). (B) Confocal microscopic analysis of cyclin D1 in both sham and TBI samples. The data shows that cyclin D1 level was increased after TBI. Scale bar 20μΜ. (C) Confocal microscopic analysis to monitor neuronal and microglial expression of CD1 following TBI. We found that higher expression of CD1 in microglial cells were increased after TBI. Neuronal cells were labelled with NeuN and glial cells were labelled with Iba1. The brown arrowhead indicated CD1 in NeuN positive cells and white arrowheads indicates cyclin D1 in glial cells. Scale bar 10μΜ. (D) The interaction between NRF1 and Cyclin D1 was monitored in both sham and TBI samples by immunoprécipitation analysis using NRF1 antibody and western blotting using Cyclin D1 antibody. Actin was kept as input. The data shows that the interaction between NRF1 and cyclin D1 was increased significantly after TBI (n=3–5; one-way ANOVA, *p<0.005). (E) Confocal microscopic analysis of the interaction between NRF1 and Cyclin D1 in both sham and TBI samples. The data shows that the interaction between NRF1 and cyclin D1 was increased significantly after TBI. The arrowhead shows co-localization and interaction between NRF1 and Cyclin D1 in sham and TBI samples. Scale bar 10μΜ.
To see whether CD1 can interact with NRF1 in the nucleus, we performed the immunoprecipitation assay and found that the interaction between NRF1 and CD1 was increased significantly after TBI (Fig. 3D). This was further confirmed by confocal microscopy analysis where NRF1 and CD1 was co-localized in the nucleus (Fig. 3E). The interaction between NRF1 and CD1 was further evaluated using an antibody that can monitor both cytosolic and nuclear pool of NRF1. We found that the interaction was restricted only inside the nucleus (supplementary Figure 1C).
3.4. Depletion of CD1 rescues transcriptional activation of NRF1, mitochondrial mass, and mtDNA content
To understand whether depletion of CD1 can rescue the mitochondrial mass, we administered CD1-RNAi through intranasal delivery within 30 mins of TBI; although the expression level of CD1 was shown to be increased within 4h of TBI. Western blot analysis monitored the level of CD1. We found that intranasal administration of cyclin D1-RNAi causes a significant decrease in CD1 level in the cortex either after sham or TBI (Fig. 4A, Input). To see whether depletion of CD1 has any influence on either acetylation of NRF1 or the interaction between NRF1 and p300 after TBI, we performed immunoprecipitation assay and found that the TBI- induced decrease in the level of interaction between NRF1 and p300 was rescued in mice administered with CD1-RNAi compared to control RNAi (Fig. 4A, B). Consistent with this result, we found that acetylation of NRF1 was recovered after depletion of Cyclin D1 before TBI in mice (Fig 4A, C).
Figure 4: Depletion of Cyclin D1 rescue the loss of mitochondrial mass following TBI.
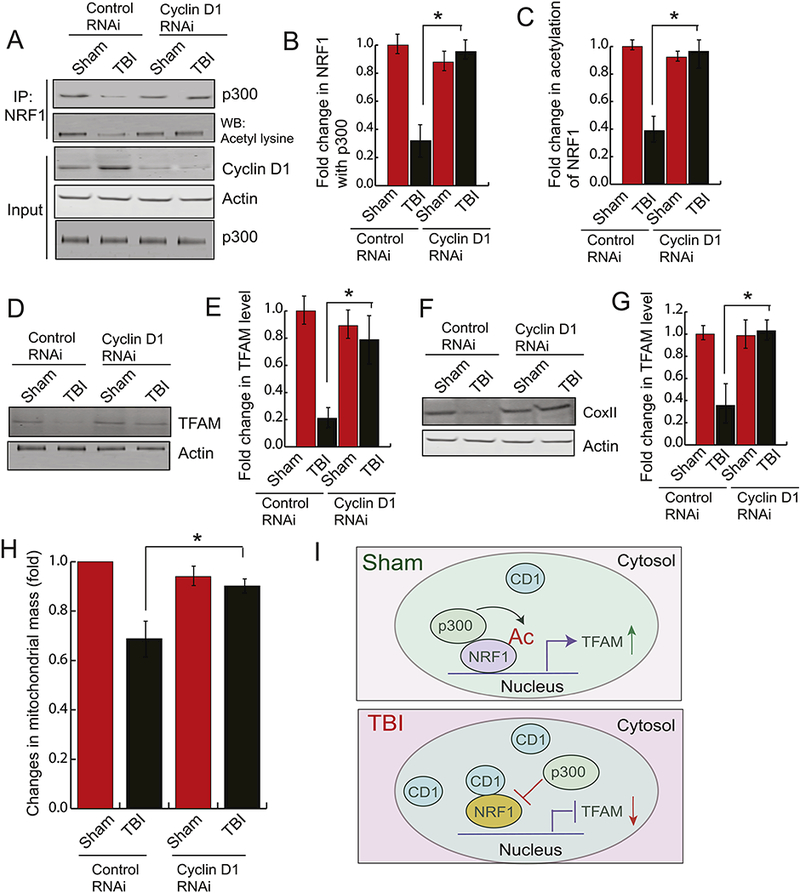
