

Abstract
α-Ketoglutarate (α-KG) is a citric acid cycle intermediate and a glutamine catabolism product. It is also the natural ligand of 2-oxoglutarate receptor 1 (OXGR1), a Gq protein-coupled receptor expressed on the apical membrane of intercalated cells. In the cortical collecting duct (CCD), Cl−/ exchange increases upon α-KG binding to the OXGR1. To determine the signaling pathway(s) by which α-KG stimulates Cl− absorption, we examined α-KG-stimulated Cl− absorption in isolated perfused mouse CCDs. α-KG increased electroneutral Cl− absorption in CCDs from wild-type mice but had no effect on Cl− absorption in pendrin knockout mice. Because Gq protein-coupled receptors activate PKC, we hypothesized that α-KG stimulates Cl− absorption through PKC. If so, PKC agonists should mimic, whereas PKC inhibitors should abolish, α-KG-stimulated Cl− absorption. Like α-KG, PKC agonist (phorbol-12,13-dibutyrate, 500 nM) application increased Cl− absorption in wild-type but not in pendrin null CCDs. Moreover, PKC inhibitors (2.5 mM GF109203X and 20 nM calphostin C), Ca2+ chelators (BAPTA, 10–20 μM), or PKC-α or -δ gene ablation eliminated α-KG-stimulated Cl− absorption. We have shown that STE20/SPS-1-related proline-alanine-rich protein kinase (SPAK) gene ablation increases urinary α-KG excretion, renal pendrin abundance, and CCD Cl− absorption. However, in SPAK null CCDs, Cl− absorption was not activated further by luminal α-KG application nor was Cl− absorption reduced with the PKC inhibitor GF109203. Thus SPAK gene ablation likely acts through a PKC-independent pathway to produce a chronic adaptive increase in pendrin function. In conclusion, α-KG stimulates pendrin-dependent Cl−/ exchange through a mechanism dependent on PKC and Ca2+ that involves PKC-α and PKC-δ.
INTRODUCTION
While the distal convoluted tubule, the connecting tubule (CNT), and the collecting duct mediate only ~10% of total renal NaCl absorption, these segments are critical to the fine tuning of NaCl balance (6, 35). As such, inhibitors of distal NaCl transporters, such as the thiazide-sensitive NaCl cotransporter of the distal convoluted tubule, are powerful antihypertensives and commonly used in clinical practice today. STE20/SPS-1-related proline-alanine-rich protein kinase (SPAK) represents the key WNK-activated kinase responsible for maintaining and activating NCC on demand (10, 11, 20, 24, 37).
In more distal segments, such as the cortical collecting duct (CCD), NaCl absorption occurs through an electrogenic, amiloride-sensitive pathway and an electroneutral pathway, which in some studies has been shown to be thiazide sensitive (29). The former occurs through the principal cell epithelial Na+ channel (ENaC) acting in parallel with either a Cl− channel or with paracellular Cl− transport (22, 29). Electroneutral, NaCl absorption occurs across the type B intercalated cell through electroneutral NaCl absorption and secretion mediated by the apical plasma membrane Cl−/exchanger pendrin (encoded by Slc26a4), which has been reported to act in tandem with the Na+-dependent Cl−/ exchanger NDCBE (encoded by Slc4a8) (18, 29). The driving force for apical Cl−/ exchange increases with enhanced net NaCl and H+ exit across the basolateral membrane, which occurs through stimulation of type B intercalated cell basolateral plasma membrane transporters, such as AE4 (3), H+-ATPase (3), and ClC-K2/barttin (13, 23). The importance of these pathways has been demonstrated in a number of studies showing that blood pressure falls in the absence of either type B intercalated cell apical Cl−/ exchange or principal cell ENaC-mediated Na+ absorption (28, 32).
While NCC is an important mediator of renal NaCl absorption, little change in blood pressure or salt balance is observed following either NCC or SPAK gene ablation (10). These data indicate that powerful compensatory pathways maintain fluid and electrolyte balance and blood pressure in these knockout (KO) mice. In particular, NCC gene and SPAK gene ablation greatly increases B intercalated cell transporter abundance and function and increases the production and excretion of α-ketoglutarate (α-KG) (10).
