

Abstract
Myocardial fibrosis and the resultant increases in left ventricular stiffness represent pivotal consequences of chronic pressure overload (PO) that impact both functional capacity and the rates of morbid and mortal events. However, the time course and cellular mechanisms that underlie PO-induced fibrosis have not been completely defined. Secreted protein acidic and rich in cysteine (SPARC) is a matricellular protein that has been shown to be required for insoluble collagen deposition and increased myocardial stiffness in response to PO in mice. As macrophages are associated with increases in fibrillar collagen, the hypothesis that macrophages represent a source of increased SPARC production in the PO myocardium was tested. The time course of changes in the myocardial macrophage population was compared with changes in procollagen type I mRNA, production of SPARC, fibrillar collagen accumulation, and diastolic stiffness. In PO hearts, mRNA encoding collagen type I was increased at 3 days, whereas increases in levels of total collagen protein did not occur until 1 wk and were followed by increases in insoluble collagen at 2 wk. Increases in muscle stiffness were not detected before increases in insoluble collagen content (>1 wk). Significant increases in myocardial macrophages that coincided with increased SPARC were found but did not coincide with increases in mRNA encoding collagen type I. Furthermore, immunohistochemistry and flow cytometry identified macrophages as a cellular source of SPARC. We conclude that myocardial macrophages play an important role in the time-dependent increases in SPARC that enhance postsynthetic collagen processing, insoluble collagen content, and myocardial stiffness and contribute to the development of fibrosis.
NEW & NOTEWORTHY Myocardial fibrosis and the resultant increases in left ventricular and myocardial stiffness represent pivotal consequences of chronic pressure overload. In this study a murine model of cardiac fibrosis induced by pressure overload was used to establish a time course of collagen expression, collagen deposition, and cardiac macrophage expansion.
Keywords: collagen, extracellular matrix, matricellular proteins, postsynthetic collagen processing, secreted protein acidic and rich in cysteine
INTRODUCTION
Secreted protein acidic and rich in cysteine (SPARC) is a collagen-binding matricellular protein that has been shown to increase significantly in response to left ventricular (LV) pressure overload (PO) (7). SPARC is necessary for processing soluble procollagen into insoluble fibrillar collagen and is necessary for the development of LV myocardial fibrosis and increased myocardial stiffness that results from LV PO (7). However, SPARC and procollagen processing are among several potential mechanisms that influence PO-induced fibrosis (13). These mechanisms include collagen synthesis, processing, and degradation; changes in each of these processes follow a differential time course during the development of PO-induced fibrosis. For example, collagen synthesis, as reflected by transcriptional regulation of mRNA encoding collagen type I, has been shown to increase within days after the induction of PO (6, 8, 13). In contrast, a significant accumulation of myocardial fibrillar collagen in the extracellular matrix (ECM) was detected weeks after the onset of PO (7). SPARC is also significantly increased weeks after the induction of PO (7). We hypothesized that mechanisms beyond transcriptional regulation, such as those mediated by SPARC, control collagen deposition into the ECM and represent key regulators of the myocardial fibrosis associated with PO. To examine this hypothesis, a time course of collagen deposition, SPARC expression, and the cellular sources of SPARC that contribute to myocardial fibrosis after PO must be defined.
Among the sources of myocardial SPARC are myocardial fibroblasts (14). In addition, SPARC expression by cell types such as macrophages has also been reported (2, 3, 22). Macrophages may be both resident within the myocardium and recruited from hematopoietic sources as monocytes. Resident macrophages are thought to contribute to normal ECM homeostasis in healthy myocardium (15, 20). However, under pathological stress, macrophage numbers within the myocardium (whether resident and/or recruited) increase (9, 21). These changes in macrophage number and phenotype have been hypothesized to constitute one mechanism responsible for the changes in ECM homeostasis that lead to PO-induced myocardial fibrosis (26). For example, in an angiotensin II infusion model of moderate LV fibrosis, the number of myocardial macrophages increased, and depletion of myocardial macrophages by clodronate liposome injection resulted in decreased fibrosis (10). In the present study, we hypothesized that, in murine PO [produced by transverse aortic constriction (TAC)], the time course of the development of severe myocardial fibrosis would be associated with an increase in the myocardial macrophage population and that macrophage-dependent SPARC production would be concordant with an increase in myocardial ECM fibrillar collagen accumulation and increased myocardial diastolic stiffness.
METHODS
Experimental protocol.
C57BL/6 mice (3–4 mo of age, male and female) underwent TAC surgery to induce LV PO, as previously described (7), or were used as normal control mice. All experiments were reviewed and approved by the Ralph H. Johnson Veterans Affairs Institutional Animal Care and Use Committee. Mice were studied 3 days, 1 wk, 2 wk, and 4 wk after LV PO. At baseline (before LV PO) and at a selected time point after LV PO, LV structure and function were characterized by echocardiography (Table 1). After LV PO and completion of echocardiographic experiments, animals were euthanized and LV myocardial experiments, including histomorphometric analyses of collagen content using picrosirius red (PSR) assessment of collagen volume fraction (CVF), immunohistochemical staining for macrophages and SPARC, biochemical assessment of collagen (hydroxyproline assay), quantification of myocardial monocytes/macrophages (FACS), measurement of myocardial stiffness, and RT-PCR analysis, were performed. The number of animals assigned to each time point and each end point measurement is shown in a consort diagram (Fig. 1).