Cyclin Dl-siRNA was administered into the mouse brain by intranasal delivery before TBI. (A-C) The pericontusional cortex was used to perform immunoprecipitation assay to detect acetylation of NRFl and the interaction between NRF1 and p300. The expression level of p300, cyclin D1 and Actin was monitored by western blot hybridization. The band intensity of the interaction between p300-NRF1 (B) and the level of acetylated NRF1 (C) was monitored by Image J. (n=3–5; one-way ANOVA, *p<0.005). The data shows that depletion of CD1 rescues acetylation of NRF1 and the interaction between p300 and NRF1. (D-E) The pericontusional cortex was used to perform western blot analysis of TFAM (D) and the band intensity of TFAM was monitored by Image J. (n=3–5; one-way ANOVA, *p<0.005) (E) in both sham and TBI mice administered with either control RNAi or CD1 RNAi. The data shows that depletion of CD1 rescues TFAM level after TBI. (F-G) The pericontusional cortex was used to perform western blot analysis of COXI (F), and the band intensity of COXII was monitored by Image J. (n=3–5; one-way ANOVA, *p<0.005) (G) in both sham and TBI mice administered with either control RNAi or CD1 RNAi. The data shows that depletion of CD1 rescues COXII level after TBI. (I) Fold change in mitochondrial mass was monitored in both sham and TBI mice administered with either control RNAi or CD1 RNAi (n=3–5; one-way ANOVA, *p<0.005). The data shows that depletion of CD1 rescues mitochondrial mass after TBI.
To see whether depletion of CD1 can rescue the transcriptional activity of NRF1, we measured the TFAM level by western blot analysis after Cyclin D1-RNAi and found that protein level of TFAM was significantly increased compared to control RNAi (Fig. 4D, E). Similarly, the expression of COXII, encoded by mt-DNA was also rescued after depletion of cyclin D1 in mice before TBI (Fig. 4F, G). This result suggests that depletion of cyclin D1 can rescue the mtDNA content by rescuing the transcriptional activity of NRF1. We also have measured the mitochondrial mass in mice administered either with control RNAi or CD1-RNAi using MitoTracker Green. We found that TBI-induced loss of mitochondrial mass was rescued in mice where CD1 was depleted following TBI (Fig. 4H).
4. Discussion:
Previous studies have linked CD1 with neuronal death and neurodegeneration in models of cerebral ischemia (Li et al., 1997; Small et al., 2001), spinal cord injury (Byrnes and Faden, 2007), and Alzheimer’s disease (Raina et al., 2000). Our study suggests that activation of CD1 following TBI affects mitochondrial mass through impairment of a key transcription factor, NRF1 in the nucleus. TBI leads to a decrease in acetylation of NRF1 due to a reduced interaction between NRF1 and p300. An increase in the level of CD1 blocks the interaction between NRF1 and p300 in the nucleus, and as a result, the transcriptional activity of NRF1 was reduced. Administration of RNAi for CD1 rescues the interaction between p300 and NRF1 and recovers the transcriptional activity of NRF1 following TBI (Fig. 4I).
