

Abstract
Mitofilin is an inner membrane protein that has been defined as a mitochondria-shaping protein in controlling and maintaining mitochondrial cristae structure and remodeling. We determined the role of mitofilin in cell survival by investigating the mechanism underlying mitofilin knockdown-induced cell death by apoptosis. Cultured H9c2 myoblasts and HEK 293 cells were treated with mitofilin siRNA or scrambled siRNA for 24 h. Cell death (apoptosis), caspase 3 activity and cell cycle phases were assessed by flow cytometry, while cytochrome c release and intracellular ATP production were measured by ELISA. Mitofilin, apoptosis-inducing factor (AIF) and poly(ADP-ribose) polymerase (PARP) expression were measured by Western blot analysis and calpain activity was assessed using a calpain activity kit. Mitochondrial images were taken using electron microscopy. We found that mitofilin knockdown increases apoptosis mainly via activation of the AIF-PARP pathway leading to nuclear fragmentation that is correlated with S phase arrest of the cell cycle. Knockdown of mitofilin also led to mitochondrial swelling and damage of cristae that is associated with the increase in reactive oxygen species production and mitochondrial calpain activity, as well as a marked decrease in intracellular ATP production and mitochondrial membrane potential. Together, these results indicate that mitofilin knockdown by siRNA increases calpain activity that presumably leads to mitochondrial structural degradation resulting in a critical reduction of mitochondrial function that is responsible for the increase in cell death by apoptosis via an AIF-PARP mechanism and associated with nuclear fragmentation, and S phase arrest of the cell cycle.
Keywords: AIF-PARP-pathway, apoptosis, cell cycle arrest, cell death, H9c2 cardiomyoblast, HEK 293 cell line, mitochondrial inner membrane protein (mitofilin), mitochondria cristae morphology and function
INTRODUCTION
Mitochondria are organelles found in large numbers in most cells and are the center of cellular energy production and essential metabolic reactions. Mitochondria have a double-membrane structure, with the inner bilayer being folded inward to form layers (cristae). Recently, several proteins resident in both the outer and the inner membrane have been found to have a role in the molecular mechanisms regulating cristae biogenesis and architecture. Mic12 and Mic27 have been identified as important factors that play an essential role in mitochondrial contact site (MICOS) complex formation (61). Coiled-coil-helix domain 6 (CHCHD6, also known as CHCM1) has been reported to harbor a coiled coil helix-domain at its COOH-terminal end and is predominantly localized to the mitochondrial inner membrane. CHCHD3 has been found to play a key role in maintaining cristae integrity and mitochondrial function as knockdown of CHCHD3 led to a complete loss of both mitofilin and Sam50 proteins (12), while CHCHD10 mutations led to MICOS complex disassembly, loss of mitochondrial cristae, decrease in nucleoid number, and nucleoid disorganization (15). In the inner mitochondrial membrane, CHCHD3, 6, and 10 physically interact with mitofilin and Sam50 in MICOS (13). Mitochondrial inner membrane protein (IMMT), also referred to as mitofilin, is a ubiquitously expressed mitochondrial membrane protein (42) that was initially characterized as a heart muscle protein based on the high abundance of its mRNA in rat embryonic heart (25). Mitofilin has been proposed to be a critical organizer of mitochondrial cristae morphology and thus indispensable for normal mitochondrial function (26). In humans, two cDNAs have been isolated that encode different isoforms of mitofilin and both isoforms are derived by alternative splicing, and encode two proteins, immt-1 and immt-2 of 88 and 90 kDa (16). In Saccharomyces cerevisiae mitochondria, von der Malsburg et al. (56) have shown that mitofilin is part of a large multisubunit protein complex in the inner membrane, termed mitochondrial inner membrane organizing system (MINOS) that controls cristae morphology. The same team has further shown that mitofilin interacts with the translocase of the outer membrane complex and promotes protein import into the intermembrane space via the mitochondrial intermembrane space assembly pathway and that mitofilin/Fcj1 functions as a multifunctional regulator of mitochondrial architecture and protein biogenesis (56). Using complexome profiling, the Reichert group has identified apolipoprotein O (APOO) and apolipoprotein O-like protein (APOOL) as putative components of the mitofilin/MINOS protein complex (58). Furthermore, it has been proposed that MINOS integrity is required for the maintenance of the characteristic morphology of the inner mitochondrial membrane (IMM), with an inner boundary region closely opposed to the outer membrane and cristae membranes (62). It has been shown that transgenic overexpression of mitofilin preserves mitochondrial structure, leading to restoration of mitochondrial function in the diabetic heart (54) while also promoting cardiac hypertrophy (63). Conversely, in Caenorhabditis elegans, mutation of either immt-1 or immt-2 produced defects in germline development and egg-laying. These defects were exacerbated by the double mutation, which seriously reduced motility, increased levels of reactive oxygen species (ROS), and decreased mitochondrial mass (39). Additionally, knockdown of mitofilin in HeLa cells led to fragmentation of the mitochondrial network and disorganization of the cristae, which caused cytochrome c release and apoptosis (59). However, the mechanism by which mitofilin downregulation induces cell apoptosis is unknown.
Apoptosis, or programmed cell death, plays an important role in cardiovascular disease (1, 18, 32, 34). Mitochondria have a central role in the induction of cell death (3) by apoptosis by releasing various apoptotic factors into the cytosol. Although caspases are thought to be central elements in the apoptotic program, recent data indicate that apoptosis may also be mediated by a caspase-independent mechanism involving proapoptotic mitochondrial factors, such as apoptosis-inducing factor (AIF) and poly(ADP-ribose) polymerase (PARP) (9, 10, 29, 36). In the heart, exposing cardiomyocytes to oxidative stress (e.g., hydrogen peroxide) releases both AIF and cytochrome c from mitochondria (6), which indicates that both caspase-independent and -dependent apoptotic pathways are activated.
In this study, using cultured H9c2 cardiomyoblasts and human embryonic kidney (HEK 293) cells treated with mitofilin siRNA, we investigated the mechanism by which mitofilin knockdown induces apoptosis. We report here that mitofilin knockdown increases mitochondrial dysfunction and cell death by apoptosis as compared with scrambled siRNA. The mechanism of apoptosis involves the AIF-PARP cleavage axis through activation of both PARP and calpain, leading to nuclear fragmentation and S phase cell cycle arrest, which occurs independent of caspase activation.
MATERIALS AND METHODS
Experimental protocol.
Protocols followed the Guide for the Care and Use of Laboratory Animals (NIH, Bethesda, MD) and received University of Texas Health Science Center at San Antonio Institutional Animal Care and Use Committee (IACUC) institutional approval.
Cell culture and reagents.
Rat H9c2 cardiomyoblast line was purchased from the American Type Culture Collection (ATCC no. CRL-1446), and HEK 293 cell line was obtained from ATCC (no. CRL-3216). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen Life Technologies) supplemented with 10% fetal bovine serum (FBS; GIBCO-BRL, Grand Island, NY), 100 U/ml penicillin-streptomycin and grown in an atmosphere of 5% CO2-95% humidified air at 37°C. The culture medium was changed every second day. Cells were used between passage 4 and 7, at 70–80% confluence. The cells were selected randomly, and the data analysis was performed by a blinded investigator.
siRNA transfection.
The siRNAs against rat IMMT (siRNA1 and siRNA2) and scrambled siRNA were purchased from Life Technologies (siRNA1, catalog no. AM16708, siRNA ID: 281926; siRNA2, catalog no. 4390771, siRNA ID: s161806; and scrambled siRNA, catalog no. AM16708, siRNA ID: 4390843). H9c2 and HEK 293 cells passage 4–7, at 70–80% confluence were transfected with IMMT siRNAs (200 nM; siRNA-1, sense sequence GGUGGUGUCUCAAUAUCAUtt and antisense AUGAUAUUGAGACACCACCtt; siRNA-2, sense sequence GGAACAUCUACAUUCGUAAtt and antisense UUACGAAUGUAGAUGUUCCtt) and scrambled siRNA (sense, GGGAGUUGGCAUUUCAAAtt and antisense UUUGAAACUGCCAACUCCCtc) using Lipofectamine 2000 (ThermoFisher Scientific, catalog no. 11668027) according to the manufacturer’s instructions.
Flow cytometry analysis of cell apoptosis.
Cell apoptosis was determined using Annexin V-FITC/PI Apoptosis Detection Kit (BD Bioscience, BD PharMingen, catalog no. 556547) according to the manufacturer's instructions. Briefly, the harvested cells were washed twice in PBS and resuspended in 500 μl binding buffer. The cells were treated with 5 μl FITC-conjugated Annexin V (BD PharMingen, catalog no. 51-65875X) and 5 μl propidium iodide (PI, BD PharMingen, catalog no. 556547) or 7-aminoactinomycin-D (7-AAD; BD PharMingen, catalog no. 559763) and immediately analyzed using a BD LSR II flow cytometer (BD Biosciences, San Jose, CA). Annexin V staining precedes the loss of membrane integrity which accompanies the latest stages of cell death resulting from either apoptotic or necrotic processes. Therefore, staining with Annexin V was used in conjunction with a vital dye such as PI or 7-AAD to identify early apoptotic cells (PI or 7-AAD negative and Annexin V positive). Viable cells with intact membranes exclude PI or 7-AAD, whereas the membranes of dead and damaged cells are permeable to PI or 7-AAD. Cells considered viable are PE Annexin V and 7-AAD negative; cells that are in early apoptosis are Annexin V positive and PI or 7-AAD negative; and cells that are in late apoptosis or already dead are both PE Annexin V and PI or 7-AAD positive.