Table 1.
Echocardiographic characterization of LV structure and function of mice at baseline and at 3 days, 1 wk, 2 wk, and 4 wk of LV PO
| LV PO |
|||||
|---|---|---|---|---|---|
| Baseline Control | 3 day | 1 wk | 2 wk | 4 wk | |
| Heart rate, beats/min | 510 ± 29 | 508 ± 27 | 488 ± 61 | 486 ± 34 | 522 ± 45 |
| LV ejection fraction, % | 62 ± 3 | 61 ± 4 | 57 ± 5 | 58 ± 8 | 56 ± 9 |
| LV mass/body mass, mg/g | 3.4 ± 0.1 | 3.7 ± 0.2 | 4.1 ± 1.1 | 5.7 ± 0.3* | 6.1 ± 1.0* |
| LV mass/tibia length, mg/mm | 4.2 ± 0.5 | 4.7 ± 0.7 | 5.6 ± 1.1 | 7.0 ± 0.9* | 8.7 ± 1.0* |
| LV end-diastolic volume/tibia length, ml/mm | 3.0 ± 0.4 | 2.8 ± 0.4 | 3.2 ± 0.5 | 3.4 ± 0.8 | 3.3 ± 0.9 |
| Left atrial dimension/tibia length, mm/mm | 0.10 ± 0.01 | 0.10 ± 0.03 | 0.12 ± 0.02 | 0.19 ± 0.04* | 0.20 ± 0.03* |
Values are means ± SD. Left ventricular (LV) ejection fraction was calculated as follows: (end-diastolic volume − end-systolic volume)/end-diastolic volume × 100. PO, pressure overload.
P < 0.05 vs. baseline control.
Fig. 1.

Consort diagram summarizing measured end points and sample sizes. Echo, echocardiography; LV, left ventricular; PO, pressure overload; Pap Ms, papillary muscle testing; rtPCR, reverse transcription polymerase chain reaction; CVF, collagen volume fraction; FACS, fluorescence-activated cell sorting.
Collagen quantification and measurements of stiffness.
For the hydroxyproline analysis, frozen LV tissue was lyophilized, weighed (dry weight), pulverized, and resuspended in PBS. Samples underwent complete acid hydrolysis with 6 N HCl for 18 h at 120°C and were then neutralized to pH 7 with 4 N NaOH. After the addition of 1 ml of chloramine T to 2-ml volumes of sample, samples were incubated at room temperature for 20 min. Then, 1 ml of Ehrlich’s reagent (60% perchloric acid, 15 ml of l-propanol, and 3.75 g of p-dimethylaminobenzaldehyde in 25 ml) was added, and samples were incubated at 60°C for 20 min. Hydroxyproline concentration was quantified as absorbance of each sample at 558-nm wavelength.CVF was measured in sections from formalin-fixed, paraffin-embedded LVs stained with PSR and visualized under polarized light. Five random fields from each heart were scanned using SigmaScan software. CVF was calculated as the area stained by PSR divided by the total area of interest.
Myocardial stiffness was measured as described elsewhere (8). Briefly, mice (baseline control and LV PO) were anesthetized, and heparin (200 units) was administered intraperitoneally. The LV was isolated, the aorta was cannulated, the LV was perfused with 2,3-butanedione monoxime, and the papillary muscle was isolated. For examination of passive diastolic stiffness, the muscle was stretched very slowly (1 mm/min) beginning from near slack length (very lightly preloaded muscle at 0.1 g) to a muscle length 15% greater than slack length (equivalent to Lmax preload). The myocardial stress versus strain relationship during this muscle stretch was used to calculate the passive stiffness constant (β) as follows: stress = A × e(β × strain) + C, where A and C are constant coefficients. Myocardial stress was calculated as muscle force divided by muscle cross-sectional area, and strain was calculated as (L − L0)/L0, where L is muscle length during stretch and L0 is muscle length at 0.1 g preload.
Immunoblot analysis.
Immunoblot analysis was carried out as described elsewhere (23). Briefly, LV samples were thawed, transferred to cold extraction/homogenization buffer [buffer volume 1:6 (wt/vol); 10 mmol/l cacodylic acid (pH 5.0), 0.15 mol/l NaCl, 10 mmol/l ZnCl2, 1.5 mmol/l NaN3, and 0.01% Triton X-100 (vol/vol)], and homogenized. Twenty micrograms of the supernatant were fractionated on a 4–12% bis-Tris gradient gel. Proteins were transferred to nitrocellulose membranes and incubated in antiserum generated against murine SPARC (catalog no. AF942, R&D Systems, Minneapolis MN). Antisera were diluted in 5% nonfat dry milk-PBS. Secondary peroxidase-conjugated antibody (1:5,000 dilution; 5% nonfat dry milk-PBS) was applied, and signals were detected with a chemiluminescent substrate. Band intensity was quantified using Gel-Pro Analyzer software (Media Cybernetics, Silver Spring, MD) and reported as the percent change from non-PO control homogenates.