Although the influence of CD 1 has been established in the neuronal cell survival or death, the influence of CD1 in mitochondrial function has not been shown yet. This study provides evidence in support of the fact that augmentation in cyclin D1 can directly influence the mitochondrial mass via modulating the transcriptional activity of NRF1. Mitochondrial mass is one of the critical factors to maintain the function of mitochondria including energy metabolism. The mitochondrial oxidative phosphorylation (OXPHOS) is critical for energy (ATP) production in eukaryotic cells (Chaban et al., 2014). The OXPHOS enzymes are multimeric complexes (Chaban et al., 2014), and NRF1 mostly transcribes genes coding for mitochondrial proteins involved in energy production (Reinecke et al., 2009). Thus, either depletion or inactivation of NRF1 will lead to an impairment in OXPHOS which ultimately leads to mitochondrial dysfunction and oxidative stress inside cells. TBI-induced, a decrease in transcriptional activation of NRF1, can explain how a loss of mitochondrial mass contributes to compromise in the mitochondrial function and induce oxidative stress. As a part of the mechanism, we have shown that reducing the loss of mitochondrial mass by reducing the level of cyclin D1 would be a novel strategy to rescue mitochondrial function following TBI.
The current literature mostly focuses on the fact that inactivation of PGC1α contributes to the loss of mitochondrial mass in several neurodegenerative diseases (Austin and St-Pierre, 2012; Corona and Duchen, 2015; Cui et al., 2006). Here we have shown that TBI causes a loss of mitochondrial mass independent of activation of PGClα instead, it depends on the activation of another transcription factor, NRF1. Also, we have provided evidence that NRF1 interacts with an acetyltransferase p300 and acetylation of NRF1 is indispensable for its catalytic activity. Our study may provide the first-time evidence where activation of NRF1 regulates mitochondrial mass independent of PGClα. Because impairment in mitochondrial mass contributes to several neurodegenerative diseases such as Alzheimer’s Diseases (AD) and Parkinson’s Disease (PD), the elucidation of the role of NRFI to mitochondrial mass can be extended to understand the pathology and develop the therapeutic strategy against these disorders.
The blood-brain barrier (BBB) is known to protect the brain from unwanted systemic circulatory toxins and pathogens by blocking the entry of molecules owing to the tight junctions (Mathupala, 2009). Under the physiological condition, the integrity of the BBB is critical to protecting the CNS; however, under the pathological condition, the integrity of BBB functions as a barrier to delivering drugs to the injured brain. Interestingly, intranasal delivery of drugs is a non-invasive and cost-effective method to deliver drugs without affecting the BBB fimction(Fukuda and Badaut, 2013). The exact mechanisms involved in intranasal drug delivery to the CNS are not fully understood. Anatomical and functional studies have suggested that this method works because of the unique connection which the olfactory and trigeminal nerves provide between the brain and external environments. The neural connections between the nasal mucosa and the brain provide a unique pathway for the non-invasive delivery of therapeutic agents to the CNS (Dhuria et al., 2010; Ilium, 2000; Mathison et al., 1998; Tayebati et al., 2013; Thome and Frey, 2001). The olfactory neural pathway provides both an intraneuronal and extraneuronal pathway into the brain (Tayebati et al., 2013; Thome et al., 1995). In fact, Dr. Frey and his associates have shown that intranasal delivery of compounds such as insulin-like growth factor and deferoxamine to brain diseases such as stroke and Alzheimer’s disease, provided efficient improvements in recovery with no significant side-effects (Danielyan et al., 2010; Hanson and Frey, 2008; Hanson et al., 2009; Liu et al., 2001a; Liu et al., 2001b; Wei et al., 2007). One of the primary reason for having promising results is that the intranasal cavity has access to the brain and is unhindered by the BBB (Chauhan and Chauhan, 2015; Djupesland et al., 2013). SiRNA, in general, cannot cross the BBB because of its high charge and molecular weight (Pardridge, 2007; Upadhyay, 2014). However, intranasal delivery of siRNA can reach the brain cells and provided beneficial effects against stroke (Kim et al., 2012). In this study, we have shown that intranasal delivery of CD1-RNAi rescues the loss of mitochondrial mass by rescuing the transcriptional activation of NRF1. Thus, our study provides a novel approach to deliver drugs to the brain following TBI.