Cell viability.
The H9c2 myoblast viability was assessed spectrofluorometrically using a calcein AM assay (ThermoFisher Scientific, catalog no. C3100MP) according to the manufacturer’s instructions. H9c2 cells seeded at varying densities (2k, 4k, 6k, and 8k cells) were collected in DMEM and centrifuged at 250 g for 5 min. The supernatant was discarded while the cell pellet was resuspended in 2 ml of 1× calcein AM in DW buffer and centrifuged at 250 g for 5 min to remove all traces of phenol red. The pelleted cells were then resuspended in 1 ml of the same buffer then 1 ml of freshly prepared 2× calcein AM Working Solution was added. Samples were incubated for 30 min at 37°C in a CO2 incubator. At the end of this incubation, the fluorescence was measured using 490/520 nm excitation/emission filters and the graph of fluorescence intensity (in arbitrary units) as a function of the number of viable cells was plotted (see Fig. 2D).
Fig. 2.

Knockdown of mitofilin in rat H9c2 myoblasts increases cell apoptosis and reduces viability. A, top: flow cytometry with unstained (blank), Annexin V only (early apoptosis, pink cells), and propidium iodide (PI) only (dead cells and/or late apoptosis, blue cells) used for calibration. Bottom: flow cytometry analysis with Annexin V/PI was used to assess cell death after mitofilin siRNA-1 treatment versus scrambled siRNA. B: immunolabeling of H9c2 myoblasts with DAPI (blue) and Annexin V (green) showing a decrease in Annexin V staining in cells treated with mitofilin siRNA-1 versus scrambled siRNA. C: graph showing an increase in cell death by apoptosis (Annexin V+/PI+) in mitofilin knockdown with mitofilin siRNA-1 compared with scrambled siRNA. Both Annexin V- and PI-positive cells were counted for cell death estimation. Values are expressed as means ± SE. *P < 0.001 H9c2 cells treated with mitofilin siRNA-1 versus cells treated with scrambled siRNA (n = 12/group). D: graph showing a reduction of H9c2 myoblast viability assessed spectrofluorometrically using calcein AM assay in cells transfected with mitofilin siRNA-1 (blue line) versus scrambled siRNA (black line). Values are expressed as means ± SE. *P < 0.01 H9c2 cells treated with mitofilin siRNA-1 versus cells treated with scrambled siRNA (n = 12/group).
Western blot analysis.
Equal amounts of protein were loaded in each well of 4–20% Tris-glycine gels (Bio-Rad) as recently described in (30). After electrophoresis for 90 min at 125 V of constant voltage, the gel was blotted onto a nitrocellulose membrane by electrophoretic transfer at 70 V of constant voltage for 1–2 h. The membrane was washed, blocked with 5% blocking solution, and probed with various primary antibodies: anti-mitofilin (Abcam, catalog no. ab110329), anti-AIF [Cell Signaling Technology (CST), catalog no. 4642], anti-PARP (CST, catalog no. 9532), and anti-GAPDH (CST, catalog no. 2118S) at 4°C overnight. The immunoreactive bands were visualized using secondary Li-Cor antibodies (LI-COR Biotechnologies, Lincoln, NE): Ire 800CW goat anti-rabbit antibody, catalog no. 926-32211 and IRDye 680RD goat anti-mouse antibody, catalog no. 926-68070).
Immunofluorescence staining.
The cells at passage 4–7 were plated on eight-well glass chamber slides and allowed to reach 70–80% confluence. The medium was changed every other day. Twenty-four hours before the mitofilin siRNA transfection, the medium was changed to OPTI-MEM serum and antibiotic-free media (Life Technologies). The next day, the cells were transfected with mitofilin siRNA or scrambled siRNA according to manufacturer’s protocol. After 24 h, the cells were fixed and stained with various antibodies: anti-mitofilin antibody (Abcam, catalog no. ab110329), anti-AIF antibody (CST, catalog no. 4642), and MitoTracker Red (Molecular Probes, Life Technologies, catalog no. M7512). Secondary antibodies used were Alexa Fluor 488 goat anti-rabbit (Abcam, catalog no. ab150077), Alexa Fluor 488 goat anti-mouse (Abcam, catalog no. ab150113), and Alexa Fluor 647 goat anti-mouse (Abcam, catalog no. ab150119). Imaging was done on a Zeiss Axiovert 200M inverted motorized fluorescence microscope (Carl Zeiss Microscope, Jena, Germany).
Cytochrome c ELISA.
Cytochrome c production was measured using a commercially available kit (Abcam, catalog no. ab110172), according to the manufacturer’s protocol specific for cultured cells. Briefly, H9c2 cardiomyoblasts were transfected with mitofilin siRNA or scrambled siRNA as control. At the 24-h time point, cells were lysed and the total protein concentration of each sample was measured by Pierce BCA protein assay kit (ThermoFisher, catalog no. 23227). Equal concentrations of protein were loaded into a 96-well plate (200 μl) and incubated for 3 h at room temperature with the cytochrome c detector antibody provided. After 3 h, the wells were washed and incubated with solution A for 1 h followed by incubation with solution B for another hour followed by incubation with the horseradish peroxidase (HRP)-labeled development solution. The absorbance of each well was measured at 600 nm at room temperature at 5, 15, and 30 min, using kinetic mode. The change in the absorbance is expressed as change in Optical Density per minute and normalized to the total protein content of each sample.
ATP assay.
Intracellular ATP levels were quantified using an ATP Bioluminescence Assay Kit (Roche Applied Science, catalog no. A22066) following the standard protocol provided by the vendor. Briefly, the cells were lysed with the Cell Lysis Reagent on ice, and 50 µl of the lysate was mixed with 50 µl of the Luciferase Reagent. Then the luminescence of the samples was measured using a plate reader. The ATP concentrations of the samples were calculated using an ATP standard curve and normalized to the protein concentrations of the samples, which were determined using the BCA assay.
Mitochondrial ROS production.
Mitochondrial ROS production was assessed as described previously (38). Mitochondrial ROS generation was measured spectrofluorometrically (560-nm excitation and 590-nm emission) in 125 μg/ml of mitochondrial protein incubated in a solution containing: 20 mM Tris, 250 mM sucrose, 1 mM EGTA, 1 mM EDTA, and 0.15% bovine serum albumin adjusted to pH 7.4 at 30°C with continuous stirring. ROS were measured with the H2O2-sensitive dye Amplex red (10 μM) according to the manufacturer’s instructions (ThermoFisher, catalog no. A12222). H2O2 levels were measured from a calibration curve obtained from the fluorescence emission intensity as a function of H2O2 concentration. The sodium salt of glutamate/malate (3 mM) was used to activate complex I of the mitochondrial electron transport chain.
Caspase 3/7 activity assay.
Caspase 3/7 activity was measured using a Vibrant FAM Caspase 3 and 7 Assay kit (ThermoFisher, catalog no. V35118) according to the manufacturer’s protocol. Briefly, cells were transfected with mitofilin siRNA as previously specified, for 24 h. After 24 h, cells were harvested and incubated for 1 h at 37°C and 5% CO2 with the Fluorescent-Labeled Inhibitor of Caspases (FLICA) reagent followed by propidium iodide staining. Cells were analyzed on a flow cytometer at 488 nm of excitation and 585 nm emission.
Calpain activity assay.
To measure calpain activity, we used the Calpain Activity Assay kit (Abcam, catalog no. ab65308) following the supplier’s instructions. We transfected cells with mitofilin siRNA as described above and collected them after 24 h. Cells were suspended in Extraction buffer, lysed and the concentration was determined. Equal concentrations of lysates were incubated with Reaction buffer, calpain substrate and inhibitor for 1 h at 37°C. Reading were then taken on a fluorescence spectrophotometer at excitation/emission wavelengths of 400/505 nm.
MMP assay.
Mitochondrial membrane potential (MMP) was assessed using a MitoProbe DilC1 (5) assay kit (ThermoFisher Scientific, catalog no. M34151) and MitoTracker Red CMXROS assay kit (ThermoFisher Scientific, catalog no. M7512) for flow cytometry according to manufacturer’s protocol. H9c2 cells were plated in six-well plates and allowed to reach 70–80% confluence. At that time, the cells were transfected with scrambled siRNA or 200 nM mitofilin siRNA. After 24 h, H9c2 cells were harvested and incubated with DilC1 (5) or MitoTracker Red for 1 h at 37°C. CCCP (supplied with the kit) was used as a control to confirm that the DilC1 (5) response is sensitive to changes in membrane potential. A total of 1 × 106 cells per sample were used. Additionally, DilC1 (5)-stained cells were incubated with Annexin V (Abcam, catalog no. ab150113) and propidium iodide as apoptosis markers. Cells were analyzed on a flow cytometer with filters appropriate for Alexa Fluor 488 and Alexa Fluor 647.
Electron microscopy.
