

Abstract
Environmental stressors that encounter in early-life and cause abnormal fetal and/or neonatal development may increase susceptibility to non-communicable diseases such as diabetes. Maternal exposure to ambient fine particulate matter (PM2.5) is associated with various fetal abnormalities, suggesting that it may program offspring’s susceptibility to diabetes. In the present study, we therefore examined whether maternal exposure to diesel exhaust PM2.5 (DEP), one of the major sources of ambient PM2.5 in urban areas, programs adult offspring’s glucose metabolism. Female C57Bl/6J mice were intratracheally instilled with DEP or vehicle throughout a 7-wk preconceptional period, gestation, and lactation, and the glucose homeostasis of their adult male offspring was assessed. Intraperitoneal glucose tolerance test (IPGTT) revealed that the maternal exposure to DEP significantly impaired adult male offspring’s glucose tolerance. Unexpectedly, it did not influence their insulin sensitivity, whereas it significantly decreased their glucose-induced insulin secretion (GIIS). This deficit in insulin secretion was corroborated by their significant decrease in arginine-induced insulin secretion. Histological analysis demonstrated that the deficit in insulin secretion was accompanied by the decrease in pancreatic islet and β cell sizes. To differentiate the effects of maternal exposure to DEP before birth and during lactation, some offspring were cross-fostered once born. We did not observe any significant effect of cross-fostering on the glucose homeostasis of adult male offspring and the function and morphology of their β cells. Prenatal exposure to DEP programs the morphology and function of β cells and thus homeostatic regulation of glucose metabolism in adult male offspring.
Keywords: β cells, diesel exhaust PM2.5, developmental programming, insulin, pancreatic islets
INTRODUCTION
Diabetes has become an uncontrolled global epidemic and a burgeoning cause of morbidity and mortality (17). This is believed to be primarily attributable to the global epidemic of obesity/overweight (17). Notably, numerous studies have also demonstrated that many noncommunicable diseases, encompassing diabetes, may originate from early-life exposure to environmental stressors that cause inappropriate fetal and/or neonatal development, known as the developmental programming of health and diseases (DPoHaD) paradigm (10). Ambient fine particulate matter (PM2.5) pollution is one of the leading preventable threats to global public health (16). Epidemiological studies have increasingly demonstrated that PM2.5 pollution is associated with various abnormal fetal development such as abortion, placental dysfunction, low birth weight, and preterm birth (5, 8, 9, 12, 20–23, 26). Therefore, as per the DPoHaD paradigm, maternal exposure to ambient PM2.5 is likely to be a risk factor for the DPoHaD. In agreement with this, we recently demonstrated that maternal exposure to diesel exhaust PM2.5 (DEP, one of the major sources of ambient PM2.5 in urban areas) persistently influences male offspring’s development and increases their adulthood adiposity (6). As obesity is one of the major risk factors for diabetes, how maternal exposure to DEP influences offspring’s glucose homeostasis has to be determined.
Insulin is central in the maintenance of glucose homeostasis. Its hypoglycemic action is collectively determined by its circulating level and the body’s sensitivity. As such, diabetes is indeed reflective of relative insulin insufficiency that is subsequent to insulin resistance and/or impaired insulin secretion by pancreatic islets. Although obesity is well known to cause diabetes primarily through induction of insulin resistance (13), many studies have indicated that the morphology and function of pancreatic islets is particularly vulnerable to the developmental programming by maternal exposure to various environmental stressors (11). Therefore, it is noteworthy that in addition to the potential effects on the insulin sensitivity subsequent to increased adiposity, maternal exposure to DEP may also predispose offspring to diabetes through the developmental programming of islet function.
We recently showed that dams’ prenatal or postnatal exposure to DEP differentially impacts the growth trajectory of offspring (6). In the present study, we systemically examined their long-term effects on offspring’s glucose homeostasis. Our results showed that while postnatal fostering by DEP-exposed dams did not significantly impact adult male offspring’s glucose homeostasis, adult male offspring born by DEP-exposed dams developed glucose intolerance. Unexpectedly, a marked decrease in glucose-induced insulin secretion (GIIS) but not insulin resistance accompanied this glucose intolerance. The impaired GIIS was corroborated by significant decreases in pancreatic insulin content and β cell area. The present study thus provides compelling evidence that prenatal exposure to ambient PM2.5 may contribute to the pathogenesis of diabetes through programming of β cell function.
MATERIALS AND METHODS
Animals.
University of Maryland, Baltimore (UMB) is an AAALAC-accredited institution. All procedures of this study were approved by the Institutional Animal Care and Use Committee (IACUC) at UMB, and all animals were treated humanely and with regard for alleviation of suffering. All dams and sires used in the present study (C57Bl/6J mice, 4-wk-old, 12 male and 12 female) were purchased from the Jackson Laboratories (stock no. 000664) and were housed in animal facilities at UMB, which maintained the 12-h:12-h light/dark light cycle and the temperature and humidity within the recommended limits. All mice used in the present study were fed with Teklad global 16% protein (cat. no. 2916).
Maternal DEP intratracheal instillation, breeding, and cross-fostering of offspring.