α-KG is a citric acid cycle intermediate and a product of glutamine catabolism that is involved in a variety of metabolic pathways (30). It is also a ligand of the G coupled-protein receptor GPR99 better known as the 2-oxoglutarate receptor 1 (OXGR1) (12). α-KG is produced in the proximal tubule and then secreted into the luminal fluid where it acts downstream on the OXGR1 receptor that localizes to the type B intercalated cell apical plasma membrane (10). As such, α-KG acts as an intrarenal paracrine signaling molecule that mediates cross talk between the proximal and distal nephron. The increase in α-KG synthesis and secretion into the luminal of the proximal tubule seen in SPAK null mice might act downstream on the OXGR1 receptor of type B intercalated cells to increase transporter abundance in this cell type.
Because α-KG has an EC50 of 32–69 μM for human OXGR1 (12), and because urinary α-KG concentration in people is ~25–2,000 μM (10, 12, 21), luminal α-KG concentration in vivo should be sufficient for receptor activation. Tokonami et al. (30) observed that when mice ingest 0.3 M NH4Cl in their drinking water for 2 days, urinary α-KG concentration is 3 μM. However, urinary α-KG concentration rises to 36,000 μM (36 mM) after ingestion of drinking water containing 0.28 M NaHCO3 for 2 days. Other studies have reported rodent urinary α-KG concentrations between 24 and 490 μM (10, 31). Since the final urine is ~10–20 times more concentrated than that of the luminal fluid found in the CCD or CNT, Tokonami et al. (30) predicted that the α-KG concentration in the luminal fluid of the CCD and CNT in vivo should be between ~0.15 and 3,600 μM. Thus luminal α-KG concentration varies widely. These data also suggest that under some treatment conditions, luminal α-KG concentration in vivo should be high enough to fully saturate the receptor. At these levels, an additional increase in α-KG concentration should not further increase receptor activity. However, under other treatment conditions, luminal α-KG concentration is orders of magnitude lower, making α-KG levels in the range of the EC50 for the OXGR1. Because the OXGR1 is not saturated at these lower concentrations, increases in α-KG concentration should increase receptor activity.
Of the organ systems studied, α-KG-OXGR1-mediated signaling in kidney is the most significant since the proximal tubule produces high levels of α-KG (10) and since OXGR1 expression is greatest in kidney (12). Within kidney, this receptor localizes to the apical membrane of type B intercalated cells, where it modulates intercalated cell function (30). Tokonami et al. (30) observed that NaCl absorption as well as electroneutral Cl−/ exchange is stimulated in mouse CCDs perfused in vitro following the application of 1 mM α-KG to the luminal fluid. However, α-KG application had no effect on Cl− absorption in CCDs from mice lacking the Gq protein-coupled receptor (GPCR) OXGR1, i.e., OXGR1 null mice.
GPCRs are coupled to heterotrimeric G proteins, which consist of Gα, Gβ, and Gγ subunits (2, 4). Upon ligand binding, the GPCR is stabilized in an active signaling confirmation, which leads to heterotrimeric G protein dissociation into GTP-bound Gα and also Gβγ subunits (2, 4). Many agonists of GPCRs activate phospholipase C, which catalyzes the hydrolysis of the membrane lipid phosphotidyl inositol-4,5-bisphosphate to generate diacylglycerol (DAG) and inositol trisphosphate (IP3) (2, 4). Ca2+ is then mobilized from the endoplasmic reticulum, which, together with DAG, activates protein kinase C (2, 4). Because α-KG is a ligand of a Gq protein receptor (OXGR1), and since GPCRs typically activate protein kinase C (PKC), the purpose of this study was therefore to determine if α-KG stimulates pendrin-dependent Cl− absorption and if this occurs through protein kinase C-mediated signaling.
METHODS
Animals.