The cells were imaged by electron microscopy to observe mitochondrial quality and morphology. Cells from both groups were fixed in 2.5% (wt/vol) glutaraldehyde (Fluka) and stored in the same solution at 4°C overnight. Cells were washed with PBS, postfixed in 2% (wt/vol) osmium tetroxide for 2 h at room temperature. Fixed cells were dehydrated in a graded alcohol series and embedded in Eponate 12 medium. The blocks were cured at 60°C for 48 h, and sections (70 nm) were cut with an RMC ultramicrotome and mounted on Formvar-coated grids. The sections were double-stained with uranyl acetate and lead citrate, and finally examined and imaged with a 100CX JEOL transmission electron microscope.
Mitofilin overexpression in H9c2 myoblasts.
Transfection of H9c2 cells was carried out following the manufacturer’s instructions (Lipofectamine 2000, Invitrogen, Carlsbad, CA). Briefly, cells in 5 ml complete DMEM per plate were grown at 37°C in a CO2 incubator until the cells reached 70–80% confluence. pCMV6-Immt or pCMV6 (vector control) plasmids (Origene Technologies, catalog no. MR216091 and catalog no. PS100001) and Lipofectamine Reagent were diluted in Opti-MEM medium separately. Diluted DNA was added to dilute Lipofectamine Reagent (1:1 ratio), at 2.5 μg DNA per 10 μl Lipofectamine, then incubated for 5 min at room temperature. The resulting DNA-lipid complex was added to cells. Following transfection, H9c2 cells were cultured in DMEM for 2 days.
Statistical analysis.
Error bars are the standard errors of the mean (±SE) for a minimum of three independent trials (n ≥ 3). Under the ANOVA model, pairwise mean comparisons were judged significant using the Tukey studentized range criterion. SPSS (version 13.0, SPSS, Chicago, IL) was used to carry out the computations. Because all outcomes were continuous, results are summarized with means ± SE. P < 0.05 was considered statistically significant.
RESULTS
Transfection of H9c2 and HEK293 cells with mitofilin siRNA reduces mitofilin expression.
To determine the role of mitofilin knockdown in cell death, we transfected cultured rat H9c2 myoblasts with 200 nM of two different mitofilin siRNAs (siRNA-1 and siRNA-2) or scrambled siRNA. Figure 1A shows the immunostaining of H9c2 cells labeled with DAPI, anti-mitofilin antibody and the overlay of both signals. We found that the level of mitofilin in cells transfected with mitofilin siRNA-1 was dramatically reduced compared with cells transfected with scrambled siRNA, with the efficiency reaching 40%. These immunolabeling results were confirmed by Western blot analysis performed on the whole cell lysate that showed a significant reduction of mitofilin in cells transfected with mitofilin siRNA versus scrambled siRNA (Fig. 1, A and B). We also checked the efficiency of a different mitofilin siRNA (named siRNA-2) by transfecting H9c2 myoblasts with mitofilin siRNA-2. Figure 1C shows images of H9c2 myoblasts labeled with DAPI (blue) and anti-mitofilin (red). We found that the mitofilin expression was similarly reduced after transfection with mitofilin siRNA-2 (Fig. 1D) as with siRNA1 (Fig. 1B) versus scrambled siRNA (~40% reduction of mitofilin). To confirm the impact of the mitofilin deletion, we transfected another cell line (HEK 293 cells) with the mitofilin siRNA-1. Western blot analysis showed the reduction in mitofilin expression in HEK 293 cells after transfection with mitofilin siRNA-1 compared with scrambled siRNA (see Fig. 8). Together, these results indicate that transfection with mitofilin siRNA reduces mitofilin expression in cultured H9c2 myoblasts and HEK 293 cells. As both mitofilin siRNAs induce similar effects, we decided to only use siRNA-1 in our further experiments.
Fig. 1.

Transfection of rat H9c2 myoblasts with two different mitofilin siRNAs reduces mitofilin expression. A, top: image of H9c2 myoblasts labeled with DAPI (blue) and anti-mitofilin (green) and the overlay of both signals showing the reduction of mitofilin expression after transfection with mitofilin siRNA-1 versus scrambled siRNA. Bottom: graph showing the level of mitofilin in H9c2 myoblasts transfected with mitofilin siRNA-1 and scrambled siRNA. B: Western blot analysis showing the reduction in expression of mitofilin in H9c2 myoblasts after transfection with mitofilin siRNA-1 versus scrambled siRNA. C, top: image of H9c2 myoblasts labeled with DAPI (blue) and anti-mitofilin (red) and the graph showing the reduction of mitofilin expression after transfection with the second mitofilin siRNA (mitofilin siRNA-2) versus scrambled siRNA. Bottom: graph showing the level of mitofilin in H9c2 myoblasts transfected with mitofilin siRNA-2 and scrambled siRNA. Values are expressed as means ± SE. *P < 0.001 H9c2 cells treated with mitofilin siRNA-1 and 2 versus cells treated with scrambled siRNA (n = 10/group).
Fig. 8.

Transfection of human embryonic kidney (HEK) 293 cells with mitofilin siRNA reduces mitofilin expression. A: Western blot analysis showing the reduction in expression of mitofilin in HEK 293 cells after transfection with mitofilin siRNA-1 versus scrambled siRNA. Values are expressed as means ± SE. *P < 0.001 mitofilin siRNA-treated cells versus cells treated with scrambled siRNA (n = 10/group). B: immunoblot and graph showing an increase in apoptosis-inducing factor (AIF) expression in HEK 293 cells transfected with mitofilin siRNA-1 versus scrambled siRNA. C: immunoblot and graph showing no change in cleaved caspase 3 in HEK 293 cells transfected with mitofilin siRNA-1 versus scrambled siRNA. PARP, poly(ADP-ribose) polymerase. D: immunoblot and graph showing an increase in cleaved PARP in HEK 293 cells transfected with mitofilin siRNA-1 versus scrambled siRNA. Values are expressed as means ± SE. *P < 0.05 HEK 293 cells treated with mitofilin siRNA-1 versus cells treated with scrambled siRNA (n = 3/group).
Mitofilin knockdown increases cell death by apoptosis.
To determine whether mitofilin knockdown promotes cell death, rat H9c2 myoblasts were transfected with 200 nM mitofilin siRNA or scrambled siRNA. Cells were stained with Annexin V/PI and analyzed by flow cytometry. Annexin V- and PI-positive cells were counted as an index of cell death. Figure 2A shows the flow cytometry analysis and Fig. 2B shows the immunostaining of cells with Annexin V (green) as well as DAPI (blue, nucleus marker) in both groups. We found that mitofilin knockdown increased H9c2 cell death by apoptosis compared with control (42.06 ± 2% vs. 16.9 ± 1%, n = 12/group, P < 0.05; Fig. 2C). We also found that the H9c2 myoblast viability assessed spectrofluorometrically using the calcein AM assay was reduced in cells transfected with mitofilin siRNA (blue line) versus scrambled siRNA (black line) (Fig. 2D). These results indicate that mitofilin knockdown enhances cell death by apoptosis. Interestingly, mitofilin overexpression in H9c2 myoblasts led to reduction in cell death (see Fig. 11).
Fig. 11.

Mitofilin overexpression in rat H9c2 myoblasts reduces cell death. A, top: flow cytometry with unstained (blank, purple cells), Annexin V only (early apoptosis, orange cells), and 7-AAD only (dead cells and/or late apoptosis, green cells) used for calibration. Bottom: Annexin V/7-AAD staining followed by flow cytometry analysis used to assess cell death. B: graph showing a decrease in cell death (dead cells and/or late apoptosis, green cells) measured with Annexin and propidium iodide dye (Annexin+/7-AAD+) in mitofilin overexpressing cells compared with untreated cells. C and D: note that the number of cells in early apoptosis (Annexin+/7-AAD−, orange cells) was reduced, but not statistically different between both groups (C) and the number of live cells (unstained cells, Annexin−/7-AAD−, purple cells) increased in cells overexpressing mitofilin as compared with cells untreated cells (D). Values are expressed as means ± SE. *P < 0.05 H9c2 cells treated with pCMV6-Immt versus untreated cells (n = 3/group).
Mitofilin knockdown induces cell death independent of caspase 3 activation, but via an apoptosis-inducing factor mechanism.
Caspases are well known to be important mediators of programmed cell death by apoptosis. Caspase 3 is a frequently activated death protease, catalyzing the specific cleavage of many key cellular proteins. We therefore determined the involvement of caspase 3 activity in the mechanism of apoptosis observed in H9c2 myoblasts transfected with mitofilin siRNA. Caspase 3 activity was assessed by flow-cytometry with FLICA reagent in H9c2 cells transfected with mitofilin siRNA or scrambled siRNA (Fig. 3A). We found that, although there was a threefold increase in cell death (Fig. 3B) measured by PI staining (yellow cells) in mitofilin siRNA-transfected cells, there was no difference in caspase 3 activity between both groups (Fig. 3C, blue cells). Note that the number of live cell (purple cells) was reduced in mitofilin siRNA transfected cells as compared with scrambled siRNA (Fig. 3D). These observations indicate that mitofilin knockdown induces apoptosis via a caspase 3-independent mechanism. To define the mechanism by which mitofilin deletion in H9c2 myoblasts transfected with mitofilin siRNA induces apoptosis, we determined the role of apoptosis inducing factor (AIF) using immunolabeling and Western blot analysis. We found that AIF expression was higher in cells treated with mitofilin siRNA compared with scrambled siRNA (Fig. 4, A and B). In fact, as shown in Fig. 4A (center), the immunostained level of AIF was increased by 60% in cells treated with mitofilin siRNA versus scrambled siRNA. Furthermore, Western blot analysis performed in both groups confirmed the increase in AIF levels in mitofilin siRNA-treated cells compared with scrambled siRNA (Fig. 4B). In addition, observation of nuclei labeled with DAPI shows that cells treated with mitofilin siRNA have increased in fragmented nuclei as compared with those treated with scrambled siRNA (Fig. 4C). All together, these results indicate that treatment of H9c2 cells with mitofilin siRNA induces apoptosis in an AIF-dependent mechanism, which is associated with nuclear (DNA) fragmentation.