These were previously described (6), and the scheme is shown (Fig. 1A). Briefly, DEP was obtained from the National Institute of Standards and Technology (DEP; SRM 2975; NIST, Gaithersburg, MD). It was stored at 4°C and kept away from direct sunlight. To perform instillation, DEP were suspended in sterile normal saline. Prior to removal of subsamples for analysis, the contents of the bottle were mixed thoroughly. To minimize aggregation, particle suspensions were sonicated (Clifton Ultrasonic Bath, Clifton, NJ) for 20 min on the day of instillation and vortexed 30 s before each instillation. Of the breeding pair, only the dams were instilled. The instillation of DEP was performed as previously described with minor modifications (14). Briefly, to instill DEP, the mice were anesthetized with 3% isoflurane, and placed supine with extended neck on an angled board. A Becton-Dickinson 18-gauge cannula was inserted via the mouth into the trachea. DEP suspension (20 µg in 50 µl, representing an average daily dose of 8.6 μg/mouse and approximately equating to inhalational exposure to 160 μg/m3 PM2.5) (2) or saline (50 µl) were intratracheally instilled via a sterile syringe and followed by an air bolus of 150 µl. The intubation catheter was removed and the mouse was transferred to a vertical hanging position with the head up for 5 min, ensuring that the delivered material was maintained in the lung and did not block the airways. The deposition and distribution of instilled material was verified by installing Evans blue (data not shown). Either DEP or saline was instilled 3 times/week (Monday, Wednesday, and Friday) beginning at the age of 5 wk and ending once offspring were weaned, covering a 7-wk preconceptional period and the whole gestation and lactation periods.
Fig. 1.

Prenatal exposure to diesel exhaust PM2.5 (DEP) impairs glucose-induced insulin secretion (GIIS) in adulthood. A: the animal experimental scheme. Dams were exposed to Vehicle (PBS) or DEP as previously described (6), and the adult offspring were subjected to IPGTT (B and C), ITT (D and E), and GIIS (F and G). Both the response curves (B, D, and F) and the areas under curve (C, E, and G) are presented. nVV = 9, nDV = 7, nVD = 8, and nDD = 1. *P < 0.05, ANOVA. VV, born by vehicle-exposed dams and postnatally mothered by vehicle-exposed dams; VD, born by vehicle-exposed dams and postnatally mothered by DEP-exposed dams; DV, born by DEP-exposed dams and postnatally mothered by vehicle-exposed dams; DD, born by DEP-exposed dams and postnatally mothered by DEP-exposed dams.
Breeding cages were set up using one normal male and one vehicle- or DEP-instilled female (12-wk-old, n = 6/group). As both prenatal and postnatal periods have been shown to be vulnerable to developmental programming (10), approximately one-half of the offspring were cross-fostered once born. There were four groups of offspring: VV (offspring born by vehicle-treated dams and postnatally fostered by vehicle-treated dams, 9 male and 3 female), DV (offspring born by DEP-treated dams and postnatally fostered by vehicle-treated dams, 7 male and 10 female), VD (offspring born by vehicle-treated dams and postnatally fostered by DEP-treated dams, 8 male and 5 female), and DD (offspring born by DEP-treated dams and postnatally fostered by DEP-treated dams, 11 male and 7 female). As we did not observe the developmental programming by exposure to concentrated ambient PM2.5 (CAP) in female offspring (7), the number of pups fostered by each dam was culled to 4–6 through euthanasia of female pups to prevent the confounding by the litter size, and all female offspring were euthanized when weaning. Therefore, only male offspring (nVV = 9, nDV = 7, nVD = 8, and nDD = 11) were analyzed in the present study.
Intraperitoneal glucose tolerance test (IPGTT).
Before testing, mice (15- to 17-wk-old male offspring) were fasted for 16 h. On the day of experiments, the basal glucose levels of the tail-vein blood were determined using an automatic glucometer (Glucotrend 2, Roche Diagnostics), and then mice were intraperitoneally injected with glucose (20%, 2 g/kg body wt). The glucose levels of tail vein blood at 15, 30, 60, and 120 min after injection were measured as described above.
Insulin tolerance test (ITT).
Before testing, mice (16- to 18-wk-old male offspring) were fasted for 4 h. The basal glucose levels of tail vein blood were determined using an automatic glucometer (Glucotrend 2, Roche Diagnostics) and then mice were intraperitoneally injected with insulin (0.5 U/kg body wt). The glucose levels of tail vein blood at 15, 30, 60, and 120 min after injection were then measured as described above.
Arginine tolerance test (ATT).
Before testing, mice (19- to 21-wk-old male offspring) were fasted for 16 h. The basal glucose levels of tail-vein blood were determined using an automatic glucometer (Glucotrend 2, Roche Diagnostics) and then mice were intraperitoneally injected with arginine (8.3%, 1 mg/g body wt). The glucose levels of tail vein blood at 5, 15, and 30 min after injection were then measured as described above.
Insulin assessments.
Insulin levels in the plasma and the tissue lysates of pancreatic tails were determined per manufacturer’s instruction (Ultra Sensitive Mouse Insulin ELISA Kit, Crystal Chemical), and expressed as nanograms per milliliter plasma (plasma) or nanograms per microgram protein (tissue lysates).
Histological analysis of pancreatic islets.