Unless otherwise stated, mice on a C57Bl/6J background (Jackson Laboratories) were studied. Slc26a4+/− mice on a C57Bl/6J background were generated by breeding Slc26a4−/− mice on a 129SvEvTac background with wild-type C57Bl/6J mice over 10 generations. Homozygous SPAK null mice on a C57Bl/6J background, reported previously (8), were bred to obtain SPAK KO mice and compared with wild-type mice on the same background. PKC-α null mice on a 129 SV background were kindly made available by Dr. Jeff Molkentin at the University of Cincinnati (1, 5). PKC-δ mice on a mixed C57/129 background were purchased from Jackson Laboratories (stock no. 028055). We bred male and female Slc26a4+/−, PKCα+/−, and PKCδ+/− heterozygotes to generate Slc26a4−/− (pendrin null), PKCα−/− (PKC-α null), and PKCδ−/− (PKC-δ null) mice, which were compared with their wild-type littermates. Mice ate a balanced diet (Envigo TS150355) and drank water ad libitum for 5–7 days before death (10). The Institutional Animal Care and Use Committee at Emory University approved all treatment protocols.
In vitro perfusion of isolated CCDs.
CCDs were dissected from medullary rays and perfused and bathed at flow rates of 2–3 nl/min in the presence of a symmetric, -buffered physiological solution containing the following (in mM): 125 NaCl, 24 NaHCO3, 2.5 K2HPO4, 2 CaCl2, 1.2 MgSO4, and 5.5 glucose. Tubules were equilibrated at 37°C for 30 min before the collections were started. All chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Measurement of net transepithelial Cl− flux.
Cl− concentration was measured in perfusate and collected samples using a continuous-flow fluorimeter and the Cl− sensitive fluorophore 6 methoxy-N-(3-sulfopropyl) quinolinium (SPQ; Molecular Probes, Eugene, OR), as described previously (7). Transepithelial Cl− flux, JCl, was calculated according to the equation: JCl = (Co – CL)Q/L, where Co and CL are perfusate and collected fluid Cl− concentrations, respectively. Q is flow rate in nanoliters per minute. L is tubule length. Net fluid transport was taken to be zero since net fluid flux has not been observed in CCDs when perfused in vitro in the presence of symmetric solutions and in the absence of vasopressin (16, 17). JCl is expressed in picomoles per millimeters per minutes.
Transepithelial voltage.
Transepithelial voltage (VT) was measured in the perfusion pipette connected to a high-impedance electrometer through an agar bridge saturated with 0.16 M NaCl and a calomel cell as described previously (33). The reference was an agar bridge from the bath to a calomel cell.
RESULTS
α-KG-stimulated Cl− absorption is pendrin dependent.
Previous studies have shown that type B intercalated cell apical Cl−/ exchange increases following α-KG application to the luminal fluid of mouse CCDs perfused in vitro (30). Since pendrin is the major mechanism of apical Cl−/ exchange in the CCD (26, 34), we asked if α-KG-stimulated Cl− absorption is pendrin dependent. To do so, we compared the effect of α-KG on Cl− absorption in CCDs from pendrin null and their wild-type littermates (Fig. 1). As shown, α-KG increased Cl− absorption in CCDs from wild-type mice without changing transepithelial voltage, VT, as reported previously (30). However, α-KG did not change Cl− flux in CCDs from pendrin null mice. We conclude that α-KG stimulates Cl− absorption in the mouse CCD through a pendrin-dependent mechanism.
Fig. 1.

α-Ketoglutarate (α-KG) increases Cl− absorption in wild-type but not in pendrin null mice. In each cortical collecting duct (CCD) studied, Cl− absorption (JCl), and transepitheial voltage (VT), were measured under basal conditions and then 15 min after the application of 1.5 mM α-KG to the luminal fluid. As shown, α-KG increased Cl− absorption without changing voltage in CCDs from wild-type (n = 4) but not from pendrin null mice (n = 4). The squares indicate means ± SE under each condition.
SPAK gene ablation increases total, but not α-KG-stimulated, Cl− absorption.
Because SPAK gene ablation increases urinary α-KG, pendrin abundance, and Cl− absorption in the mouse CCD (10), we asked if SPAK gene ablation magnifies the increment in Cl− absorption observed with α-KG application in vitro. Figure 2A shows that baseline Cl− absorption is higher in CCDs from SPAK null than from wild-type mice, similar to our previous observations (10). This figure also shows that while α-KG application to the luminal fluid increased Cl− absorption in CCDs from wild-type mice (P < 0.05), it produced no statistically significant increment in Cl− absorption with in CCDs from SPAK KO mice (P = NS). Figure 2B compares the change in Cl− absorption that follows α-KG application in CCDs from wild-type and SPAK null mice. As shown, the change in Cl− absorption that follows α-KG application was not higher in CCDs from SPAK null than in wild-type mice. We conclude that while SPAK gene ablation increases pendrin abundance and increases the basal level of Cl− absorption in mouse CCDs perfused in vitro, SPAK gene ablation does not amplify the α-KG-induced increment in Cl− absorption.