Fig. 3.

Knockdown of mitofilin in rat H9c2 myoblasts increases apoptosis in caspase 3 cleavage-independent pathway. A, top: flow cytometry analysis with unstained dye [blank, live cells, FLICA−/propidium iodide (PI)−, purple cells], FLICA only (caspase 3 activity, FLICA+, blue cells), and PI only (dead cells and/or late apoptosis, FLICA+/PI+ orange cells) used for calibration. Bottom: caspase 3 activity was assessed by flow cytometry using FLICA dye in H9c2 cells transfected with mitofilin siRNA-1 or scrambled siRNA. B and C: although there was a threefold increase in cell death (late apoptosis, orange cells) in cells transfected with mitofilin siRNA-1 compared with scrambled siRNA measured by propidium iodide dye (PI+) (B), there was no difference in caspase 3 positive cells (FLICA staining, blue cells) between both groups (C). D: note that the number of live cells (unstained cells, FLICA−/PI−, purple cells) was reduced in H9c2 cells transfected with mitofilin siRNA-1 or scrambled siRNA (D) confirming the increase in cell death. Values are expressed as means ± SE. *P < 0.05 H9c2 cells treated with mitofilin siRNA-1 versus cells treated with scrambled siRNA (n = 6/group).
Fig. 4.

Knockdown of mitofilin in rat H9c2 myoblasts increases apoptosis via apoptosis-inducing factor (AIF). A: confocal images showing H9c2 cells transfected with mitofilin siRNA-1 or scrambled siRNA labeled with DAPI (blue) and AIF (red) as well as the overlay. B: immunoblot (top) and graph (bottom) showing an increase in AIF in cells transfected with mitofilin siRNA-1 versus scrambled siRNA. Values are expressed as means ± SE. *P < 0.05 H9c2 cells treated with mitofilin siRNA-1 versus cells treated with scrambled siRNA (n = 6/group). C: graph showing an increase in the number of fragmented nuclei in cells transfected with mitofilin siRNA-1 versus scrambled siRNA. The number of fragmented nuclei was obtained in 100 cells randomly chosen from images. Values are expressed as means ± SE. *P < 0.01 H9c2 cells treated with mitofilin siRNA-1 versus cells treated with scrambled siRNA (n = 6/group).
Mitofilin knockdown increases PARP cleavage as well as S phase cell cycle arrest.
Knowing that mitofilin siRNA transfected into H9c2 myoblasts induces apoptosis via an AIF-dependent mechanism, we examined whether the mechanism of this apoptosis involves the AIF-PARP mechanism that is known to be associated with cell cycle dysfunction. To this end, we performed Western blot analysis in the whole cell lysate and found that the level of cleaved PARP was increased in cells treated with mitofilin siRNA compared with scrambled siRNA (Fig. 5A). This result certainly indicates that the increase in AIF expression with mitofilin siRNA treatment leads to PARP cleavage in H9c2 cells. To further reveal the impact of the cell cycle dysfunction in H9c2 cell death caused by mitofilin knockdown with siRNA, H9c2 cells were stained with propidium iodide (PI) and cell cycle analysis was performed by flow cytometry. We found that, whereas the number of cells in the G1 phase was similar in both groups, the number of cells in S phase of the cell cycle was increased in the mitofilin knocked down cell group compared with control (cells transfected with scrambled siRNA, 18 ± 0.85% vs. 34 ± 2.8%) (Fig. 5B). This result indicates that mitofilin knockdown induces H9c2 cell death via an AIF-PARP mechanism and is associated with S phase arrest of the cell cycle.
Fig. 5.

Knockdown of mitofilin in rat H9c2 myoblasts increases poly(ADP-ribose) polymerase (PARP) cleavage and induces cell cycle arrest. A: immunoblot and graph showing an increase in cleaved PARP in H9c2 cells transfected with mitofilin siRNA-1 versus scrambled siRNA. B: H9c2 cells transfected with mitofilin siRNA-1 or scrambled siRNA were stained with propidium iodide (PI) for cell cycle analysis. The result indicates that mitofilin-deficient H9c2 cells have much longer S and G2 phases compared with scrambled siRNA indicating arrest in the cell cycle. Values are expressed as means ± SE. *P < 0.05 H9c2 cells treated with mitofilin siRNA-1 versus cells treated with scrambled siRNA (n = 3/group).
Mitofilin knockdown decreases mitochondrial membrane potential and increases ROS production.
To assess whether the mitochondrial membrane potential (MMP) is impacted by mitofilin knockdown, transfected cells were harvested and the MMP was measured by flow cytometry using two different dyes, MitoProbe DilC1 (5) and MitoTracker Red. We found that mitofilin siRNA-treated cells exhibited a 46% reduction in mitochondrial membrane potential compared with control (scrambled siRNA) (Fig. 6A). In fact, the DilC1 (5) mean fluorescent intensity was reduced from 100% in scrambled siRNA-treated cells to 54% in cells transfected with mitofilin siRNA. Using MitoTracker Red dye, a red fluorescent dye that stains mitochondria in live cells and whose accumulation is dependent on membrane potential, we found a reduction in the MMP in cells transfected with mitofilin siRNA versus scrambled siRNA. In fact, the number of MitoTracker Red positive (orange bars) cells was decreased, while the number of MitoTracker Red negative (green) was increased in cells transfected with mitofilin siRNA versus scrambled siRNA (Fig. 6B). Together, these results indicate the dissipation of MMP with mitofilin deletion in H9c2 myoblasts. To define whether mitofilin knockdown in cells induces increases in ROS production, ROS generation was measured spectrofluorometrically using Amplex red and HRP in the presence of glutamate-malate in transfected cells. Figure 6C shows the increase in ROS production in mitofilin siRNA-treated cells versus scrambled siRNA. In fact, ROS production in cells treated with scrambled siRNA was 52.73 ± 7.13 pmol·min−1·mg protein−1 versus 123.94 ± 6.14 pmol·min−1·mg protein−1 in mitofilin siRNA. These results indicate that mitofilin knockdown by siRNA increases mitochondrial ROS production.
Fig. 6.

Knockdown of mitofilin in rat H9c2 myoblasts reduces mitochondrial membrane potential and increases reactive oxygen species (ROS) production. A, top left: flow cytometry with unstained (blank), and DiLC1 (5) only (mitochondrial membrane potential: MMP) used for calibration. Bottom left: mitochondrial membrane potential (MMP) was measured by flow cytometry using DiLC1 (5). Right: corresponding graph showing the reduction of the MMP in cells transfected with mitofilin siRNA-1 (blue) versus scrambled siRNA (black). MFI, mean fluorescent intensity. B, top left: flow cytometry with unstained (blank), and MitoTracker Red only (mitochondrial membrane potential: MMP) used for calibration. Bottom left: mitochondrial membrane potential (MMP) was measured by flow cytometry using MitroTracker red. Right: corresponding graph showing the reduction in MitoTracker Red positive (orange) and increase in MitoTracker Red negative (green) cells transfected with mitofilin siRNA-1 versus scrambled siRNA. C, left: recording of mitochondria reactive oxygen species (ROS) production using Amplex red in the presence of horseradish peroxidase after stimulation of complex I with glutamate/malate. Right: graph showing increase in ROS production in H9c2 cells transfected with mitofilin siRNA-1 versus scrambled siRNA. Values are expressed as means ± SE. *P < 0.05 H9c2 cells treated with mitofilin siRNA-1 versus cells treated with scrambled siRNA (n = 6/group).
Mitofilin knockdown induces disruption of mitochondrial cristae, decreases intracellular ATP production, and increases cytochrome c release.
To define the structural changes in mitochondrial cristae morphology related to mitofilin knockdown, transfected H9c2 cells in both groups were collected and processed for electron microscopy imaging. Figure 7A shows that mitochondria from cells transfected with mitofilin siRNA have fragmented cristae morphology versus those transfected with scrambled siRNA, in which cristae were mostly normal and continuous. This observation indicates that mitofilin knockdown increases mitochondrial cristae damage and presumably reduces mitochondrial membrane potential observed in Fig. 6, A and B, and subsequently mitochondrial function.
Fig. 7.