Supplementary Material
Supplementary Figure 1: TBI does not affect total level of PGC1α, NRF1 and interaction between NRF1 and CD1. (A-B) Confocal microscopic analysis of PGC1α and NRF1 in the pericontusional following either Sham or TBI. Red fluorescence indicates the level of either PGC1α (A) and NRF1 (B). The data shows that the expression level of both proteins remain unaltered in both sham and TBI conditions. (C) the interaction between CD1 and NRF1 was increased in the nucleus after TBI.
Highlights.
Mitochondrial mass was compromised after TBI.
The level of NRF1, a key transcription factor to maintain mitochondrial mass remains unaltered after TBI.
A decrease in acetylation of NRF1 is responsible for attenuation of its transcriptional activity.
An increase in interaction between NRF1 and cyclin D1 in the nucleus prevents acetylation of NRF1.
Acknowledgment:
This work was partly supported by the National Institutes of Health (Grants RO1NS094516 and RO1EY025622 to N.S., R.G., P.S and T.S.), funding from University of Pittsburgh and Copeland Foundation to R.G, T.S, and N.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aigner A (2006). Delivery systems for the direct application of siRNAs to induce RNA interference (RNAi) in vivo. J Biomed Biotechnol 2006, 71659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin S, and St-Pierre J (2012). PGC1alpha and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci 125, 4963–4971. [DOI] [PubMed] [Google Scholar]
- Ballista-Hernandez J, Martinez-Ferrer M, Velez R, Climent C, Sanchez-Vazquez MM, Torres C, Rodriguez-Munoz A, Ayala-Pena S, and Torres-Ramos CA (2017). Mitochondrial DNA Integrity Is Maintained by APE1 in Carcinogen-Induced Colorectal Cancer. Mol Cancer Res 15, 831–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitko V, and Barik S (2008). Nasal delivery of siRNA. Methods Mol Biol 442, 75–82. [DOI] [PubMed] [Google Scholar]
- Byrnes KR, and Faden AI (2007). Role of cell cycle proteins in CNS injury. Neurochem Res 32, 1799–1807. [DOI] [PubMed] [Google Scholar]
- Byrnes KR, Stoica BA, Fricke S, Di Giovanni S, and Faden AI (2007). Cell cycle activation contributes tc post-mitotic cell death and secondary damage after spinal cord injury. Brain 130, 2977–2992. [DOI] [PubMed] [Google Scholar]
- Chaban Y, Boekema EJ, and Dudkina NV (2014). Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim Biophys Acta 1837, 418–426. [DOI] [PubMed] [Google Scholar]
- Chauhan MB, and Chauhan NB (2015). Brain Uptake of Neurotherapeutics after Intranasal versus Intraperitoneal Delivery in Mice. J Neurol Neurosurg 2. [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Hou Y, and Mattson MP (2010). Mitochondria and neuroplasticity. ASN Neuro 2, e00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Wan R, Yang JL, Kamimura N, Son TG, Ouyang X, Luo Y, Okun E, and Mattson MP (2012). Involvement of PGC-1alpha in the formation and maintenance of neuronal dendritic spines. Nat Commun 3, 1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona JC, and Duchen MR (2015). Impaired mitochondrial homeostasis and neurodegeneration: towards new therapeutic targets? J Bioenerg Biomembr 47, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, and Krainc D (2006). Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127, 59–69. [DOI] [PubMed] [Google Scholar]
- Danielyan L, Klein R, Hanson LR, Buadze M, Schwab M, Gleiter CH, and Frey WH (2010). Protective effects of intranasal losartan in the APP/PS1 transgenic mouse model of Alzheimer disease. Rejuvenation Res 13, 195–201. [DOI] [PubMed] [Google Scholar]
- de la Monte SM, Luong T, Neely TR, Robinson D, and Wands JR (2000). Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer’s disease. Lab Invest 80, 1323–1335. [DOI] [PubMed] [Google Scholar]