Macrophage immunohistochemistry.
For immunohistochemistry, myocardial sections were fixed in zinc formalin. After antigen retrieval, primary antibodies against macrophages (F4/80; catalog no. ab16911, Abcam) or SPARC (rabbit polyclonal against recombinant human SPARC; kind gift of Dr. E. Helene Sage) were incubated on tissue sections and detected with appropriate secondary antibodies linked to biotin or distinct fluorophores. Biotin antibodies were detected with avidin linked to horseradish peroxidase and reacted with 3,3′-diaminobenzidine.
FACS.
Control and LV PO hearts were flushed with saline and removed at designated experimental times. Atria were removed, and LVs were minced in Hanks’ buffered saline solution. LV tissue was digested with 0.5 mg/ml collagenase type II in Hanks’ buffered saline solution until fully digested. Single cells were filtered through a 70-μm cell filter into buffer containing 20% FCS to stop digestion. Cells were then washed in PBS (ThermoFisher Scientific, Rochester, NY) and stained with Live/Dead fixable near-IR dye (ThermoFisher Scientific). To block nonspecific interactions, cells were incubated with mouse Fc receptor (Miltenyi Biotec, San Diego, CA) before incubation with antibodies against the following specific cell surface markers: CD45-PE (clone 30-F11, BD Biosciences, San Jose CA), CD11b-FITC (clone M1/70, BD Biosciences), and F4/80-BV421 (clone BM8, BioLegend, San Diego, CA). CD45 is indicative of cells of bone marrow origin, CD11b is a monocyte marker, and F4/80 is a marker of mature macrophages. Cells were then fixed and permeabilized (BD Cytofix/Cytoperm kit, ThermoFisher Scientific) and subsequently stained for intracellular SPARC (catalog no. AF942, R&D Systems) conjugated to Alexa 647 (ThermoFisher Scientific) according to the manufacturer’s protocol. Intracellular antibody incubation and subsequent wash steps were performed in BD Perm/Wash buffer. Before analysis, cells were washed and resuspended in PBS. Samples were assayed on a cell analyzer (model LSRFortessa X-20, BD Bioscience) and analyzed using FlowJo v10 software (Tree Star, Ashland, OR). Gates were set based on fluorescence − 1 (FMO) controls. The sequential gating strategy was as follows: sized, single, live, CD45+, CD11b+/F480+, and SPARC+.
Statistics.
Temporal changes of all measured end points were compared between LV PO and control groups using one-way repeated-measures ANOVA followed by all pair-wise multiple-comparison procedures by means of Fisher’s least-significant-difference method (SigmaStat 3.5, Systat Software, Richmond, CA). Values are means ± SE and medians ± interquartile ranges.
RESULTS
Time course of collagen accumulation and tissue stiffness in PO hearts.
To assess the time course of changes in collagen expression in LV PO mice, levels of mRNA encoding the α1-subunit of collagen type I in TAC hearts was assessed at 3 days, 1 wk, and 4 wk of PO by RT-PCR. As shown in Fig. 2A, compared with controls, levels of mRNA encoding the α1-subunit of collagen type I were increased at 3 days and 1 wk of LV PO and began to decline at 4 wk, although levels at 4 wk remained increased compared with control. In contrast, total (soluble + insoluble) collagen (measured by hydroxyproline analysis) was not increased until 1 wk of LV PO (Fig. 2B). Elevated levels of total myocardial collagen remained increased at 2 and 4 wk of LV PO. Insoluble fibrillar collagen (measured as CVF; Fig. 2C) was unchanged at 3 days and 1 wk of LV PO but was increased at 2 and 4 wk of LV PO. We concluded that early remodeling events that occur with TAC during the first wk involving myocyte hypertrophy and ECM turnover did not result in a net accumulation of insoluble collagen and suggested that cellular mechanisms initiated between 1 and 2 wk of LV PO regulated the accumulation of insoluble collagen.
Fig. 2.

Time course of procollagen synthesis and processing from soluble to insoluble collagen in response to left ventricular (LV) pressure overload (PO) induced by transverse aortic constriction. A: collagen transcriptional activation measured by mRNA expression of the α1-subunit of collagen type I (Col1a1) increased rapidly at 3 days of LV PO. Sustained elevations in Col1a1 expression were detected at 1 wk; levels decreased by 4 wk and remained greater than baseline. B: total (soluble + insoluble) myocardial collagen protein production measured by hydroxyproline assay increased at 1 wk of LV PO and reached a plateau that was maintained throughout 4 wk of LV PO. C: insoluble fibrillar collagen accumulation and deposition measured as collagen volume fraction did not increase until 2 wk of LV PO and increased further at 4 wk of LV PO. These data show an increase in procollagen synthesis immediately (as early as 3 days) after the imposition of PO; however, deposition of fibrillar collagen was not synchronous with increased transcription but was delayed until 2 wk of LV PO, when procollagen processing was increased. Values are means ± SE. *P < 0.05 vs. control (no LV PO); #P < 0.05 vs. 3 days of LV PO.