Fig. 2.

STE20/SPS-1-related proline-alanine-rich protein kinase (SPAK) gene ablation does not augment the increment in Cl− absorption observed with α-ketoglutarate (α-KG) application. A: Cl− absorption (JCl) was measured in wild-type and SPAK null mice before and after α-KG application to the perfusate. While α-KG increased Cl− absorption, JCl, in wild-type mice (n = 4), no statistically significant increase in Cl− absorption was observed following α-KG application in CCDs from SPAK null mice (n = 5). Solid lines indicate experiments where vehicle was added in the 1st period and α-KG in the 2nd period. Dashed lines indicate increment experiments where the order was reversed, i.e., α-KG added in the 1st period and then removed in the 2nd period. Squares indicate means ± SE under each condition. KO, knockout. B: comparison of the increment in Cl− absorption that followed α-KG application to the perfusate in cortical collecting ducts (CCDs) from wild-type and SPAK null mice.
α-KG-stimulated Cl− absorption is PKC dependent.
α-KG acts on the apical plasma membrane OXGR1 in type B intercalated cells to stimulate Cl− absorption by the CCD (30). Because OXGR1 is a GPCR (30), and since activated GPCRs stimulate PKC (2, 4), we asked if α-KG stimulates Cl− absorption in the mouse CCD through a PKC-dependent pathway. If so, PKC agonists, such as phorbol esters, should mimic, whereas PKC inhibitors should block, the α-KG-induced increment in Cl− absorption. To test this hypothesis, we examined the effect of a phorbol ester on JCl in CCDs from wild-type and pendrin null mice. As shown (Fig. 3), phorbol esters increased Cl− absorption in CCDs from wild-type mice. However, phorbol ester application did not increase Cl− absorption in CCDs from pendrin null mice (Fig. 3). We conclude that PKC agonists stimulate Cl− absorption through a pendrin-dependent pathway. In so doing, phorbol esters mimic the effect of α-KG on Cl− absorption in the mouse CCD.
Fig. 3.

Phorbol esters increase Cl− absorption (JCl) in cortical collecting ducts (CCDs) from wild-type but not from pendrin null mice. Shown is Cl− absorption in CCDs from wild-type (n = 3) and pendrin null mice (n = 3) before and then 30 min after the application of the PKC agonist phorbol-12,13-dibutyrate (PDBu, 500 nM) to the bath. As shown, in wild-type mice PDBu significantly increased Cl− absorption. However, in CCDs from pendrin null mice, we observed no change in Cl− absorption with PDBu application. Squares indicate means ± SE under each condition.
To explore the role of PKC on α-KG-stimulated Cl− absorption further, we examined the effect of α-KG on Cl− absorption in the presence of two structurally distinct PKC inhibitors or their vehicle. As shown, while α-KG stimulated Cl− absorption in the presence of vehicle (Fig. 4), α-KG was without effect when either of the PKC inhibitors GF109203X (2.5 mM) or Calphostin C (20 nM) were present in the bath solution (Fig. 4). We conclude that α-KG stimulates pendrin-dependent Cl− absorption through a PKC-dependent mechanism.
Fig. 4.

PKC inhibitors prevent the α-ketoglutarate (αKG)-induced increase in Cl− absorption (JCl). In wild-type mice, application of α-KG increased Cl− absorption with vehicle present in the bath [perfusate solution, n = 5 (A) or DMSO, n = 4 (B)]. However, α-KG had no effect when PKC inhibitors, such as GF109203X [2.5 μM, n = 5 (A)] or calphostin C [20 nM, n = 5 (B)] were present in the bath solution. Solid lines indicate that α-KG was added in the 2nd period, whereas dashed lines indicate that α-KG was present in the 1st period and then washed out in the 2nd. The squares indicate means ± SE under each condition.
a-KG-stimulated Cl− absorption is Ca2+ dependent.