Offspring mice were euthanized when they were 20–22 wk old. The heads of pancreas were fixed in 4% paraformaldehyde, embedded in paraffin, cut into 5-μm sections, and subjected to hematoxylin and eosin staining. Quantitative planimetric analyses of islet number and islet size were performed on three successive sections per slide, and at least 9 sections from 3 consecutive slides per mouse were examined. Each image was digitized using a digital camera and analyzed under a research microscope (Zeiss Axioscope with Spot I digital camera, Jena, Germany), using NIH ImageJ software (version 1.61). Islet number was expressed as islets per square micrometer pancreatic tissue, and islet size was presented in square micrometers. All analyses were performed by an investigator blinded to group assignment.
Immunohistochemistry and quantitation of apoptotic cells.
Immunostaining for insulin (1:100 dilution, rabbit anti-insulin monoclonal antibody cat. no. 3014, Cell Signaling Technology), glucagon (1:200 dilution, mouse anti-glucagon monoclonal antibody cat. no. 565859, BD Biosciences), and Ki-67 (1:100, rat anti-Ki-67 monoclonal antibody cat. no. 652401, Biolegend) in pancreas was performed as previously reported (27). Visualization of the staining was achieved using the species-appropriate fluorescent secondary antibody (Santa Cruz Biotechnology). The DeadEnd Fluorometric TUNEL System (Promega) was used for the quantitation of apoptotic cells in pancreatic islets per manufacturer’s instruction. Quantitative planimetric analyses were performed on three successive sections per slide, and at least six sections from three consecutive slides per mouse were examined. Each image was digitized using Zeiss LSM 510 Meta Confocal Microscope. All analyses were performed by an investigator blinded to group assignment.
Quantitative RT-PCR (qPCR).
Total RNA was extracted and purified from the pancreatic tissues using the Trizol reagent (Invitrogen). The quality of RNA was assessed by determination of the ratio of absorbance at 260 nm to absorbance at 280 nm by nanodrop. Two micrograms of total DNase-treated RNA was reverse transcribed into cDNA using High Capacity cDNA Reverse Transcription Kits (Applied Biosystem) per manufacturer’s instruction. qPCR was performed using LightCycler 480 SYBR Green I Master in the LightCycler (Roche, German). Reactions were performed in a total volume of 10 μl containing 1 μl cDNA, 0.2 μM of each primer, and 5 μl of the SYBR Green reaction mix. The amplification protocol was as follows: 95°C/5 min (95°C/10 s, 60°C/20 s, and 72°C/30 s) × 50. Following amplification, a dissociation curve analysis was performed to ensure purity of PCR product. The specific sense and antisense primers were Glut2, 5′-TCA GAA GAC AAG ATC ACC GGA-3′ and 5′-GTC ATA GCC GAA CTG GAA GGA-3′; Nkx6.1, 5′-CTG CAC AGT ATG GCC GAG ATG-3′ and 5′-CCG GGT TAT GTG AGC CCA A-3′; MafAl, 5′-AGG AGG AGG TCA TCC GAC TG-3′ and 5′-CTT CTC GCT CTC CAG AAT GTG-3′; insulin, 5′-GCT TCT TCT ACA CAC CCA TGT C-3′ and 5′-AGC ACT GAT CTA CAA TGC CAC-3′; and GAPDH, 5′-GCA GTG GCA AAG TGG AGA TTG TTG C-3′ and 5′-CCC GTT GAT GAC AAG CTT CCC ATT C-3′. The relative expression levels were determined using Pfaffl methods as previously described (19).
Statistics.
All data are expressed as means ± SE unless noted otherwise. Statistical tests were performed using one-way or two-way analysis of variance (ANOVA) followed by Bonferroni correction or unpaired t-test using GraphPad Prism (version 5; GraphPad Software, La Jolla, CA). The significance level was set at P < 0.05.
RESULTS
Prenatal exposure to DEP results in glucose intolerance in adult male offspring.
We recently reported that dams’ prenatal or postnatal exposure to DEP differentially programs mouse energy metabolism and adiposity in adulthood (6). As adiposity is well known to correlate to glucose homeostasis, we performed glucose homeostasis assessments on these adult male offspring. Figure 1, B and F, reveals that the fasting glucose and insulin levels in all groups were comparable. IPGTT assessments revealed that prenatal (VV vs. DV and VD vs. DD) but not postnatal (VV vs. VD and DV vs. DD) exposure to DEP resulted in glucose intolerance in adult male offspring (Fig. 1, B and C). In contrast to their marked effects on adiposity (6), we did not observe any significant effect of dams’ prenatal and postnatal exposure to DEP on offspring’s sensitivity to insulin (Fig. 1, D and E). Glucose tolerance is collectively determined by insulin sensitivity and glucose-induced insulin secretion (GIIS). We therefore assessed the levels of circulating insulin during IPGTT (Fig. 1, F and G). Consistent with the IPGTT and ITT results above, we observed that dams’ prenatal but not postnatal exposure to DEP significantly reduced GIIS of adult male offspring, strongly suggesting that prenatal exposure to DEP impacts glucose metabolism primarily through impairment of GIIS.
Prenatal exposure to DEP results in decrease in arginine-induced insulin secretion (AIIS) in adult male offspring.