Myocardial stiffness.
To determine whether the time course of insoluble collagen accumulation was associated with increased myocardial stiffness, myocardial stress versus strain was measured in LV papillary muscle isolated at 3 days, 1 wk, 2 wk, and 4 wk of LV PO (Fig. 3). Coincident with an increase in insoluble ECM fibrillar collagen, myocardial stiffness was not changed at 3 days and 1 wk of LV PO but was significantly increased at 2 and 4 wk of LV PO. Concurrent with increases in myocardial stiffness, significant increases in left atrial dimension were also detected at 2 and 4 wk of LV PO by echocardiography (Table 1).
Fig. 3.

Time course of changes in myocardial stiffness in response to the imposition of left ventricular (LV) pressure overload (PO). A: myocardial stress versus strain curves determined by passive stretch of LV papillary muscle from control hearts compared with hearts subjected to 3 days, 1 wk, 2 wk, and 4 wk of LV PO. B: myocardial stiffness measured by the passive stiffness constant β did not increase until 2 wk of LV PO. Values are means ± SE. *P < 0.05 vs. control (no LV PO).
Time course of SPARC production after PO.
Levels of SPARC protein were assessed by immunoblot analysis at baseline and at 3 days, 1 wk, and 4 wk after the induction of PO to assess whether production of SPARC followed a time course similar to collagen production and deposition. As shown in Fig. 4, compared with control, SPARC production was not significantly changed at 3 days of LV PO. SPARC was significantly increased at 1 and 4 wk after the induction of PO.
Fig. 4.
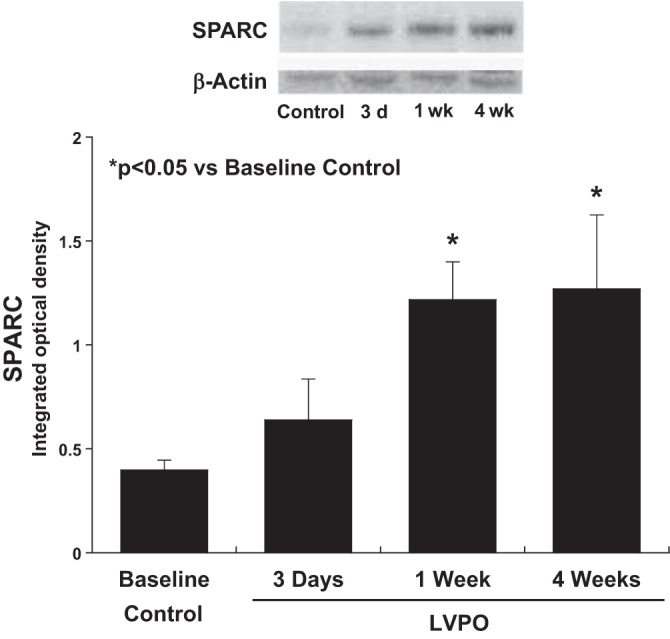
Time course of changes in secreted protein acidic and rich in cysteine (SPARC) expression in response to the imposition of left ventricular (LV) pressure overload (PO). Immunoblot analysis of SPARC showed that levels were significantly increased at 1 wk of LV PO and remained increased at 4 wk of LV PO. Values are means ± SE. *P < 0.05 vs. control (no LV PO).
SPARC expression by myocardial macrophages.
To determine whether myocardial macrophages produced SPARC after the induction of PO, cell populations isolated at 1 wk of LV PO were analyzed by flow cytometry. Cells were analyzed for the expression of CD45 to mark hematopoietic cells, CD11b to label monocytic cells, and F4/80 to identify mature macrophages (Fig. 5, A–C). Notably, the myocardial population of CD45+/CD11b+/F4/80+ cells was positive for SPARC expression, and the number of CD45+/CD11b+/F4/80+/SPARC+ cells was significantly increased in LV PO versus baseline control at 1 wk (Fig. 5D).
Fig. 5.

Quantitative assessment of total number of macrophages and number of secreted protein acidic and rich in cysteine (SPARC)-producing macrophages in left ventricle (LV) after the induction of pressure overload (PO). FACS analysis at 1 wk of LV PO (red) versus control (blue) used a sequential gating strategy. A: Live/Dead fixable near-IR (APC-Cy7), depicted as side scatter (SSC) versus APC-Cy7; sized, single, live cells gated. B: SSC versus CD45-PE; sized, single, live, CD45+ (hematopoietic) cells gated. C: CD11b-FITC (myeloid) versus F4/80-BV421 (macrophage); sized, single, live, CD45+/CD11b+/F4/80+ cells gated. D: cell count versus SPARC-APC, indicated by the gate; sized, single, live, CD45+/CD11b+/F4/80+/SPARC+ cells. Note the significant increase in percentage of live CD45+/CD11b+/F4/80+ cells, i.e., an increase in the number of macrophages in LV PO versus control and an increase in the number of macrophages expressing SPARC [red (LV PO sample) versus blue (baseline control sample) counts]. Gates were set based on fluorescence − 1 (FMO) controls (gray peak in D).