Since PKC-mediated signaling events can be either Ca2+ dependent or independent, we examined the effect of α-KG on Cl− absorption in the presence of a Ca2+ chelator or its vehicle. As shown (Fig. 5), while α-KG stimulates Cl− absorption in the presence of vehicle, it had no effect in the presence of the Ca2+ chelator BAPTA. We conclude that α-KG-stimulates Cl− absorption through a Ca2+-dependent mechanism.
Fig. 5.

α-Ketoglutarate (α-KG)-stimulated Cl− absorption (JCl) is eliminated with a Ca2+ chelator present in the bath solution. Cortical collecting ducts (CCDs) were perfused in vitro in the presence of BAPTA (n = 6) or its vehicle (perfusate, n = 4) in the bath solution. α-KG increased Cl− absorption in CCDs from wild-type mice in the absence but not in the presence of the Ca2+ chelator BAPTA. Open circles indicate a BAPTA concentration of 10, whereas solid circles indicate a concentration of 20 μM. Solid lines indicate that α-KG was added in the 2nd period, whereas dashed lines indicate that α-KG was present in the 1st period and then washed out in the 2nd. Squares indicate means ± SE under each condition.
α-KG-stimulated Cl− absorption is mediated by the α- and δ-isoforms of PKC.
Further experiments explored the PKC isoform(s) that mediate α-KG-stimulated Cl− absorption. In the mouse CCD, intercalated cells express the α-, βI-, and δ-isoforms of PKC (15, 25). Of these, the α- and βI-isoforms are activated by Ca2+, whereas the δ-isoform is Ca2+ insensitive (9). Because α-KG-stimulated Cl− absorption is Ca2+ dependent, we examined α-KG-stimulated Cl− absorption following PKC-α gene ablation. Figure 6 shows that while α-KG application to the perfusate stimulated Cl− absorption in CCDs from wild-type mice, in CCDs from PKC-α KO mice, α-KG was without effect. We conclude that elimination of the PKC-α prevents α-KG-stimulated Cl− absorption.
Fig. 6.

Genetic ablation of the PKC-α isoform eliminates α-ketoglutarate (α-KG)-stimulated Cl− absorption (JCl). Cortical collecting ducts (CCDs) from PKC-α null (n = 3) and their wild-type littermates (n = 3) were perfused in vitro in the presence and absence of α-KG in the luminal fluid. As shown, PKC-α gene ablation eliminated the increase in Cl− absorption observed with α-KG application to the perfusate. Squares indicate means ± SE under each condition.
Because PKC-δ is also expressed in intercalated cells (15, 25), we examined the effect of PKC-δ on α-KG-stimulated Cl− absorption. As shown in Fig. 7, α-KG increased Cl− absorption in CCDs from wild-type mice but had no effect on JCl in CCDs from PKC-δ null mice. We conclude that α-KG stimulates Cl− absorption in the mouse CCD through a mechanism dependent on the α- and δ-isoforms of PKC.
Fig. 7.

Ablation of the PKC-δ isoform eliminates α-ketoglutarate (α-KG)-stimulated Cl− absorption (JCl). Cortical collecting ducts (CCDs) from PKC-δ (n = 3) and their wild-type littermates (n = 4) were perfused in vitro in the presence and absence of α-KG in the luminal fluid. As shown, PKC-δ gene ablation eliminated the increase in Cl− absorption observed with α-KG application to the perfusate. Squares indicate means ± SE under each condition.
SPAK gene ablation does not increase Cl− absorption by stimulating PKC activity.
Since SPAK gene ablation increases Cl− absorption in the CCD, we hypothesized that SPAK gene ablation increases Cl− absorption through enhanced PKC activity. If so, PKC inhibitor application should reduce Cl− absorption in CCDs from the SPAK null mice. To test this hypothesis, we measured Cl− absorption in CCDs from SPAK null mice before and after the application of the PKC inhibitor GF109203 to the bath. Robust Cl− absorption was again observed in CCDs from the SPAK null mice (Fig. 8), consistent with our previous observations (10). However, PKC inhibitor (GF109203) application to the bath failed to reduce Cl− absorption in these tubules. We conclude that the increment in Cl− absorption observed in CCDs from SPAK null mice does not occur through enhanced PKC signaling. Therefore, SPAK gene ablation in vivo and α-KG application in vitro increase in Cl− absorption in the mouse CCD through different mechanisms.