Arginine is a membrane-depolarizing agent that promotes calcium influx and subsequently GIIS of pancreatic β cells (25). To further document the effect of the maternal exposure to DEP on the pancreatic β cell function of offspring, we assessed their plasma glucose and insulin responses to arginine. Figure 2, A and B, shows that arginine administration slightly but significantly increased plasma glucose levels in offspring born by DEP-exposed dams (VV vs. DV and VD vs. DD). In contrast, fostering by DEP-exposed dams (VV vs. VD and DV vs. DD) did not significantly influence offspring’s plasma glucose response to arginine. In agreement with their effects on the plasma glucose response to arginine, dams’ prenatal but not postnatal exposure to DEP significantly reduced offspring’s AIIS (Fig. 2, C and D).
Fig. 2.
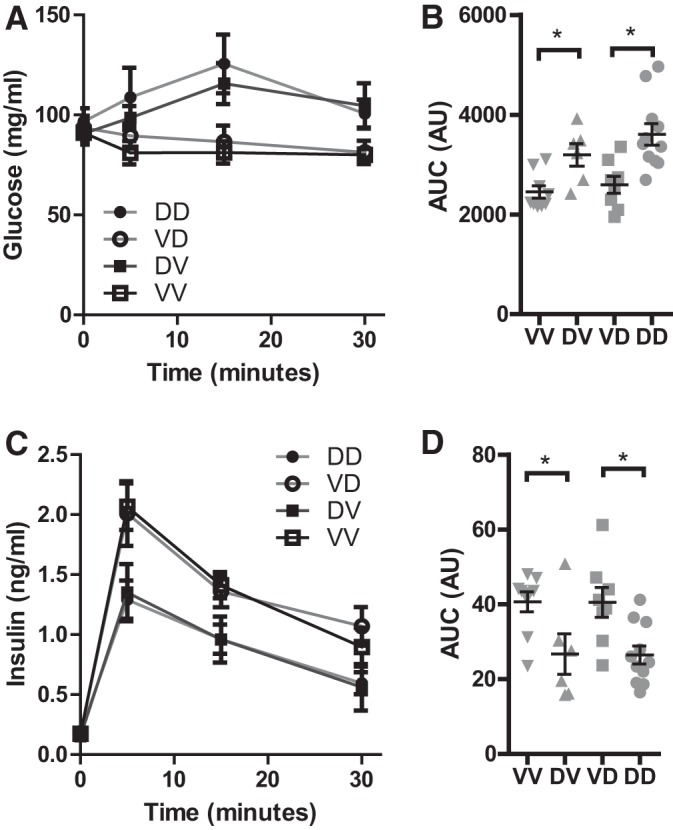
Prenatal exposure to DEP impairs arginine-induced insulin secretion (AIIS) in adulthood. The adult offspring were injected ip with arginine (1 mg/g body wt), and plasma glucose (A and B) and insulin (C and D) levels were assessed. Both the response curves (A and C) and the areas under curve (B and D) are presented. nVV = 9, nDV = 7, nVD = 8, and nDD = 11. *P < 0.05, ANOVA.
Prenatal exposure to DEP reduces the area of pancreatic islets in adult male offspring.
To delineate the mechanism by which prenatal exposure to DEP programs the dysfunction of pancreatic islets, we harvested the pancreas of male offspring and performed histological analysis. Figure 3A shows that there was no any significant difference in pancreatic weight between any groups. In contrast, histological analysis revealed that dams’ prenatal but not postnatal exposure to DEP significantly reduced the relative area of pancreatic islets (Fig. 3, B and C). This reduction in islet areas appeared to be primarily due to a decrease in the size but not the number of pancreatic islets (Fig. 3, D–F). Furthermore, consistent with the decrease in the area of pancreatic islets, Fig. 3, G and H, demonstrates that dams’ prenatal but not postnatal exposure to DEP significantly reduced the insulin content of pancreas in adult male offspring.
Fig. 3.

Prenatal exposure to DEP reduces the size of pancreatic islets in adulthood. A: the pancreas and body of offspring were weighed after necropsy, and the pancreatic weight relative to body weight is presented. B–F: the pancreas of offspring were subjected to hematoxylin and eosin (H&E) staining and histological analysis. B: representative images. Scale bar, 250 µm. Insets, 10× magnified islets. C: islet areas expressed as the percentage of pancreas tissue. D: islet density expressed as number per µm2 pancreas. E: islet sizes. F: frequency distribution of islet size. G: tissue lysates of pancreas were prepared using RIPA buffer, and the insulin concentrations were determined with the ELISA kit. The results were then normalized by total protein concentrations. H: insulin mRNA expression in the pancreas by quantitative RT-PCR. nVV = 9, nDV = 7, nVD = 8, and nDD = 11. *P < 0.05, ANOVA.
Prenatal exposure to DEP decreases the size but not the number of β cells in adult male offspring.
To further document the effects of prenatal exposure to DEP on the pancreatic islets of adult male offspring, the α and β cells in the islets were visualized with anti-insulin and anti-glucagon respectively. Figure 4, A–C, show that the number and size of α cells between any groups were comparable. In contrast, whereas prenatal exposure to DEP did not significantly altered the number of β cells in the pancreatic islets (Fig. 4E), it significantly decreased the area of β cells (Fig. 4D). Consistently, Fig. 4F reveals that prenatal exposure to DEP significantly decreased the size of β cells. To examine if maternal prenatal exposure to DEP impacts the maturation of β cells, we assessed the pancreatic expression of β cell maturation/differentiation markers by qPCR. Figure 4, G–I, shows that maternal prenatal exposure to DEP significantly decreased the expression of Glut2 but not MafA and Nkx6.1.