Anti-SPARC and anti-F4/80 antibodies were used for immunofluorescence imaging of tissue sections from control hearts and hearts subjected to 1 wk of PO. Coincident staining of SPARC and F4/80 in individual cells was observed in hearts at 1 wk of LV PO but not in control hearts (Fig. 6). These results confirm the flow cytometry data demonstrating that myocardial macrophages in PO myocardium represent an important cellular source of SPARC.
Fig. 6.

Control left ventricular (LV) tissue (A) and LV tissue after 1 wk of pressure overload (PO; B) stained with anti-SPARC (red) and anti-F4/80 macrophage (green) antibodies. Arrows indicate cells that costained with anti-SPARC and anti-F4/80 antibodies (yellow). Scale bar = 20 μm.
Levels of myocardial macrophages increase after PO.
Immunohistochemical analysis of F4/80+ macrophages at 3 days, 1 wk, and 4 wk of LV PO was performed to assess the time course of increases in myocardial macrophages in response to LV PO and sites of localization. The number of macrophages in hearts at 3 days of LV PO did not differ from that in control hearts (6.8 ± 2.2% and 8.5 ± 3.5% F4/80+ cells of total nuclei in control hearts and at 3 days of LV PO, respectively; Fig. 7, A and B). In contrast, macrophage numbers were increased substantially at 1 wk (23.5 ± 7.1%) and remained increased at 4 wk (18.2 ± 6.6%) of LV PO (Fig. 7, C and D). As a staining control, sections from hearts at 1 wk of LV PO were stained with secondary antibody only (Fig. 7E). Primarily, F4/80+ cells were associated with vascular structures at each time point (Fig. 7, arrows).
Fig. 7.

Increases in total macrophages (F4/80+) occur with time in the left ventricle (LV) after LV pressure overload (PO) induced by transverse aortic constriction (TAC). A: control (no LV PO) LV tissue. B: LV tissue at 3 days of LV PO. C and D: increase in total macrophages (F4/80+) at 1 and 4 wk of LV PO. E: myocardium stained with secondary antibodies only. Arrows indicate F4/80+ cells. Scale bar = 50 μm.
Quantification of CD45+/CD11b+/F4/80+ cells positive for SPARC expression at each time point was performed by flow cytometric analysis (Fig. 8). Myocardial populations of CD45+ and CD11b+ cells peaked at 3 days after PO (Fig. 8, A and B). In contrast, the highest levels of myocardial CD45+/CD11b+/F4/80+ cells (mature macrophages) were observed at 1 wk of PO (Fig. 8C). Elevated levels of CD45+/CD11b+/F4/80+ myocardial cells persisted at 4 wk of PO compared with control. The percentage of CD45+/CD11b+/F4/80+ cells expressing SPARC was similar in the control condition and after PO (Fig. 8); however, CD45+/CD11b+/F4/80+ cells were present in greater quantities in PO myocardium and, therefore, very likely contributed to increased amounts of myocardial SPARC.
Fig. 8.

Flow cytometric analysis of cell markers and secreted protein acidic and rich in cysteine (SPARC) over time after left ventricular (LV) pressure overload (PO). Cells positive for CD45 and CD11b, denoting hematopoietic origin and monocyte lineage, respectively, demonstrated increases in myocardial number at 3 days of LV PO (A and B). Cells positive for mature macrophages (F4/80+) showed greatest levels at 1 wk in LV PO myocardium (C and D). SPARC expression by each individual population peaked at 1 wk of LV PO. Sequential gating of cells positive for CD45, CD11b, and F4/80 cells (C and D) showed sustained expression of SPARC at 4 wk of LV PO. Box-and-whisker graphs show medians and interquartile ranges. Temporal changes of cell markers were compared between control and LV PO groups by one-way repeated-measures ANOVA followed by all pair-wise multiple-comparison procedures by Fisher’s least-significant-difference method (SigmaStat 3.5). *P < 0.05 vs. baseline; #P < 0.05 vs. 3-day transverse aortic constriction (TAC); +P < 0.05 vs. 1-wk TAC.
DISCUSSION
Data from the present study support the following novel findings: 1) the time course of fibrillar collagen deposition (fibrosis) and myocardial stiffness in the PO myocardium is divergent from the transcriptional increases in mRNA encoding collagen type I and synchronous with the postsynthetic processing of procollagen, the expression of SPARC, and the expansion of myocardial macrophages and 2) because myocardial macrophages serve as a cellular source of SPARC expression, macrophage-derived SPARC might play a pivotal role in PO-induced myocardial fibrosis.
Postsynthetic processing of procollagen in the PO myocardium.