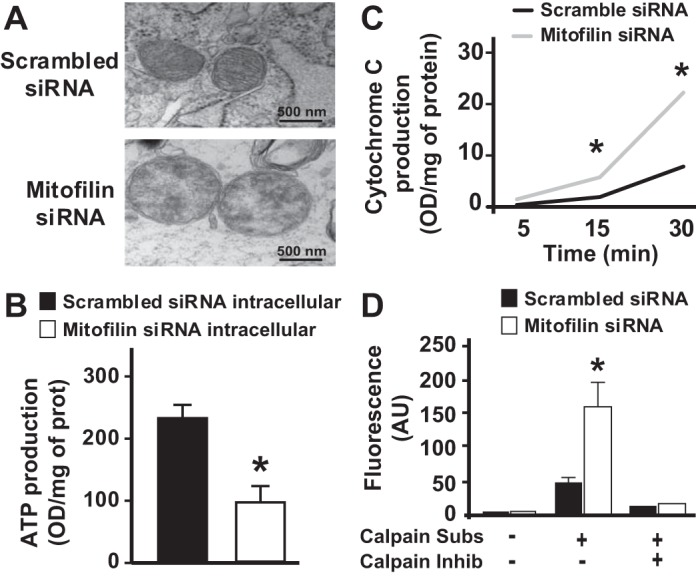
Knockdown of mitofilin in rat H9c2 myoblasts reduces damage to mitochondrial structure and intracellular ATP production as well as increases calpain activity and cytochrome c release. A: electron microscopy images of mitochondria in cells showing damaged cristae in cells transfected with mitofilin siRNA-1 compared with those transfected with scrambled siRNA. B: graph showing the reduction of intracellular ATP production measured by an ATP assay in H9c2 cells transfected with mitofilin siRNA-1 versus scrambled siRNA. C: graph showing the increase in cytochrome c release in H9c2 myoblasts transfected with mitofilin siRNA-1 versus scrambled siRNA control. Cytochrome c release was measured in lysed cells at 5, 15, and 30 min. Cells treated with mitofilin siRNA-1 showed an increase in cytochrome c release from the 5 min time point and much more at 15 min and 30 min time points as compared with cells treated with scrambled siRNA. D: graph showing the increase in calpain activity in H9c2 myoblasts transfected with mitofilin siRNA-1 versus scrambled siRNA control. Note that calpain activity was measured using a kit in the presence or absence of calpain substrate and inhibitor. Values are expressed as means ± SE. *P < 0.05 H9c2 cells treated with mitofilin siRNA versus cells treated with scrambled siRNA (n = 6/group).
To determine whether mitofilin knockdown in H9c2 cells affects the ATP production, transfected cells were lysed and the ATP production was measured by quantification of luciferase activity. The ATP concentration of the samples was calculated using an ATP standard curve and normalized by the total protein content of each sample. Mitofilin siRNA-transfected H9c2 cells showed a marked decrease in ATP production compared with control [OD/mg protein for control was 224.57 ± 19.15 vs. 82.86 ± 20.20 for mitofilin siRNA-transfected cells (Fig. 7B), n = 3/group, P < 0.05). This result indicates that mitofilin knockdown by siRNA induces mitochondrial damage that results in reduction of intracellular ATP production.
To further investigate whether mitofilin knockdown induces an increase in cytochrome c release from mitochondria, H9c2-transfected cardiomyoblasts were lysed and cytochrome c activity was measured at 5, 15, and 30 min. We found that mitofilin siRNA-treated cells displayed an increase in cytochrome c production (Fig. 7C). In fact, at the 5 min time point, cytochrome c production was 2.21 OD·min−1·total protein−1 mitofilin siRNA-transfected cells versus 0.935 OD·min−1·total protein−1 for control (scrambled RNA). At 15 min, cytochrome c production was 7.85 935 OD·min−1·total protein−1 content for transfected cells versus 4.05 OD·min−1·total protein−1 content for control. Finally, at the 30 min time point, cytochrome c production in treated cells was 29.53 OD·min−1·total protein−1 versus 16.18 OD·min−1·total protein−1 content for control. These results indicate that mitofilin knockdown by siRNA increases mitochondrial cytochrome c production in H9c2 cells.
Mitofilin knockdown increases calpain activity.
To assess whether calpain activity was affected by mitofilin knockdown in H9c2 cells, transfected cells were harvested and calpain activity was assessed using a calpain activity assay kit. As shown in Fig. 7D, calpain activity was dramatically increased in cells transfected with mitofilin siRNA versus scrambled siRNA. In fact, the level of calpain activity was threefold higher in cells treated with mitofilin siRNA versus scrambled siRNA (Fig. 7D). This result indicates that mitofilin knockdown by siRNA increases calpain activity that presumably can lead to mitochondrial protein fragmentation resulting in mitochondria cristae damage and subsequent reduction of mitochondrial function that is responsible for cell death by apoptosis.
Mitofilin knockdown in HEK 293 cells induces similar results to those observed in H9c2 myoblasts.
To determine whether mitofilin knockdown induces similar effects to those observed in H9c2 myoblasts when using another cell line, we transfected HEK 293 cells with the same mitofilin siRNA-1 (200 nM) and or scrambled siRNA. We found that the expression of mitofilin in HEK 293 cells was also reduced in cells transfected with mitofilin siRNA-1 as compared with those transfected with scrambled siRNA, with the efficiency of 35–40% (Fig. 8A). We also found that mitofilin knockdown in HEK 293 cells led to increased cell death (Fig. 9, A–C). The mechanism of this cell death was associated with an increase in AIF expression (Fig. 8B) and PARP cleavage (Fig. 8C), but independent of caspase 3 activation (Fig. 8D).
Fig. 9.

Mitofilin knockdown in human embryonic kidney (HEK) 293 cells increases cell death. A, top: flow cytometry with unstained (blank, purple cells), Annexin V only (early apoptosis, brown cells), and 7-aminoactinomycin-D (7-AAD) only (dead cells and/or later apoptosis, green cells) used for calibration. Bottom: Annexin V/propidium iodide (PI) staining followed by flow cytometry analysis was used to assess cell death. B: bar graph showing that the number of cell death (dead and in late apoptosis, green cells, Annexin V+/7-AAD+ measured with PI dye increased in cells with mitofilin siRNA as compared with those treated with scrambled siRNA. C: note that the number of cells in early apoptosis (Annexin V+/7-AAD−, brown cells) was similar in both groups, indicating that cells treated with mitofilin siRNA transit quickly to the late apoptosis and then death. D: the number of live cells (unstained, Annexin V−/7-AAD− purple cells) was slightly reduced in HEK 293 cells transfected with mitofilin siRNA-1 versus scrambled siRNA. Values are expressed as means ± SE. *P < 0.05 HEK 293 cells treated with mitofilin siRNA-1 versus cells treated with scrambled siRNA (n = 3/group).
Together, these results indicate that mitofilin knock down by siRNA induces similar effects in H9c2 myoblasts and HEK 293 cells.
Mitofilin overexpression in H9c2 myoblasts reduces cell death.
To further confirm the role of mitofilin expression in cell death, we overexpressed mitofilin in H9c2 myoblasts using a pCMV6-Immt or pCMV6 (vector control) plasmid and Lipofectamine Reagent. After 48 h transfection, cells were treated with H2O2 for 4 h. In Fig. 10A are images of H9c2 myoblasts transfected with mitofilin (pCMV6-Immt) and labeled with anti-mitofilin antibody as well as DAPI. The overexpression efficiency was confirmed by Western blot analysis (Fig. 10B). We found a twofold increase in mitofilin expression in H9c2 myoblasts transfected with mitofilin (pCMV6-Immt) versus untreated/vector-only cells (pCMV6). Using flow cytometry analysis with Annexin V/PI, we found that mitofilin overexpression protected myoblasts against cell death (Fig. 11, A–D). In fact, mitofilin overexpression was associated a significant decrease in cell death (Fig. 11A, green cells) and a 25% reduction of the number of annexin-positive cells (Fig. 11B, orange cells), suggesting a reduction in apoptosis.
Fig. 10.
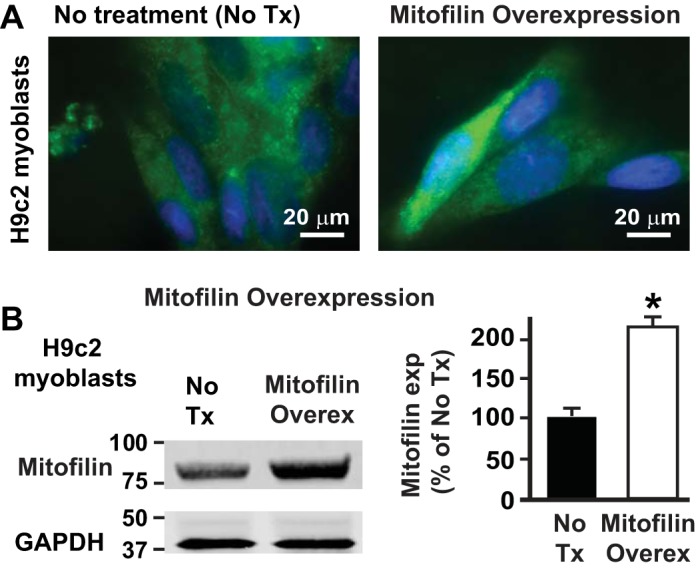
Mitofilin overexpression in H9c2 myoblasts increases mitofilin level. A: images of H9c2 myoblasts transfected with mitofilin pCMV6-Immt vector and labeled with anti-mitofilin antibody (green) and DAPI (blue) showing increase in mitofilin expression in some cells as compared with untreated/vector control. B: Western blot analysis showing the increase in expression of mitofilin in pCMV6-Immt treated cells versus untreated. Values are expressed as means ± SE. *P < 0.01 H9c2 overexpressing cells versus untreated-cells (n = 3/group).