Fig. 4.

Prenatal exposure to DEP reduces the size of β cells but not α cells in adulthood. The sections of offspring pancreas were subjected to immune staining with anti-insulin (green, β cells) and anti-glucagon (red, α cells). A: representative images. Scale bar, 25 µm. B: average α cell number per islet. C: average α cell area per islet. D: average β cell number per islet. E: average β cell area per islet. F: average β cell size (β cell area/β cell number). G–I: pancreatic expression of the indicated mRNA by quantitative RT-PCR. nVV = 9, nDV = 7, nVD = 8, and nDD = 11. *P < 0.05, ANOVA.
Prenatal exposure to DEP does not alter the proliferation and apoptosis of islet cells in adult male offspring.
It have been demonstrated that the proliferation and apoptosis of pancreatic islet β cells may be subjected to dynamic regulation (18). To determine the mechanism whereby prenatal exposure to DEP programs pancreatic islet structure and function, we assessed the proliferating and apoptotic cells in the pancreas through TUNEL assay and immunostaining of Ki67, respectively. Figure 5, A and B, shows that the apoptotic cells in the islets between any groups were comparable. In addition, neither maternal prenatal nor postnatal exposure to DEP altered the proliferation of islet cells (Fig. 5, C and D).
Fig. 5.

Prenatal exposure to DEP does not impact apoptosis and proliferation of islet cells. A and B: sections of offspring pancreas were subjected to apoptotic cells assessment using the DeadEnd Fluorometric TUNEL System (Promega). Representative pictures (A) and quantitation (normalized by the total islet cells; B) are presented. Scale bar, 25 µm. Insets, 5× magnified apoptotic cells. C and D: proliferating cells in the pancreas were visualized through immunostaining with anti-Ki-67 (green) and anti-insulin (red) antibodies. C: representative pictures of proliferative cells outside the islet (top panel) and in the islet (bottom panel). Scale bar, 25 µm. Insets, 5× magnified proliferative cells. D: quantitation (normalized by the total β cells) are presented. nVV = 9, nDV = 7, nVD = 8, and nDD = 11.
DISCUSSION
In the present study, we investigated the long-term effects of maternal exposure to DEP on the homeostatic regulation of glucose metabolism in adult male offspring. The major findings include that 1) maternal prenatal but not postnatal exposure to DEP decreases male offspring’s glucose tolerance in adulthood; 2) this glucose intolerance was primarily due to decreased GIIS but not insulin resistance; 3) the decreased GIIS was accompanied by decreases in the insulin content of pancreas and the sizes but not numbers of pancreatic islets and β cells. Taken together, these results have strongly suggested that maternal prenatal exposure to DEP may predispose male offspring to diabetes through programming of pancreatic islet function, and thus aroused a new and potentially important health concern over maternal exposure to ambient particulate matter pollution.
Several studies have shown that maternal exposure to DEP through either inhalation or intratracheal instillation during pregnancy does not alter the glucose tolerance of offspring when fed with normal diet but predisposes them to glucose intolerance and insulin resistance when fed with a high-fat diet (HFD) (3, 4). In the present study, we demonstrated that maternal exposure to DEP covering a 7-wk preconceptional period and the whole pregnancy period was sufficient to result in glucose intolerance in adult male offspring fed with normal diet. To our best knowledge, this is the first study showing that maternal exposure to ambient fine particulate matter results in glucose intolerance in adult offspring fed with standard rodent diet. Along with the previous studies mentioned above (3, 4), the present study has implicated maternal exposure to ambient fine particulate matter pollution in the pathogenesis of diabetes. Since ambient fine particulate matter pollution will continue to be one of the leading preventable threats to global public health in the foreseeable future, this study has thus raised attention to the potentially major influences of maternal exposure to ambient particulate matter pollution on the development of diabetes.
Notably, although both the previous (3, 4) and present studies have supported the association between maternal exposure to DEP and the pathogenesis of diabetes, they indeed presented remarkably different effects of maternal exposure to DEP on offspring’s homeostatic regulation of glucose metabolism. Whereas the previous studies showed that maternal exposure to DEP predisposes offspring to diabetes primarily though exacerbation of HFD-induced insulin resistance (3, 4), the present study revealed that maternal exposure to DEP induced glucose intolerance primarily through a decrease in GIIS. The reason for this discrepancy remains to be determined. It is noteworthy that exposure to DEP in the previous studies covered the gestation period only, whereas exposure to DEP in the present study covered an additional 7-wk preconceptional period. Most recently, we reported that the 7-wk preconceptional period is a vulnerable window for the developmental programming of offspring’s adulthood adiposity by exposure to concentrated ambient PM2.5 (CAP) (7). Therefore, the discrepancy above may be a reflection of their difference in the timing of exposure. As the determination of vulnerable windows is obviously public policy relevant, further studies are undergoing to verify this proposed role of the 7-wk preconceptional exposure.