Previously, the time course of collagen deposition in a feline model of right ventricular (RV) cardiac PO was reported (4). Similar to the LV PO murine model, a distinct time course of collagen deposition in the myocardium was found in which significant accumulation of insoluble collagen was not detected until hypertrophic growth of the myocardium reached a plateau. Like the murine LV PO model, increases in soluble collagen in the feline RV PO model were detected before increases in insoluble collagen accumulation. However, cellular sources of proteins that drive insoluble collagen deposition were not addressed in these RV PO experiments. Together, previous and present data suggest that increased collagen accumulation in the myocardium is dependent on postsynthetic processing steps that occur independent of transcriptional regulation of collagen and likely are driven in the extracellular space by factors that enhance incorporation of soluble collagen into insoluble collagen fibers. As these processes are extracellular, secreted proteins from different cell types are able to influence fibrotic deposition of collagen. As SPARC also binds to collagen type III, the second-most-represented fibrillar collagen in the myocardium after collagen type I, we predict that SPARC might serve to regulate processing of procollagen type III as well. The identification of cellular sources of proteins that influence collagen incorporation is critical to understanding mechanisms of fibrosis.
Myocardial macrophages.
Recent studies using lineage-tracing and transgenic reporter technologies have strongly suggested that the primary cell type expressing mRNA encoding collagen type I in PO is the resident cardiac fibroblast (1, 16). Other cell types, such as monocytes and macrophages, are not considered to be primary contributors to fibrillar collagen production. Specifically, Moore-Morris et al. (16) concluded that cells of hematopoietic origin (CD45+) in the PO myocardium did not demonstrate collagen type I promoter activity (as monitored by green fluorescent protein expression driven by the collagen promoter). However, a clear increase in recruited hematopoietic cells was evident in the PO myocardium (16). These CD45+ cells were found in close proximity to collagen-producing fibroblasts at sites of collagen accumulation. Several studies in other tissues support a functional role for recruited monocytes/macrophages, derived from CD45+ cells, in making a significant contribution to fibrosis (11). The present study demonstrates that myocardial macrophages produce SPARC, a matricellular protein shown to promote procollagen processing and maturation to insoluble fibrillar collagen.
Data from the present study are largely concurrent with previous studies examining the changes in myocardial monocyte/macrophage number and location after the onset of PO. Myocardial monocytes have been shown to increase as early as 2 days (18) and macrophages as early as 6 days (25) after the imposition of PO. Myocardial macrophages have also been shown to persist to variable degrees for as long as 4 wk after the imposition of PO (12, 19). The specific phenotype (pro- vs. anti-inflammatory) and the origin (resident vs. recruited) of myocardial macrophages after the onset of PO remain to be clearly defined (17). Importantly, the present study significantly advances our understanding of the pathophysiological role of macrophages in the development of PO-induced fibrosis by demonstrating that macrophages are an important source of SPARC and that the time course of changes in fibrosis is synchronous with the increase in macrophages and macrophage-dependent SPARC production. In this study, CD45, CD11b, and F4/80 were used to identify hematopoietic cells, monocytes, and mature macrophages, respectively. Future studies that further identify subsets of macrophage populations that specifically contribute to ECM deposition and fibrosis will be insightful. For example, markers such as CD115, CD62, and CD206 have been used to distinguish proinflammatory from anti-inflammatory macrophages.
Clinical implications.
In both patients with LV PO and animal models of LV PO, myocardial fibrosis is a critical development in the progression to chronic heart failure (5, 7, 24, 27). In studies that examined LV epicardial myocardial biopsies from patients undergoing open heart surgery, myocardial collagen-dependent stiffness and CVF were increased in patients with hypertensive heart disease and heart failure, whereas in patients with hypertensive heart disease who did not have heart failure, myocardial collagen or collagen-dependent stiffness was unchanged compared with age- and sex-matched control subjects (27). The transition from PO-induced hypertrophy to heart failure was dependent, at least in part, on the development of fibrosis. Similar results have been found in murine models of PO. While these animal models do not precisely recapitulate all aspects of heart failure in patients, the cellular and molecular mechanisms that contribute to increased collagen and collagen-dependent stiffness in murine models of PO, such as those reported here, offer an opportunity to define cellular mechanisms driving fibrotic deposition of collagen. These mechanistic insights may be fundamental to the development of novel therapies.
Limitations.
The strategy used in generating a time course for measurements of collagen production and deposition is represented as following a linear temporal course. For practical purposes, measurements could not be made daily; thus, an arbitrary discontinuous timeframe was examined. Hence, changes were examined in a binary manner to characterize differences in the time course of procollagen synthesis versus procollagen processing to insoluble collagen and to characterize recruitment of macrophages to the myocardium and maturation of macrophages. However, it is recognized that such a binary assessment strategy has some clear limitations.
We did not specifically define the change in SPARC production by cardiac fibroblasts after LV PO in these experiments. As resident fibroblasts also produce SPARC, fibroblast-dependent SPARC production could also influence time-dependent fibrosis in the PO myocardium. Future studies to assess cell-specific SPARC production will address differential contribution of SPARC from specific sources to fibrotic deposition of collagen through the use transgenic cell-specific deletion of SPARC.