DISCUSSION
In this study, we report that mitofilin knockdown by siRNA in both H9c2 myoblasts and HEK 293 cells increases mitochondrial structural degradation by damaging cristae. This results in a severe reduction of mitochondrial function highlighted by reductions in mitochondrial membrane potential and ATP production, as well as increases in ROS production and calpain activity. Mitochondrial dysfunction is therefore responsible for the increase in cell death by apoptosis via an AIF-PARP mechanism that triggers nuclear fragmentation and S phase arrest of the cell cycle.
Mitochondria are well-known organelles that play an important role in the generation of cellular energy and are required for normal physiological functions. In cardiomyocytes, healthy mitochondria provide the energy to support cardiac contraction, whereas diseased and disabled mitochondria are a source of cardiomyocyte damage (20, 35, 40). Hence, despite their essential role in cellular function, mitochondria have also been implicated in the process of cell death by apoptosis (23). Apoptosis is a regulated cellular suicide mechanism characterized by nuclear condensation and fragmentation, cell shrinkage, membrane bulging, and DNA fragmentation. In the intramitochondrial pathway of apoptosis, mitochondrial damage and depolarization in response to stimuli release proapoptotic proteins such as Smac/Diablo, AIF, HtrA2, and Endo G, in addition to cytochrome c, which triggers both caspase-dependent and/or -independent cell death (46, 47).
Mitofilin is an important component of the inner mitochondrial membrane (IMM) that is part of the MINOS complex, which plays a key role in controlling mitochondrial cristae morphology. In this study, we report that the protein mitofilin, besides its role as a structural constituent of the IMM, has a functional impact on cellular activity. In fact, we show here that loss of mitofilin triggers the activation of mitochondrial-dependent apoptosis pathways involving apoptosis-inducing factor (AIF), a nuclear-encoded protein synthesized as a 67 kDa precursor (49, 53). The mature form of AIF (62 kDa) is located within the mitochondrial intermembrane space following import and removal of the mitochondrial localization signal (41, 49). AIF has a prosurvival role when it is located within mitochondria, whereas release of AIF from mitochondria into the cytosol followed by nuclear translocation activates caspase-independent cell death signaling (41, 49). It has been also reported that, when activated following birth, AIF deletion in heart and skeletal muscle leads to profound dilated cardiomyopathy and muscle wasting due to a marked decrease in electron transport complex I activity concomitant with an increase in reactive oxygen species (ROS) production from mitochondria (28, 48). In this study, we found that mitofilin knockdown with two different siRNAs in H9c2 myoblasts as well as in HEK 293 cells led to dramatically reduced mitofilin expression. In both cell lines, we found that this reduction of mitofilin level increases AIF release from mitochondria to the cytosol and this effect was associated with an increase in ROS production from respiratory complex I (Fig. 6C) that is known to have deleterious effects in the myocardium (38). The generation of ROS has been implicated in the pathogenesis of myocardial, lung, and renal ischemia-reperfusion (I/R) injury, and many other pathological conditions (19, 55). DNA feature breaks caused by ROS lead to the activation of Poly(ADP-ribose) polymerase (PARP), a member of a family of proteins involved in several cellular processes involving DNA repair and programmed cell death (22). Excessive activation of PARP results in the depletion of both NAD and ATP (45) and this process may explain the increase in PARP cleavage observed in both cells (H9c2 and HEK 293 cells) treated with mitofilin siRNA in our study. Excessive poly ADP-ribosylation in response to DNA damage is known to lead to cell death observed in stroke, traumatic brain injury, and I/R injury in various organs (24). When DNA damage is mild, the poly ADP-ribosylation (PAR) response mechanism repairs the injury. However, when DNA damage is extensive, cells cannot repair the injury and the disproportionate activation of PARP depletes the cellular pools of NAD+ and ATP, and cell death ensues. This mechanism might justify the significant reduction in intracellular ATP production associated with mitofilin knockdown observed in our study. It has been proposed that PARP hyperactivation contributes to cell death, with the generation of PAR triggering the nuclear translocation of AIF, resulting in DNA condensation and fragmentation, in a caspase-independent manner (11, 60). Lozano and Elledge (37) have reported that cell cycle arrest in response to DNA damage from a variety of stimuli allows time for repair or direct cell apoptosis. After DNA damage, the cell cycle is arrested at the transition from G1 to S phase or from G2 to M phase of the cell cycle. Interestingly, we observed that H9c2 cells treated with mitofilin siRNA have condensed and fragmented nuclei (Fig. 4C), and this effect was associated with long S phase of cell cycle (Fig. 5B). As increasing evidence indicates a central role for p53 in mediating cell cycle arrest or apoptosis (5, 8), it will be interesting to investigate the role p53 in the S phase arrest of cell cycle induced by mitofilin knockdown in future studies. In addition, it has been shown in primary cortical neurons that exposure to the N-methyl-d-aspartate (NMDA) activates PARP-1 resulting in Ca2+ dysregulation, subsequent calpain activation and AIF release from mitochondria (57). The increase in mitochondrial Ca2+, associated with the increase in ROS production, disruption of ATP production, and dissipation of MMP is therefore proposed to promote the mitochondrial permeability transition (MPT) pore opening that is well known to contribute to cell death (4, 21) (Fig. 12).
Fig. 12.

Scheme depicting the mechanism underlying mitofilin knockdown-induced increase in cell death by apoptosis. Mitofilin knockdown by siRNA in cultured H9c2 myoblasts and human embryonic kidney (HEK) 293 cells induces mitochondrial structural degradation that causes the reduction of ATP production and dissipation of membrane potential. Disruption of mitochondrial cristae promotes the release of apoptosis-inducing factor (AIF) from mitochondria into the cytosol. AIF interacting with poly(ADP-ribose) polymerase (PARP) facilitates translocation into the nucleus, and together with the reactive oxygen species (ROS) produced in response to the increase in calpain activity are responsible for the DNA condensation and fragmentation that causes cell cycle arrest and subsequent apoptosis by a caspase 3-independent pathway. On the other hand, PARP is released into the cytosol and translocates into mitochondria causing Ca2+ dysregulation. The increase in intramitochondrial Ca2+, together with the increase in ROS production in the electron transport chain (ETC) and dissipation of mitochondrial membrane potential (MMP) caused by an increase in calpain activity results in the mitochondrial permeability transition pore (mPTP) opening, which promotes cell death. Note that the increase in intracellular Ca2+ may also potentiate the activation of calpain catalytic activity to promote more AIF release and ROS production that will exacerbate this harmful mechanism.
Calpains are proteins belonging to the family of Ca2+-dependent, nonlysosomal cysteine proteases (proteolytic enzymes) expressed ubiquitously in mammals and many other organisms (52). It is well known that calpains play an important role in the induction of MPT (2, 17) and in the necrotic/apoptotic cell death pathways (31, 33, 44). In addition, several studies have reported that calpain inhibition during I/R reduces apoptosis and infarct size (33, 44). Moreover, calpains have been reported to be proapoptotic proteases mediating the cleavage of AIF after Ca2+ overload (7, 43) and excessive calpain activity has also been found to result in mitochondrial damage and disrupted oxidative phosphorylation during myocardial I/R injury (7, 43). Chen et al. (7) have shown in isolated heart mitochondria that addition of exogenous Ca2+ causes AIF release and that pretreatment with MD28170, a calpain inhibitor, prevented this release indicating a calpain-related mechanism for AIF release. We report here that mitofilin knockdown exacerbates calpain activity which in turn can contribute, at least in part, to the catalytic activity on the mitochondrial membranes that may therefore, result in the dissipation of MMP and release of AIF. However, the last effect still needs to be proven as Joshi et al. (27) have reported that calpastatin and PD 150606, other calpain inhibitors, pretreated before Ca2+ overload in liver mitochondria and brain mitochondria did not prevent AIF release.
Caspases, including caspase-2, -3, -8, -9, -10, -11, and -12, are a family of cysteine proteases that are the central regulators of apoptosis (14). In the caspase-dependent mechanism of apoptosis, caspase-3 interacts with caspase-8 and caspase-9 and plays a central role in the execution-phase of cell apoptosis. In our study, we did not observe changes in the levels of cleaved caspase 3 (activated) in H9c2 cells and HEK 293 cells (Fig. 8D) treated with mitofilin siRNA versus scrambled siRNA. This result indicates that mitofilin knockdown induces cell apoptosis mainly via a caspase-3 independent mechanism and more likely in an AIF dependent pathway. The effect of mitofilin deletion in promoting cell death observed in our studies is confirmed by the fact that mitofilin overexpression in H9c2 myoblasts led to reduced cell death (Figs. 10 and 11). However, because of the limitation of our transfection that led to around 40% mitofilin knockdown and the time of our transfection (24 h), it is not excluded that a more pronounced knockdown of mitofilin may also activate the caspase 3-dependent mechanism as we found an increase in cleaved caspase 9 in cells transfected with mitofilin siRNA versus scrambled siRNA (data not shown). Nevertheless, our results seem to corroborate those obtained by Sharma et al. (51) on neuronal death during oxygen-glucose deprivation (OGD) followed by reoxygenation (Reox) treatment in segregated male (XY) and female (XX) neuronal cells. In fact, they reported a sexual dimorphic nature between male and female neuronal cell death by apoptosis during the Reox phase following an OGD episode. They found that OGD/Reox induces death of XY neurons via a PARP-1-AIF-dependent mechanism, but blockade of the PARP-1-AIF pathway shifts neuronal death towards a caspase-dependent mechanism. In XX neurons, OGD/Reox caused prolonged depletion of ATP and delayed activation of caspase 8 and caspase 3 (51). Because we do not know the sex of the embryo from which H9c2 myoblasts were cloned from (50), future studies may determine whether our results are sex specific.