Fig. 8.
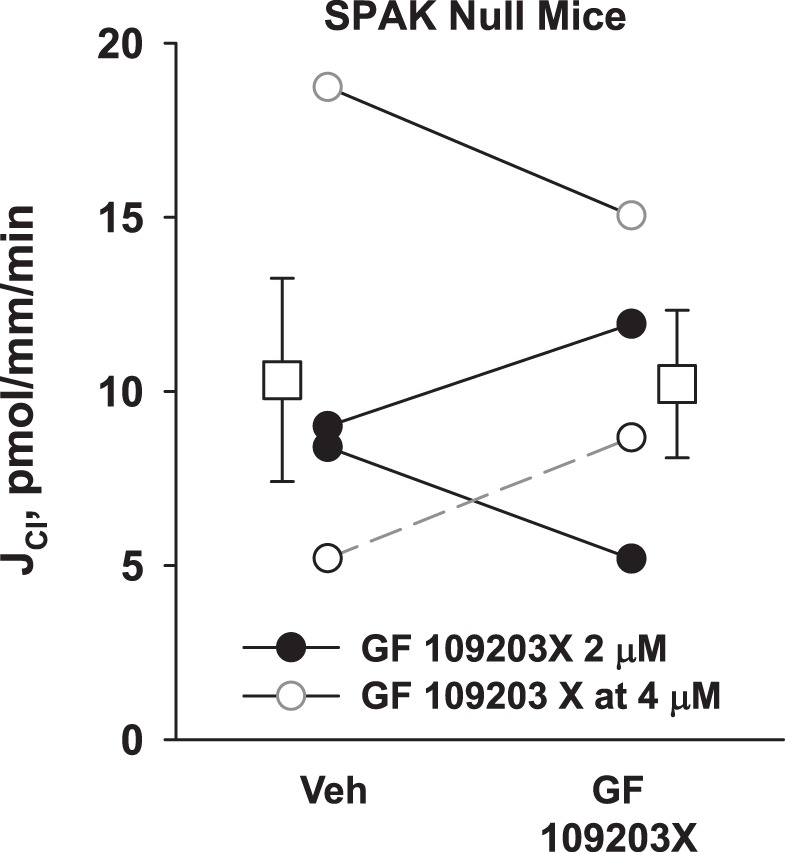
PKC inhibition does not reduce Cl− absorption (JCl) in cortical collecting ducts (CCDs) from STE20/SPS-1-related proline-alanine-rich protein kinase (SPAK) null mice. CCDs from SPAK null mice (n = 4) were perfused in vitro. Each CCD was perfused in the presence and absence of the PKC inhibitor GF109203X at a concentration of either 2 or 4 μM. Solid lines show data from tubules where the vehicle was present in the 1st period and GF109203X in the 2nd period. Dashed lines show data from tubules where GF109203 was present in the 1st period and then removed (i.e., washed out) in the 2nd. As shown, GF109203X did not change JCl in SPAK null CCDs. Squares indicate means ± SE under each condition.
DISCUSSION
α-KG-mediated signaling in renal intercalated cells occurs through its interaction with the GPCR OXGR1. Our observation that α-KG signaling transduction is dependent on PKC is consistent with previous reports showing that the primary downstream mediators of GPCRs such as OXGR1 are through serine/threonine intracellular kinases known as PKC and phospholipase C (4).
We propose that α-KG stimulates type B intercalated cell transporters through the signal transduction pathway shown in Fig. 9. α-KG binds to the OXGR1 on the apical plasma membrane of type B intercalated cells. Being a GPCR, the OXGR1 should activate phospholipase C, which catalyzes the hydrolysis of the membrane lipid phosphotidyl inositol-4,5-bisphosphate to generate DAG and IP3 (2). IP3 increases intracellular Ca2+, which acts with DAG to stimulate PKC-α. While DAG activates PKC-α through a Ca2+-dependent process, DAG activates PKC-δ independently of Ca2+. These two PKC isoforms act in concert to stimulate apical Cl−/ exchange.
Fig. 9.