Conclusions.
Myocardial macrophages play an important role in the time-dependent increase in SPARC that increases postsynthetic collagen processing, insoluble collagen content, and myocardial stiffness in PO and appears to contribute to the development of LV myocardial fibrosis. Given our previous work establishing SPARC as a key factor driving myocardial fibrosis, identification of cellular sources of SPARC provides insight into cellular mechanisms of fibrosis.
GRANTS
This work was supported by Department of Veterans Affairs Office of Research and Development Merit Awards 1I01BX001385 and 1I01-CX001608 (to M. R. Zile and A. D. Bradshaw), 1I01-BX000904-06 (to J. A. Jones), and 1I01-BX002277 and 1I01-BX000333 (to M. C. LaRue); Cell Evaluation and Therapy Shared Resource, Hollings Cancer Center, Medical University of South Carolina through National Cancer Institute Grant P30-CA-138313 (to M. C. LaRue); and National Heart, Lung, and Blood Institute Grant 1-R01-HL-123478 (to M. R. Zile).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.T.M., Y.Z., A.O.V.L., C.F.B., R.E.S., and A.D.B. performed experiments; L.T.M., M.R.Z., A.O.V.L., C.F.B., R.E.S., J.A.J., A.C.L., and A.D.B. analyzed data; L.T.M., M.R.Z., Y.Z., A.O.V.L., C.F.B., R.E.S., J.A.J., A.C.L., and A.D.B. interpreted results of experiments; L.T.M., C.F.B., and A.D.B. prepared figures; L.T.M., M.R.Z., C.F.B., J.A.J., A.C.L., and A.D.B. edited and revised manuscript; L.T.M., M.R.Z., Y.Z., A.O.V.L., C.F.B., R.E.S., J.A.J., A.C.L., and A.D.B. approved final version of manuscript; M.R.Z., C.F.B., J.A.J., A.C.L., and A.D.B. conceived and designed research; M.R.Z. and A.D.B. drafted manuscript.
REFERENCES
- 1.Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, Hojjat A, Kamran P, Müller AM, Volz KS, Tang Z, Red-Horse K, Ardehali R. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ Res 115: 625–635, 2014. doi: 10.1161/CIRCRESAHA.115.303794. [DOI] [PubMed] [Google Scholar]
- 2.Alpers CE, Hudkins KL, Segerer S, Sage EH, Pichler R, Couser WG, Johnson RJ, Bassuk JA. Localization of SPARC in developing, mature, and chronically injured human allograft kidneys. Kidney Int 62: 2073–2086, 2002. doi: 10.1046/j.1523-1755.2002.00680.x. [DOI] [PubMed] [Google Scholar]
- 3.Arnold SA, Rivera LB, Miller AF, Carbon JG, Dineen SP, Xie Y, Castrillon DH, Sage EH, Puolakkainen P, Bradshaw AD, Brekken RA. Lack of host SPARC enhances vascular function and tumor spread in an orthotopic murine model of pancreatic carcinoma. Dis Model Mech 3: 57–72, 2010. doi: 10.1242/dmm.003228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baicu CF, Li J, Zhang Y, Kasiganesan H, Cooper G IV, Zile MR, Bradshaw AD. Time course of right ventricular pressure-overload induced myocardial fibrosis: relationship to changes in fibroblast postsynthetic procollagen processing. Am J Physiol Heart Circ Physiol 303: H1128–H1134, 2012. doi: 10.1152/ajpheart.00482.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baicu CF, Zhang Y, Van Laer AO, Renaud L, Zile MR, Bradshaw AD. Effects of the absence of procollagen C-endopeptidase enhancer-2 on myocardial collagen accumulation in chronic pressure overload. Am J Physiol Heart Circ Physiol 303: H234–H240, 2012. doi: 10.1152/ajpheart.00227.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bishop JE, Rhodes S, Laurent GJ, Low RB, Stirewalt WS. Increased collagen synthesis and decreased collagen degradation in right ventricular hypertrophy induced by pressure overload. Cardiovasc Res 28: 1581–1585, 1994. doi: 10.1093/cvr/28.10.1581. [DOI] [PubMed] [Google Scholar]
- 7.Bradshaw AD, Baicu CF, Rentz TJ, Van Laer AO, Boggs J, Lacy JM, Zile MR. Pressure overload-induced alterations in fibrillar collagen content and myocardial diastolic function: role of secreted protein acidic and rich in cysteine (SPARC) in post-synthetic procollagen processing. Circulation 119: 269–280, 2009. doi: 10.1161/CIRCULATIONAHA.108.773424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chapman D, Weber KT, Eghbali M. Regulation of fibrillar collagen types I and III and basement membrane type IV collagen gene expression in pressure overloaded rat myocardium. Circ Res 67: 787–794, 1990. doi: 10.1161/01.RES.67.4.787. [DOI] [PubMed] [Google Scholar]
- 9.Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ, Mann DL. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40: 91–104, 2014. doi: 10.1016/j.immuni.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Falkenham A, de Antueno R, Rosin N, Betsch D, Lee TD, Duncan R, Légaré JF. Nonclassical resident macrophages are important determinants in the development of myocardial fibrosis. Am J Pathol 185: 927–942, 2015. doi: 10.1016/j.ajpath.2014.11.027. [DOI] [PubMed] [Google Scholar]