In summary, this study indicates that mitofilin knockdown by siRNA in cultured H9c2 myoblasts and HEK 293 cells induces mitochondrial structural degradation and functional impairment including reduction of ATP production, dissipation of membrane potential, and increases in ROS production as well as calpain activity. Together, these effects promote the release of AIF from mitochondria into the cytosol, resulting in the interaction between PARP and AIF, which facilitates translocation into the nucleus, where AIF activation concomitant with ROS effects results in DNA condensation and fragmentation that is responsible for cell cycle arrest and subsequent cell death by a caspase-independent pathway of apoptosis (Fig. 12).
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant HL-138093, American Heart Association Grant 17SDG33100000 (J. C. Bopassa), and the Voeckler fund (J. C. Bopassa).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.B.M., M.L., and J.C.B. conceived and designed research; N.B.M., Y.F., M.L., N.T., and L.L. performed experiments; N.B.M., Y.F., M.L., and J.C.B. analyzed data; N.B.M., Y.F., F.K., and J.C.B. interpreted results of experiments; N.B.M., Y.F., M.L., and J.C.B. drafted manuscript; N.B.M., F.K., and J.C.B. edited and revised manuscript; M.L., N.T., L.L., and J.C.B. prepared figures; J.C.B. approved final version of manuscript.
REFERENCES
- 1.Abraham MC, Shaham S. Death without caspases, caspases without death. Trends Cell Biol 14: 184–193, 2004. doi: 10.1016/j.tcb.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 2.Aguilar HI, Botla R, Arora AS, Bronk SF, Gores GJ. Induction of the mitochondrial permeability transition by protease activity in rats: a mechanism of hepatocyte necrosis. Gastroenterology 110: 558–566, 1996. doi: 10.1053/gast.1996.v110.pm8566604. [DOI] [PubMed] [Google Scholar]
- 3.Bopassa JC, Ferrera R, Gateau-Roesch O, Couture-Lepetit E, Ovize M. PI 3-kinase regulates the mitochondrial transition pore in controlled reperfusion and postconditioning. Cardiovasc Res 69: 178–185, 2006. doi: 10.1016/j.cardiores.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 4.Bopassa JC, Michel P, Gateau-Roesch O, Ovize M, Ferrera R. Low-pressure reperfusion alters mitochondrial permeability transition. Am J Physiol Heart Circ Physiol 288: H2750–H2755, 2005. doi: 10.1152/ajpheart.01081.2004. [DOI] [PubMed] [Google Scholar]
- 5.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282: 1497–1501, 1998. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 6.Chen M, Zsengellér Z, Xiao CY, Szabó C. Mitochondrial-to-nuclear translocation of apoptosis-inducing factor in cardiac myocytes during oxidant stress: potential role of poly(ADP-ribose) polymerase-1. Cardiovasc Res 63: 682–688, 2004. doi: 10.1016/j.cardiores.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 7.Chen Q, Paillard M, Gomez L, Ross T, Hu Y, Xu A, Lesnefsky EJ. Activation of mitochondrial μ-calpain increases AIF cleavage in cardiac mitochondria during ischemia-reperfusion. Biochem Biophys Res Commun 415: 533–538, 2011. doi: 10.1016/j.bbrc.2011.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ciciarello M, Mangiacasale R, Casenghi M, Zaira Limongi M, D’Angelo M, Soddu S, Lavia P, Cundari E. p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. J Biol Chem 276: 19205–19213, 2001. doi: 10.1074/jbc.M009528200. [DOI] [PubMed] [Google Scholar]
- 9.Cregan SP, Dawson VL, Slack RS. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene 23: 2785–2796, 2004. doi: 10.1038/sj.onc.1207517. [DOI] [PubMed] [Google Scholar]
- 10.Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, Dawson TM, Dawson VL, Park DS, Kroemer G, Slack RS. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol 158: 507–517, 2002. doi: 10.1083/jcb.200202130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 342: 249–268, 1999. doi: 10.1042/bj3420249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darshi M, Mendiola VL, Mackey MR, Murphy AN, Koller A, Perkins GA, Ellisman MH, Taylor SS. ChChd3, an inner mitochondrial membrane protein, is essential for maintaining crista integrity and mitochondrial function. J Biol Chem 286: 2918–2932, 2011. doi: 10.1074/jbc.M110.171975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding C, Wu Z, Huang L, Wang Y, Xue J, Chen S, Deng Z, Wang L, Song Z, Chen S. Mitofilin and CHCHD6 physically interact with Sam50 to sustain cristae structure. Sci Rep 5: 16064, 2015. doi: 10.1038/srep16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 35: 495–516, 2007. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Genin EC, Plutino M, Bannwarth S, Villa E, Cisneros-Barroso E, Roy M, Ortega-Vila B, Fragaki K, Lespinasse F, Pinero-Martos E, Augé G, Moore D, Burté F, Lacas-Gervais S, Kageyama Y, Itoh K, Yu-Wai-Man P, Sesaki H, Ricci JE, Vives-Bauza C, Paquis-Flucklinger V. CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol Med 8: 58–72, 2016. doi: 10.15252/emmm.201505496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gieffers C, Korioth F, Heimann P, Ungermann C, Frey J. Mitofilin is a transmembrane protein of the inner mitochondrial membrane expressed as two isoforms. Exp Cell Res 232: 395–399, 1997. doi: 10.1006/excr.1997.3539. [DOI] [PubMed] [Google Scholar]
- 17.Gores GJ, Miyoshi H, Botla R, Aguilar HI, Bronk SF. Induction of the mitochondrial permeability transition as a mechanism of liver injury during cholestasis: a potential role for mitochondrial proteases. Biochim Biophys Acta 1366: 167–175, 1998. doi: 10.1016/S0005-2728(98)00111-X. [DOI] [PubMed] [Google Scholar]
- 18.Gottlieb RA, Gruol DL, Zhu JY, Engler RL. Preconditioning rabbit cardiomyocytes: role of pH, vacuolar proton ATPase, and apoptosis. J Clin Invest 97: 2391–2398, 1996. doi: 10.1172/JCI118683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol 6: 524–551, 2015. doi: 10.1016/j.redox.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovasc Res 77: 334–343, 2008. doi: 10.1093/cvr/cvm005. [DOI] [PubMed] [Google Scholar]
- 21.Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta 1787: 1402–1415, 2009. doi: 10.1016/j.bbabio.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 22.Herceg Z, Wang ZQ. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat Res 477: 97–110, 2001. doi: 10.1016/S0027-5107(01)00111-7. [DOI] [PubMed] [Google Scholar]
- 23.Hockenbery DM, Giedt CD, O’Neill JW, Manion MK, Banker DE. Mitochondria and apoptosis: new therapeutic targets. Adv Cancer Res 85: 203–242, 2002. doi: 10.1016/S0065-230X(02)85007-2. [DOI] [PubMed] [Google Scholar]
- 24.Hong SJ, Dawson TM, Dawson VL. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol Sci 25: 259–264, 2004. doi: 10.1016/j.tips.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Icho T, Ikeda T, Matsumoto Y, Hanaoka F, Kaji K, Tsuchida N. A novel human gene that is preferentially transcribed in heart muscle. Gene 144: 301–306, 1994. doi: 10.1016/0378-1119(94)90394-8. [DOI] [PubMed] [Google Scholar]
- 26.John GB, Shang Y, Li L, Renken C, Mannella CA, Selker JM, Rangell L, Bennett MJ, Zha J. The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol Biol Cell 16: 1543–1554, 2005. doi: 10.1091/mbc.e04-08-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joshi A, Bondada V, Geddes JW. Mitochondrial micro-calpain is not involved in the processing of apoptosis-inducing factor. Exp Neurol 218: 221–227, 2009. doi: 10.1016/j.expneurol.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joza N, Oudit GY, Brown D, Bénit P, Kassiri Z, Vahsen N, Benoit L, Patel MM, Nowikovsky K, Vassault A, Backx PH, Wada T, Kroemer G, Rustin P, Penninger JM. Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy. Mol Cell Biol 25: 10261–10272, 2005. doi: 10.1128/MCB.25.23.10261-10272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, Sasaki T, Elia AJ, Cheng HY, Ravagnan L, Ferri KF, Zamzami N, Wakeham A, Hakem R, Yoshida H, Kong YY, Mak TW, Zúñiga-Pflücker JC, Kroemer G, Penninger JM. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 410: 549–554, 2001. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- 30.Kabir ME, Singh H, Lu R, Olde B, Leeb-Lundberg LM, Bopassa JC. G protein-coupled estrogen receptor 1 mediates acute estrogen-induced cardioprotection via MEK/ERK/GSK-3β pathway after ischemia/reperfusion. PLoS One 10: e0135988, 2015. doi: 10.1371/journal.pone.0135988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kakkar R, Wang X, Radhi JM, Rajala RV, Wang R, Sharma RK. Decreased expression of high-molecular-weight calmodulin-binding protein and its correlation with apoptosis in ischemia-reperfused rat heart. Cell Calcium 29: 59–71, 2001. doi: 10.1054/ceca.2000.0157. [DOI] [PubMed] [Google Scholar]