Signal transduction pathway mediating the increase in apical Cl−/ exchange that follows αKG application to the luminal fluid. The dual requirement of PKC-α and PKC-δ may reflect parallel activation of pendrin by 1 PKC isoform and activation of a basolateral transporter by the other PKC isoform. Possible synergistic couplings by PKC isoforms are shown on the right. αKG, α-ketoglutarate; OXGR1, 2-oxoglutarate receptor 1; JCl, Cl− absorption.
How the two PKC isoforms stimulate pendrin-dependent Cl− absorption remains to be determined. PKC may increase apical plasma membrane pendrin abundance directly, thereby stimulating pendrin-mediated Cl−/ exchange. PKC may also stimulate another channel or transporter that augments the driving force for apical Cl−/ exchange. In type B intercalated cells of the mouse CCD, apical Cl−/ exchange is augmented by NaCl and H+ exit across the basolateral plasma membrane that is mediated by the NaHCO3 cotransporter AE4 (3), H+-ATPase (3), and ClC-K2/barttin Cl− channels (13, 23). PKC might act on one of these or another basolateral plasma membrane channels/transporters, thereby augmenting apical Cl−/ exchange. For example, Schwiebert et al. (27) has shown that PKC stimulates a 305-pS Cl− channel in a rabbit CCD cell line. PKC may stimulate this or another Cl− channel, thereby reducing intracellular Cl−, which augments the driving force for pendrin-mediated Cl−/ exchange. The dual requirement of PKC-α and PKC-δ may reflect parallel activation of pendrin by one PKC isoform and activation of a basolateral transporter by the other PKC isoform (Fig. 9).
All PKC family members share a common structure that includes a flexible hinge segment, a COOH-terminal catalytic domain, and an NH2-terminal regulatory domain. This regulatory domain has two discrete membrane modules, C1 and C2, which bind diaclyglycerol, anionic lipids, phosphatidylserine, and calcium (4). These membrane-targeting modules are the basis of PKC isoform classification (36). Conventional PKCs, such as PKC-α, PKC-β1, and PKC-βII, contain tandem C1a/C1B motifs that bind phorbol esters or diacylglycerol and a C2 domain that binds anion phospholipids through a Ca2+-dependent mechanism (36). PKC-δ is considered to be a novel PKC since it contains a nonfunctional C2 domain, which renders it Ca2+ insensitive (36).
Of the PKC isoforms expressed in mouse intercalated cells (15), we found that α-KG-stimulated Cl− absorption is dependent on PKC-α and PKC-γ. We were unable to test the effect of PKC-β gene ablation on α-KG-mediated Cl− absorption due to the absence of ready availability of PKC-β KO mice and the absence of a specific PKC-β inhibitor (19). Nevertheless, ablation of either the PKC-α or δ-isoforms eliminated the increment in Cl− absorption that follows the application of α-KG to the luminal fluid. As such, α-KG-stimulated Cl− absorption may occur through these pathways acting in parallel. In other cells, gene ablation of either PKC-α or PKC-δ changes cellular function by blocking two separate, but parallel, downstream signal transduction pathways. For example, in hypoxic HeLa human cervical cancer cells, either PKC-α or PKC-δ blockade reduces hypoxia-inducible factor-1a mRNA expression, although each isoform acts through separate signal transduction pathways (14). Similarly, PKC-α or PKC-δ gene ablation might eliminate the same or different signaling pathways downstream of α-KG-stimulated OXGR1 activation in intercalated cells.
α-KG production by the proximal tubule is highly regulated. Urinary α-KG rises with NaHCO3 and falls with NH4Cl administration, although NaCl administration has no effect (30). As such, one could hypothesize that pendrin abundance increases in models of metabolic alkalosis due to increased distal delivery of α-KG. In a previous study, we observed that eliminating NCC activation through SPAK gene ablation increased pendrin abundance and increased Cl− absorption in the mouse CCD, while also increasing α-KG excretion (10). We hypothesized that SPAK gene ablation stimulates pendrin abundance and function by increasing distal delivery of α-KG. However, the increment in CCD Cl− absorption that follows SPAK gene ablation cannot be explained by a sustained PKC-dependent mechanism since we observed no fall in Cl− absorption with PKC inhibitor application in CCDs from SPAK null mice. Moreover, while SPAK gene ablation increases distal delivery of α-KG and increases pendrin protein abundance in vivo, it did not magnify the increment in Cl− absorption observed with α-KG administration in vivo. As such, if SPAK gene ablation magnifies pendrin abundance and function through a chronic increase in distal α-KG delivery, it is unlikely to occur through a sustained increase in PKC activity. However, in CCDs from SPAK KO mice, the acute activation of pendrin by PKC may be obscured if pendrin is already maximally activated. We speculate that longer term adaptive changes in the SPAK KO mice, which lead to increased type B intercalated cell number and increased pendrin abundance, work through different signaling pathways.