- 11.Frantz S, Nahrendorf M. Cardiac macrophages and their role in ischaemic heart disease. Cardiovasc Res 102: 240–248, 2014. doi: 10.1093/cvr/cvu025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujiu K, Shibata M, Nakayama Y, Ogata F, Matsumoto S, Noshita K, Iwami S, Nakae S, Komuro I, Nagai R, Manabe I. A heart-brain-kidney network controls adaptation to cardiac stress through tissue macrophage activation. Nat Med 23: 611–622, 2017. doi: 10.1038/nm.4326. [DOI] [PubMed] [Google Scholar]
- 13.Goldsmith EC, Bradshaw AD, Spinale FG. Cellular mechanisms of tissue fibrosis. 2. Contributory pathways leading to myocardial fibrosis: moving beyond collagen expression. Am J Physiol Cell Physiol 304: C393–C402, 2013. doi: 10.1152/ajpcell.00347.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris BS, Zhang Y, Card L, Rivera LB, Brekken RA, Bradshaw AD. SPARC regulates collagen interaction with cardiac fibroblast cell surfaces. Am J Physiol Heart Circ Physiol 301: H841–H847, 2011. doi: 10.1152/ajpheart.01247.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, Ornitz DM, Randolph GJ, Mann DL. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci USA 111: 16029–16034, 2014. doi: 10.1073/pnas.1406508111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore-Morris T, Guimarães-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, Gomez-Amaro R, Zhou B, Brenner DA, Peterson KL, Chen J, Evans SM. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest 124: 2921–2934, 2014. doi: 10.1172/JCI74783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nahrendorf M, Swirski FK. Monocyte and macrophage heterogeneity in the heart. Circ Res 112: 1624–1633, 2013. doi: 10.1161/CIRCRESAHA.113.300890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485: 251–255, 2012. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel B, Ismahil MA, Hamid T, Bansal SS, Prabhu SD. Mononuclear phagocytes are dispensable for cardiac remodeling in established pressure-overload heart failure. PLoS One 12: e0170781, 2017. doi: 10.1371/journal.pone.0170781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pinto AR, Paolicelli R, Salimova E, Gospocic J, Slonimsky E, Bilbao-Cortes D, Godwin JW, Rosenthal NA. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS One 7: e36814, 2012. doi: 10.1371/journal.pone.0036814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sager HB, Hulsmans M, Lavine KJ, Moreira MB, Heidt T, Courties G, Sun Y, Iwamoto Y, Tricot B, Khan OF, Dahlman JE, Borodovsky A, Fitzgerald K, Anderson DG, Weissleder R, Libby P, Swirski FK, Nahrendorf M. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ Res 119: 853–864, 2016. doi: 10.1161/CIRCRESAHA.116.309001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sangaletti S, Di Carlo E, Gariboldi S, Miotti S, Cappetti B, Parenza M, Rumio C, Brekken RA, Chiodoni C, Colombo MP. Macrophage-derived SPARC bridges tumor cell-extracellular matrix interactions toward metastasis. Cancer Res 68: 9050–9059, 2008. doi: 10.1158/0008-5472.CAN-08-1327. [DOI] [PubMed] [Google Scholar]
- 23.Spinale FG, Mukherjee R, Zavadzkas JA, Koval CN, Bouges S, Stroud RE, Dobrucki LW, Sinusas AJ. Cardiac restricted overexpression of membrane type-1 matrix metalloproteinase causes adverse myocardial remodeling following myocardial infarction. J Biol Chem 285: 30316–30327, 2010. doi: 10.1074/jbc.M110.158196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stroud JD, Baicu CF, Barnes MA, Spinale FG, Zile MR. Viscoelastic properties of pressure overload hypertrophied myocardium: effect of serine protease treatment. Am J Physiol Heart Circ Physiol 282: H2324–H2335, 2002. doi: 10.1152/ajpheart.00711.2001. [DOI] [PubMed] [Google Scholar]
- 25.Weisheit C, Zhang Y, Faron A, Köpke O, Weisheit G, Steinsträsser A, Frede S, Meyer R, Boehm O, Hoeft A, Kurts C, Baumgarten G. Ly6Clow and not Ly6Chigh macrophages accumulate first in the heart in a model of murine pressure-overload. PLoS One 9: e112710, 2014. doi: 10.1371/journal.pone.0112710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44: 450–462, 2016. doi: 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, Slater R, Palmer BM, Van Buren P, Meyer M, Redfield MM, Bull DA, Granzier HL, LeWinter MM. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation 131: 1247–1259, 2015. doi: 10.1161/CIRCULATIONAHA.114.013215. [DOI] [PMC free article] [PubMed] [Google Scholar]