- 32.Kang PM, Izumo S. Apoptosis and heart failure: a critical review of the literature. Circ Res 86: 1107–1113, 2000. doi: 10.1161/01.RES.86.11.1107. [DOI] [PubMed] [Google Scholar]
- 33.Khalil PN, Neuhof C, Huss R, Pollhammer M, Khalil MN, Neuhof H, Fritz H, Siebeck M. Calpain inhibition reduces infarct size and improves global hemodynamics and left ventricular contractility in a porcine myocardial ischemia/reperfusion model. Eur J Pharmacol 528: 124–131, 2005. doi: 10.1016/j.ejphar.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 34.Kim NH, Kang PM. Apoptosis in cardiovascular diseases: mechanism and clinical implications. Korean Circ J 40: 299–305, 2010. doi: 10.4070/kcj.2010.40.7.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia-reperfusion, aging, and heart failure. J Mol Cell Cardiol 33: 1065–1089, 2001. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- 36.Lockshin RA, Zakeri Z. Caspase-independent cell death? Oncogene 23: 2766–2773, 2004. doi: 10.1038/sj.onc.1207514. [DOI] [PubMed] [Google Scholar]
- 37.Lozano G, Elledge SJ. p53 sends nucleotides to repair DNA. Nature 404: 24–25, 2000. doi: 10.1038/35003670. [DOI] [PubMed] [Google Scholar]
- 38.Madungwe NB, Zilberstein NF, Feng Y, Bopassa JC. Critical role of mitochondrial ROS is dependent on their site of production on the electron transport chain in ischemic heart. Am J Cardiovasc Dis 6: 93–108, 2016. [PMC free article] [PubMed] [Google Scholar]
- 39.Mun JY, Lee TH, Kim JH, Yoo BH, Bahk YY, Koo HS, Han SS. Caenorhabditis elegans mitofilin homologs control the morphology of mitochondrial cristae and influence reproduction and physiology. J Cell Physiol 224: 748–756, 2010. doi: 10.1002/jcp.22177. [DOI] [PubMed] [Google Scholar]
- 40.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev 88: 581–609, 2008. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Natarajan SK, Becker DF. Role of apoptosis-inducing factor, proline dehydrogenase, and NADPH oxidase in apoptosis and oxidative stress. Cell Health Cytoskelet 2012: 11–27, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Odgren PR, Toukatly G, Bangs PL, Gilmore R, Fey EG. Molecular characterization of mitofilin (HMP), a mitochondria-associated protein with predicted coiled coil and intermembrane space targeting domains. J Cell Sci 109: 2253–2264, 1996. [DOI] [PubMed] [Google Scholar]
- 43.Ozaki T, Yamashita T, Ishiguro S. ERp57-associated mitochondrial μ-calpain truncates apoptosis-inducing factor. Biochim Biophys Acta 1783: 1955–1963, 2008. doi: 10.1016/j.bbamcr.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 44.Perrin C, Ecarnot-Laubriet A, Vergely C, Rochette L. Calpain and caspase-3 inhibitors reduce infarct size and post-ischemic apoptosis in rat heart without modifying contractile recovery. Cell Mol Biol (Noisy-le-grand) 49: OL497–505, 2003. [PubMed] [Google Scholar]
- 45.Pieper AA, Verma A, Zhang J, Snyder SH. Poly (ADP-ribose) polymerase, nitric oxide and cell death. Trends Pharmacol Sci 20: 171–181, 1999. doi: 10.1016/S0165-6147(99)01292-4. [DOI] [PubMed] [Google Scholar]
- 46.Polster BM, Basañez G, Etxebarria A, Hardwick JM, Nicholls DG. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J Biol Chem 280: 6447–6454, 2005. doi: 10.1074/jbc.M413269200. [DOI] [PubMed] [Google Scholar]
- 47.Polster BM, Fiskum G. Mitochondrial mechanisms of neural cell apoptosis. J Neurochem 90: 1281–1289, 2004. doi: 10.1111/j.1471-4159.2004.02572.x. [DOI] [PubMed] [Google Scholar]
- 48.Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, Esterbauer H, Kozlov A, Kahn CR, Kroemer G, Rustin P, Burcelin R, Penninger JM. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 131: 476–491, 2007. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- 49.Sevrioukova IF. Apoptosis-inducing factor: structure, function, and redox regulation. Antioxid Redox Signal 14: 2545–2579, 2011. doi: 10.1089/ars.2010.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shah K, McCormack CE, Bradbury NA. Do you know the sex of your cells? Am J Physiol Cell Physiol 306: C3–C18, 2014. doi: 10.1152/ajpcell.00281.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharma J, Nelluru G, Wilson MA, Johnston MV, Hossain MA. Sex-specific activation of cell death signalling pathways in cerebellar granule neurons exposed to oxygen glucose deprivation followed by reoxygenation. ASN Neuro 3: e00056, 2011. doi: 10.1042/AN20100032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith MA, Schnellmann RG. Calpains, mitochondria, and apoptosis. Cardiovasc Res 96: 32–37, 2012. doi: 10.1093/cvr/cvs163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397: 441–446, 1999. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 54.Thapa D, Nichols CE, Lewis SE, Shepherd DL, Jagannathan R, Croston TL, Tveter KJ, Holden AA, Baseler WA, Hollander JM. Transgenic overexpression of mitofilin attenuates diabetes mellitus-associated cardiac and mitochondria dysfunction. J Mol Cell Cardiol 79: 212–223, 2015. doi: 10.1016/j.yjmcc.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turrens JF, Freeman BA, Levitt JG, Crapo JD. The effect of hyperoxia on superoxide production by lung submitochondrial particles. Arch Biochem Biophys 217: 401–410, 1982. doi: 10.1016/0003-9861(82)90518-5. [DOI] [PubMed] [Google Scholar]
- 56.von der Malsburg K, Müller JM, Bohnert M, Oeljeklaus S, Kwiatkowska P, Becker T, Loniewska-Lwowska A, Wiese S, Rao S, Milenkovic D, Hutu DP, Zerbes RM, Schulze-Specking A, Meyer HE, Martinou JC, Rospert S, Rehling P, Meisinger C, Veenhuis M, Warscheid B, van der Klei IJ, Pfanner N, Chacinska A, van der Laan M. Dual role of mitofilin in mitochondrial membrane organization and protein biogenesis. Dev Cell 21: 694–707, 2011. doi: 10.1016/j.devcel.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 57.Vosler PS, Sun D, Wang S, Gao Y, Kintner DB, Signore AP, Cao G, Chen J. Calcium dysregulation induces apoptosis-inducing factor release: cross-talk between PARP-1- and calpain-signaling pathways. Exp Neurol 218: 213–220, 2009. doi: 10.1016/j.expneurol.2009.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weber TA, Koob S, Heide H, Wittig I, Head B, van der Bliek A, Brandt U, Mittelbronn M, Reichert AS. APOOL is a cardiolipin-binding constituent of the mitofilin/MINOS protein complex determining cristae morphology in mammalian mitochondria. PLoS One 8: e63683, 2013. doi: 10.1371/journal.pone.0063683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang RF, Zhao GW, Liang ST, Zhang Y, Sun LH, Chen HZ, Liu DP. Mitofilin regulates cytochrome c release during apoptosis by controlling mitochondrial cristae remodeling. Biochem Biophys Res Commun 428: 93–98, 2012. doi: 10.1016/j.bbrc.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 60.Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci USA 103: 18314–18319, 2006. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zerbes RM, Höß P, Pfanner N, van der Laan M, Bohnert M. Distinct roles of Mic12 and Mic27 in the mitochondrial contact site and cristae organizing system. J Mol Biol 428: 1485–1492, 2016. doi: 10.1016/j.jmb.2016.02.031. [DOI] [PubMed] [Google Scholar]
- 62.Zerbes RM, van der Klei IJ, Veenhuis M, Pfanner N, van der Laan M, Bohnert M. Mitofilin complexes: conserved organizers of mitochondrial membrane architecture. Biol Chem 393: 1247–1261, 2012. doi: 10.1515/hsz-2012-0239. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Y, Xu J, Luo YX, An XZ, Zhang R, Liu G, Li H, Chen HZ, Liu DP. Overexpression of mitofilin in the mouse heart promotes cardiac hypertrophy in response to hypertrophic stimuli. Antioxid Redox Signal 21: 1693–1707, 2014. doi: 10.1089/ars.2013.5438. [DOI] [PubMed] [Google Scholar]