Obesity is well known to cause insulin resistance and subsequently glucose intolerance (1). However, although we previously reported that prenatal exposure to DEP resulted in lower body weight but increased adiposity in adult male offspring (6), the present study unexpectedly revealed that it did not change the sensitivity of these offspring to insulin, whereas it impaired their glucose tolerance primarily through the reduction in GIIS. Our results are consistent with the well-known thrifty phenotype hypothesis proposed by Hales and Barker (11), which has pointed out that maternal influences on the developing pancreatic islets may play a role in metabolic adaptations in the progeny that promote survival under conditions of limited nutrient availability, but render them vulnerable to nutritional excess later in life. Currently, the mechanism for this apparent disconnect between increased adiposity and insulin resistance in these offspring is unknown. One possibility is that ITT may not be sensitive enough to observe the small change in insulin sensitivity induced by a slight increase in adiposity. Further studies using state-of-art techniques such as hyperinsulinemic-euglycemic clamps may be needed to provide valuable mechanistic insights into this apparent disconnect.
One more important finding in the present study is that consistent with its effects on glucose tolerance and GIIS, prenatal exposure to DEP significantly influences the mass of β cells in adult offspring, as evidenced by the significant decreases in the insulin content of pancreas and the relative areas of islets and β cells. As the sole source of endogenous insulin, β cells are well known to be central in the homeostatic regulation of glucose metabolism. Moreover, human genomewide association analysis has demonstrated that the majority of genes associated with increased risk for Type 2 diabetes mellitus are implicated in the process of insulin secretion (15), suggesting that β cells may play a more important role than previously expected in the pathogenesis of type 2 diabetes mellitus. As such, the demonstration of β cell dysfunction in offspring born by DEP-exposed dams has underscored the potential impact of maternal exposure to ambient particulate matter pollution in the prevalence of diabetes. Interestingly, the dysfunction of β cells in offspring born by DEP-exposed dams is not associated with changes in their proliferation and apoptosis. This is consistent with their normal number of islet β cells (Fig. 4). These results are consistent with recent studies showing that the reduction of β cell mass during the pathogenesis of diabetes is not only due to apoptosis, as previously believed, but also from the transcriptional silencing of insulin and other markers of functionally mature β cells (24). Notably, the present data reveal a significantly decreased pancreatic expression of Glut2 mRNA in offspring born by DEP-exposed dams (Fig. 4H), suggesting that their β cell dysfunction may be due to defective glucose sensing. However, further studies are needed to confirm this.
Although the present study provides compelling evidence that maternal exposure to ambient particulate matter may persistently influence glucose homeostasis of male offspring, it has several important limitations. These include the fact that we have not provided enough data to determine the molecular mechanisms by which prenatal exposure to DEP impact the morphology and function of β cells in adulthood. This will require the exploitation of high-throughput techniques to identify the susceptibility genes/signaling pathways. In addition, we did not assess the morphology and function of the offspring’s pancreatic islets in the early life, particularly the neonatal period, which is crucial for the development of pancreatic islets.
Conclusions.
In the present study, we provide evidence that maternal prenatal exposure to DEP persistently impacts the morphology and function of the male offspring’s β cells and thus impairs their homeostatic regulation of glucose metabolism in adulthood.
GRANTS
This work was supported by the National Institutes of Health (R01-ES-024516 to Z. Ying), the American Heart Association (13SDG17070131 to Z. Ying), and the National Natural Science Foundation of China (81500216 to M. Chen and 81770805 to Z. Ying). S. Chen was supported by the Program of training abroad of Henan medical academic leaders (2016011).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.C. and Z.Y. conceived and designed research; M.C., S.L., X.Q., L.Z., L.Q., S.C., Z.H., and W.W. performed experiments; M.C., S.L., X.Q., L.Z., L.Q., S.C., and Z.H. analyzed data; M.C., S.L., X.Q., Y.X., Y.Z., and Z.Y. interpreted results of experiments; M.C., S.L., and X.Q. prepared figures; M.C., Q.C., and Z.Y. drafted manuscript; M.C., S.L., X.Q., L.Q., S.C., Y.X., W.W., Y.Z., Q.C., and Z.Y. approved final version of manuscript; Y.X., Y.Z., Q.C., and Z.Y. edited and revised manuscript.