- Di Giovanni S, Movsesyan V, Ahmed F, Cernak I, Schinelli S, Stoica B, and Faden AI (2005). Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci U S A 102, 8333–8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djupesland PG, Mahmoud RA, and Messina JC (2013). Accessing the brain: the nose may know the way. J Cereb Blood Flow Metab 33, 793–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominy JE, and Puigserver P (2013). Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farook JM, Shields J, Tawfik A, Markand S, Sen T, Smith SB, Brann D, Dhandapani KM, and Sen,. (2013). GADD34 induces cell death through inactivation of Akt following traumatic brain injury. Cell Death Dis 4, e754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Marcos PJ, and Auwerx J (2011). Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 93, 884S–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer TD, Hylin MJ, Zhao J, Moore AN, Waxham MN, and Dash PK (2016). Altered Mitochondrial Dynamics and TBI Pathophysiology. Front Syst Neurosci 10, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frade JM, and Ovejero-Benito MC (2015). Neuronal cell cycle: the neuron itself and its circumstances. Cell Cycle 14, 712–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda AM, and Badaut J (2013). siRNA Treatment: “A Sword-in-the-Stone” for Acute Brain Injuries. Genes (Basel) 4, 435–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Hunt CP, Rooney JP, Ryde IT, Anbalagan C, Joglekar R, and Meyer JN (2016). PCR-Based Analysis of Mitochondrial DNA Copy Number, Mitochondrial DNA Damage, and Nuclear DNA Damage. Curr Protoc Toxicol 67, 20 11 21–20 11 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurd BJ (2011). Deacetylation of PGC-1alpha by SIRT1: importance for skeletal muscle function and exercise- induced mitochondrial biogenesis. Appl Physiol Nutr Metab 36, 589–597. [DOI] [PubMed] [Google Scholar]
- Hanson LR, and Frey WH 2nd, (2008). Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci 9 Suppl 3, S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson LR, Roeytenberg A, Martinez PM, Coppes VG, Sweet DC, Rao RJ, Marti DL, Hoekman JD, Matthews RB, Frey WH, 2nd, et al. (2009). Intranasal deferoxamine provides increased brain exposure and significant protection in rat ischemic stroke. J Pharmacol Exp Ther 330, 679–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrup K, and Yang Y (2007). Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat Rev Neurosci 8, 368–378. [DOI] [PubMed] [Google Scholar]
- Hill RL, Singh IN, Wang JA, and Hall ED (2017). Time courses of post-injury mitochondrial oxidative damage and respiratory dysfunction and neuronal cytoskeletal degradation in a rat model of focal traumatic brain injury. Neurochem Int 111, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton GD, Stoica BA, Byrnes KR, and Faden AI (2008). Roscovitine reduces neuronal loss, glial activation, and neurologic deficits after brain trauma. J Cereb Blood Flow Metab 28, 1845–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi H, Ohta R, Nagatani G,Ise T, Nakayama Y, Nomoto M, and Kohno K (2003). p300/CBP- associated factor (P/CAF) interacts with nuclear respiratory factor-1 to regulate the UDP-N-acetyl-alpha-d- galactosamine: polypeptide N-acetylgalactosaminyltransferase-3 gene. Biochem J 373, 713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeninga EH, Schoonjans K, and Auwerx J (2010). Reversible acetylation of PGC-1: connecting energy sensors and effectors to guarantee metabolic flexibility. Oncogene 29, 4617–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jezierska-Drutel A, Rosenzweig SA, and Neumann CA (2013). Role of oxidative stress and the microenvironment in breast cancer development and progression. Adv Cancer Res 119, 107–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jornayvaz FR, and Shulman GI (2010). Regulation of mitochondrial biogenesis. Essays Biochem 47, 69–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi SV, and Faden AI (2014). Selective CDK inhibitors: promising candidates for future clinical traumatic brain injury trials. Neural Regen Res 9, 1578–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi SV, Stoica BA, Loane DJ, Byrnes KR, Hanscom M, Cabatbat RM, Tan MT, and Faden AI (2012). Cyclin D1 gene ablation confers