REFERENCES
- 1.Abdelaal M, le Roux CW, Docherty NG. Morbidity and mortality associated with obesity. Ann Transl Med 5: 161, 2017. doi: 10.21037/atm.2017.03.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bide R, Armour SJ, Yee E; Defence Research Establishment (Suffield, Ralston, Alberta) . Estimation of Human Toxicity from Animal Inhalation Toxicity Data. I. Minute Volume-Body Weight Relationships Between Animals and Man. Accession No. ADA336351 Washington, DC: US Dept. of Defense, 1997. [Google Scholar]
- 3.Bolton JL, Auten RL, Bilbo SD. Prenatal air pollution exposure induces sexually dimorphic fetal programming of metabolic and neuroinflammatory outcomes in adult offspring. Brain Behav Immun 37: 30–44, 2014. doi: 10.1016/j.bbi.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 4.Bolton JL, Smith SH, Huff NC, Gilmour MI, Foster WM, Auten RL, Bilbo SD. Prenatal air pollution exposure induces neuroinflammation and predisposes offspring to weight gain in adulthood in a sex-specific manner. FASEB J 26: 4743–4754, 2012. doi: 10.1096/fj.12-210989. [DOI] [PubMed] [Google Scholar]
- 5.Bonzini M, Carugno M, Grillo P, Mensi C, Bertazzi PA, Pesatori AC. Impact of ambient air pollution on birth outcomes: systematic review of the current evidences. Med Lav 101: 341–363, 2010. [PubMed] [Google Scholar]
- 6.Chen M, Liang S, Zhou H, Xu Y, Qin X, Hu Z, Wang X, Qiu L, Wang W, Zhang Y, Ying Z. Prenatal and postnatal mothering by diesel exhaust PM2.5-exposed dams differentially program mouse energy metabolism. Part Fibre Toxicol 14: 3, 2017. doi: 10.1186/s12989-017-0183-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen M, Wang X, Hu Z, Zhou H, Xu Y, Qiu L, Qin X, Zhang Y, Ying Z. Programming of mouse obesity by maternal exposure to concentrated ambient fine particles. Part Fibre Toxicol 14: 20, 2017. doi: 10.1186/s12989-017-0201-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dadvand P, Parker J, Bell ML, Bonzini M, Brauer M, Darrow LA, Gehring U, Glinianaia SV, Gouveia N, Ha EH, Leem JH, van den Hooven EH, Jalaludin B, Jesdale BM, Lepeule J, Morello-Frosch R, Morgan GG, Pesatori AC, Pierik FH, Pless-Mulloli T, Rich DQ, Sathyanarayana S, Seo J, Slama R, Strickland M, Tamburic L, Wartenberg D, Nieuwenhuijsen MJ, Woodruff TJ. Maternal exposure to particulate air pollution and term birth weight: a multi-country evaluation of effect and heterogeneity. Environ Health Perspect 121: 267–373, 2013. doi: 10.1289/ehp.1205575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fleisch AF, Rifas-Shiman SL, Koutrakis P, Schwartz JD, Kloog I, Melly S, Coull BA, Zanobetti A, Gillman MW, Gold DR, Oken E. Prenatal exposure to traffic pollution: associations with reduced fetal growth and rapid infant weight gain. Epidemiology 26: 43–50, 2015. doi: 10.1097/EDE.0000000000000203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grandjean P, Barouki R, Bellinger DC, Casteleyn L, Chadwick LH, Cordier S, Etzel RA, Gray KA, Ha EH, Junien C, Karagas M, Kawamoto T, Paige Lawrence B, Perera FP, Prins GS, Puga A, Rosenfeld CS, Sherr DH, Sly PD, Suk W, Sun Q, Toppari J, van den Hazel P, Walker CL, Heindel JJ. Life-long implications of developmental exposure to environmental stressors: new perspectives. Endocrinology 156: 3408–3415, 2015. doi: 10.1210/EN.2015-1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br Med Bull 60: 5–20, 2001. doi: 10.1093/bmb/60.1.5. [DOI] [PubMed] [Google Scholar]
- 12.Hyder A, Lee HJ, Ebisu K, Koutrakis P, Belanger K, Bell ML. PM2.5 exposure and birth outcomes: use of satellite- and monitor-based data. Epidemiology 25: 58–67, 2014. doi: 10.1097/EDE.0000000000000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kammoun HL, Kraakman MJ, Febbraio MA. Adipose tissue inflammation in glucose metabolism. Rev Endocr Metab Disord 15: 31–44, 2014. doi: 10.1007/s11154-013-9274-4. [DOI] [PubMed] [Google Scholar]
- 14.Kyjovska ZO, Jacobsen NR, Saber AT, Bengtson S, Jackson P, Wallin H, Vogel U. DNA strand breaks, acute phase response and inflammation following pulmonary exposure by instillation to the diesel exhaust particle NIST1650b in mice. Mutagenesis 30: 499–507, 2015. doi: 10.1093/mutage/gev009. [DOI] [PubMed] [Google Scholar]