neuroprotection in traumatic brain injury. J Neurotrauma 29, 813–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor S, Kim SM, Farook JM, Mir S, Saha R, and Sen N (2013). Foxo3a transcriptionally upregulates AQP4 and induces cerebral edema following traumatic brain injury. J Neurosci 33, 17398–17403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ID, Shin JH, Kim SW, Choi S, Ahn J, Han PL, Park JS, and Lee JK (2012). Intranasal delivery of HMGB1 siRNA confers target gene knockdown and robust neuroprotection in the postischemic brain. Mol Ther 20, 829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranenburg O, van der Eb AJ, and Zantema A (1996). Cyclin D1 is an essential mediator of apoptotic neuronal cell death. EMBO J 15, 46–54. [PMC free article] [PubMed] [Google Scholar]
- Lanza IR, and Nair KS (2010). Mitochondrial function as a determinant of life span. Pflugers Arch 459, 277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC, and Wei YH (2005). Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol 37, 822–834. [DOI] [PubMed] [Google Scholar]
- Li Y, Chopp M, Powers C, and Jiang N (1997). Immunoreactivity of cyclin D1/cdk4 in neurons and oligodendrocytes after focal cerebral ischemia in rat. J Cereb Blood Flow Metab 17, 846–856. [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, and Sheng M (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. [DOI] [PubMed] [Google Scholar]
- Lien LM, Chiou HY, Yeh HL, Chiu SY, Jeng JS, Lin HJ, Hu CJ, Hsieh FI, and Wei YH (2017). Significant Association Between Low Mitochondrial DNA Content in Peripheral Blood Leukocytes and Ischemic Stroke. J Am Heart Assoc 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TF, Vachharajani V, Millet P, Bharadwaj MS, Molina AJ, and McCall CE (2015). Sequential actions of SIRT1-RELB-SIRT3 coordinate nuclear-mitochondrial communication during immunometabolic adaptation to acute inflammation and sepsis. J Biol Chem 290, 396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XF, Fawcett JR, Thorne RG, DeFor TA, and Frey WH 2nd, (2001a). Intranasal administration of insulin-like growth factor-I bypasses the blood-brain barrier and protects against focal cerebral ischemic damage. J Neurol Sci 187, 91–97. [DOI] [PubMed] [Google Scholar]
- Liu XF, Fawcett JR, Thorne RG, and Frey WH 2nd, (2001b). Non-invasive intranasal insulin-like growth factor-I reduces infarct volume and improves neurologic function in rats following middle cerebral artery occlusion. Neurosci Lett 308, 91–94. [DOI] [PubMed] [Google Scholar]
- Lopez-Lluch G, Hunt N, Jones B, Zhu M, Jamieson H, Hilmer S, Cascajo MV, Allard J, Ingram DK, Navas P, et al. (2006). Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc Natl Acad Sci U S A 103, 1768–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M (2014). Cyclin-dependent kinases. Genome Biol 15, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathupala SP (2009). Delivery of small-interfering RNA (siRNA) to the brain. Expert Opin Ther Pat 19, 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May-Panloup P, Vignon X, Chretien MF, Heyman Y, Tamassia M, Malthiery Y, and Reynier P (2005). Increase of mitochondrial DNA content and transcripts in early bovine embryogenesis associated with upregulation of mtTFA and NRF1 transcription factors. Reprod Biol Endocrinol 3, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir S, Sen T, and Sen N (2014). Cytokine-induced GAPDH sulfhydration affects PSD95 degradation and memory. Mol Cell 56, 786–795. [DOI] [PubMed] [Google Scholar]
- Pardridge WM (2007). shRNA and siRNA delivery to the brain. Adv Drug Deliv Rev 59, 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina AK, Zhu X, Rottkamp CA, Monteiro M, Takeda A, and Smith MA (2000). Cyclin’ toward dementia: cell cycle abnormalities and abortive oncogenesis in Alzheimer disease. J Neurosci Res 61, 128–133. [DOI] [PubMed] [Google Scholar]
- Reinecke F, Smeitink JA, and van der Westhuizen FH (2009). OXPHOS gene expression and control in mitochondrial disorders. Biochim Biophys Acta 1792, 1113–1121. [DOI] [PubMed] [Google Scholar]