- 15.Lawlor N, Khetan S, Ucar D, Stitzel ML. Genomics of islet (dys)function and Type 2 diabetes. Trends Genet 33: 244–255, 2017. doi: 10.1016/j.tig.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, Amann M, Anderson HR, Andrews KG, Aryee M, Atkinson C, Bacchus LJ, Bahalim AN, Balakrishnan K, Balmes J, Barker-Collo S, Baxter A, Bell ML, Blore JD, Blyth F, Bonner C, Borges G, Bourne R, Boussinesq M, Brauer M, Brooks P, Bruce NG, Brunekreef B, Bryan-Hancock C, Bucello C, Buchbinder R, Bull F, Burnett RT, Byers TE, Calabria B, Carapetis J, Carnahan E, Chafe Z, Charlson F, Chen H, Chen JS, Cheng AT, Child JC, Cohen A, Colson KE, Cowie BC, Darby S, Darling S, Davis A, Degenhardt L, Dentener F, Des Jarlais DC, Devries K, Dherani M, Ding EL, Dorsey ER, Driscoll T, Edmond K, Ali SE, Engell RE, Erwin PJ, Fahimi S, Falder G, Farzadfar F, Ferrari A, Finucane MM, Flaxman S, Fowkes FG, Freedman G, Freeman MK, Gakidou E, Ghosh S, Giovannucci E, Gmel G, Graham K, Grainger R, Grant B, Gunnell D, Gutierrez HR, Hall W, Hoek HW, Hogan A, Hosgood HD III, Hoy D, Hu H, Hubbell BJ, Hutchings SJ, Ibeanusi SE, Jacklyn GL, Jasrasaria R, Jonas JB, Kan H, Kanis JA, Kassebaum N, Kawakami N, Khang YH, Khatibzadeh S, Khoo JP, Kok C, Laden F, Lalloo R, Lan Q, Lathlean T, Leasher JL, Leigh J, Li Y, Lin JK, Lipshultz SE, London S, Lozano R, Lu Y, Mak J, Malekzadeh R, Mallinger L, Marcenes W, March L, Marks R, Martin R, McGale P, McGrath J, Mehta S, Mensah GA, Merriman TR, Micha R, Michaud C, Mishra V, Mohd Hanafiah K, Mokdad AA, Morawska L, Mozaffarian D, Murphy T, Naghavi M, Neal B, Nelson PK, Nolla JM, Norman R, Olives C, Omer SB, Orchard J, Osborne R, Ostro B, Page A, Pandey KD, Parry CD, Passmore E, Patra J, Pearce N, Pelizzari PM, Petzold M, Phillips MR, Pope D, Pope CA III, Powles J, Rao M, Razavi H, Rehfuess EA, Rehm JT, Ritz B, Rivara FP, Roberts T, Robinson C, Rodriguez-Portales JA, Romieu I, Room R, Rosenfeld LC, Roy A, Rushton L, Salomon JA, Sampson U, Sanchez-Riera L, Sanman E, Sapkota A, Seedat S, Shi P, Shield K, Shivakoti R, Singh GM, Sleet DA, Smith E, Smith KR, Stapelberg NJ, Steenland K, Stöckl H, Stovner LJ, Straif K, Straney L, Thurston GD, Tran JH, Van Dingenen R, van Donkelaar A, Veerman JL, Vijayakumar L, Weintraub R, Weissman MM, White RA, Whiteford H, Wiersma ST, Wilkinson JD, Williams HC, Williams W, Wilson N, Woolf AD, Yip P, Zielinski JM, Lopez AD, Murray CJ, Ezzati M, AlMazroa MA, Memish ZA. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380: 2224–2260, 2012. doi: 10.1016/S0140-6736(12)61766-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McMahon G. Cardiovascular risk reduction in diabetes: managing the metabolic milieu. Hosp Physician 5: 2–13, 2005. [Google Scholar]
- 18.Paul R, Choudhury A, Choudhury S, Mazumder MK, Borah A. Cholesterol in pancreatic β-cell death and dysfunction: underlying mechanisms and pathological implications. Pancreas 45: 317–324, 2016. doi: 10.1097/MPA.0000000000000486. [DOI] [PubMed] [Google Scholar]
- 19.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45, 2001. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rich DQ, Liu K, Zhang J, Thurston SW, Stevens TP, Pan Y, Kane C, Weinberger B, Ohman-Strickland P, Woodruff TJ, Duan X, Assibey-Mensah V, Zhang J. Differences in birth weight associated with the 2008 Beijing Olympics air pollution reduction: results from a natural experiment. Environ Health Perspect 123: 880–887, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Savitz DA, Bobb JF, Carr JL, Clougherty JE, Dominici F, Elston B, Ito K, Ross Z, Yee M, Matte TD. Ambient fine particulate matter, nitrogen dioxide, and term birth weight in New York, New York. Am J Epidemiol 179: 457–466, 2014. doi: 10.1093/aje/kwt268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schembari A, de Hoogh K, Pedersen M, Dadvand P, Martinez D, Hoek G, Petherick ES, Wright J, Nieuwenhuijsen MJ. Ambient air pollution and newborn size and adiposity at birth: differences by maternal ethnicity (the Born in Bradford Study Cohort). Environ Health Perspect 123: 1208–1215, 2015. doi: 10.1289/ehp.1408675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun X, Luo X, Zhao C, Chung Ng RW, Lim CE, Zhang B, Liu T. The association between fine particulate matter exposure during pregnancy and preterm birth: a meta-analysis. BMC Pregnancy Childbirth 15: 300, 2015. doi: 10.1186/s12884-015-0738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 150: 1223–1234, 2012. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thams P, Capito K. l-Arginine stimulation of glucose-induced insulin secretion through membrane depolarization and independent of nitric oxide. Eur J Endocrinol 140: 87–93, 1999. doi: 10.1530/eje.0.1400087. [DOI] [PubMed] [Google Scholar]
- 26.Valdiglesias V, Pásaro E, Méndez J, Laffon B. In vitro evaluation of selenium genotoxic, cytotoxic, and protective effects: a review. Arch Toxicol 84: 337–351, 2010. doi: 10.1007/s00204-009-0505-0. [DOI] [PubMed] [Google Scholar]
- 27.Vasavada RC, Cavaliere C, D’Ercole AJ, Dann P, Burtis WJ, Madlener AL, Zawalich K, Zawalich W, Philbrick W, Stewart AF. Overexpression of parathyroid hormone-related protein in the pancreatic islets of transgenic mice causes islet hyperplasia, hyperinsulinemia, and hypoglycemia. J Biol Chem 271: 1200–1208, 1996. doi: 10.1074/jbc.271.2.1200. [DOI] [PubMed] [Google Scholar]