- Rodriguez M, Lapierre J, Ojha CR, Kaushik A, Batrakova E, Kashanchi F, Dever SM, Nair M, and El- Hage N (2017). Intranasal drug delivery of small interfering RNA targeting Beclin1 encapsulated with polyethylenimine (PEI) in mouse brain to achieve HIV attenuation. Sci Rep 7, 1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney JP, Ryde IT, Sanders LH, Howlett EH, Colton MD, Germ KE, Mayer GD, Greenamyre JT, and Meyer JN (2015). PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol Biol 1241, 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC (2008). Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 88, 611–638. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC (2011). Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta 1813, 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T, Gupta R, Kaiser H, and Sen N (2017). Activation of PERK Elicits Memory Impairment through Inactivation of CREB and Downregulation of PSD95 After Traumatic Brain Injury. J Neurosci 37, 5900–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T, and Sen N (2016). Treatment with an activator of hypoxia-inducible factor 1, DMOG provides neuroprotection after traumatic brain injury. Neuropharmacology 107, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T, Sen N, Jana S, Khan FH, Chatterjee U, and Chakrabarti S (2007). Depolarization and cardiolipin depletion in aged rat brain mitochondria: relationship with oxidative stress and electron transport chain activity. Neurochem Int 50, 719–725. [DOI] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Deng Y, Mbye LH, and Hall ED (2006). Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J Cereb Blood Flow Metab 26, 1407–1418. [DOI] [PubMed] [Google Scholar]
- Small DL, Monette R, Fournier MC, Zurakowski B, Fiander H, and Morley P (2001). Characterization of cyclin D1 expression in a rat global model of cerebral ischemia. Brain Res 900, 26–37. [DOI] [PubMed] [Google Scholar]
- Upadhyay RK (2014). Drug delivery systems, CNS protection, and the blood brain barrier. Biomed Res Int 2014, 869269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura-Clapier R, Garnier A, and Veksler V (2008). Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res 79, 208–217. [DOI] [PubMed] [Google Scholar]
- Wang B, Zeng J, and Gu Q (2017a). Exercise restores bioavailability of hydrogen sulfide and promotes autophagy influx in livers of mice fed with high-fat diet. Can J Physiol Pharmacol 95, 667–674. [DOI] [PubMed] [Google Scholar]
- Wang Z, Figueiredo-Pereira C, Oudot C, Vieira HL, and Brenner C (2017b). Mitochondrion: A Common Organelle for Distinct Cell Deaths? Int Rev Cell Mol Biol 331, 245–287. [DOI] [PubMed] [Google Scholar]
- Wei G, Wang D, Lu H, Parmentier S, Wang Q, Panter SS, Frey WH 2nd, and Ying W (2007). Intranasal administration of a PARG inhibitor profoundly decreases ischemic brain injury. Front Biosci 12, 4986–4996. [DOI] [PubMed] [Google Scholar]
- Wu J, Stoica BA, and Faden AI (2011). Cell cycle activation and spinal cord injury. Neurotherapeutics 8, 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W, Signore AP, Iwai M, Cao G, Gao Y, and Chen J (2008). Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke 39, 3057–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Konstantinidis DG, Yang JQ, Mizukawa B, Kalim K, Lang RA, Kalfa TA, Zheng Y, and Guo F (2014). Gene targeting RhoA reveals its essential role in coordinating mitochondrial function and thymocyte development. J Immunol 193, 5973–5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: TBI does not affect total level of PGC1α, NRF1 and interaction between NRF1 and CD1. (A-B) Confocal microscopic analysis of PGC1α and NRF1 in the pericontusional following either Sham or TBI. Red fluorescence indicates the level of either PGC1α (A) and NRF1 (B). The data shows that the expression level of both proteins remain unaltered in both sham and TBI conditions. (C) the interaction between CD1 and NRF1 was increased in the nucleus after TBI.