In conclusion, stimulation of OXGR1 with α-KG increases pendrin-dependent Cl− absorption through a PKC-dependent pathway. It will be important for future studies to elucidate the underlying molecular mechanism further.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-110375 (project director: P. A. Welling; principal investigator: S. M. Wall). Y. Lazo-Fernandez was supported by a minority supplement to DK-104125 (to S. M. Wall).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.M.W. conceived and designed research; Y.L.-F. performed experiments; Y.L.-F., P.A.W., and S.M.W. analyzed data; Y.L.-F., P.A.W., and S.M.W. interpreted results of experiments; S.M.W. and P.A.W. prepared figures; S.M.W. drafted manuscript; Y.L.-F., P.A.W., and S.M.W. edited and revised manuscript; Y.L.-F., P.A.W., and S.M.W. approved final version of manuscript;
ACKNOWLEDGMENTS
We thank Dr. Mitsi Blount for helpful suggestions.
REFERENCES
- 1.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med 10: 248–254, 2004. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 2.Cattaneo F, Guerra G, Parisi M, De Marinis M, Tafuri D, Cinelli M, Ammendola R. Cell-surface receptors transactivation mediated by g protein-coupled receptors. Int J Mol Sci 15: 19700–19728, 2014. doi: 10.3390/ijms151119700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chambrey R, Kurth I, Peti-Peterdi J, Houillier P, Purkerson JM, Leviel F, Hentschke M, Zdebik AA, Schwartz GJ, Hübner CA, Eladari D. Renal intercalated cells are rather energized by a proton than a sodium pump. Proc Natl Acad Sci USA 110: 7928–7933, 2013. doi: 10.1073/pnas.1221496110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dowling CM, Kiely PA. Targeting protein kinase C downstream of growth factor and adhesion signalling. Cancers (Basel) 7: 1271–1291, 2015. doi: 10.3390/cancers7030836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eaton AF, Yue Q, Eaton DC, Bao HF. ENaC activity and expression is decreased in the lungs of protein kinase C-α knockout mice. Am J Physiol Lung Cell Mol Physiol 307: L374–L385, 2014. doi: 10.1152/ajplung.00040.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eaton DC, Pooler JP. Vander’s Renal Physiology. New York: McGraw-Hill, 2004. [Google Scholar]
- 7.García NH, Plato CF, Garvin JL, García NH. Fluorescent determination of chloride in nanoliter samples. Kidney Int 55: 321–325, 1999. doi: 10.1046/j.1523-1755.1999.00239.x. [DOI] [PubMed] [Google Scholar]
- 8.Geng Y, Hoke A, Delpire E. The Ste20 kinases Ste20-related proline-alanine-rich kinase and oxidative-stress response 1 regulate NKCC1 function in sensory neurons. J Biol Chem 284: 14020–14028, 2009. doi: 10.1074/jbc.M900142200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res 106: 1319–1331, 2010. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grimm PR, Lazo-Fernandez Y, Delpire E, Wall SM, Dorsey SG, Weinman EJ, Coleman R, Wade JB, Welling PA. Integrated compensatory network is activated in the absence of NCC phosphorylation. J Clin Invest 125: 2136–2150, 2015. doi: 10.1172/JCI78558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grimm PR, Taneja TK, Liu J, Coleman R, Chen YY, Delpire E, Wade JB, Welling PA. SPAK isoforms and OSR1 regulate sodium-chloride co-transporters in a nephron-specific manner. J Biol Chem 287: 37673–37690, 2012. doi: 10.1074/jbc.M112.402800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, Chen JL, Tian H, Ling L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 429: 188–193, 2004. doi: 10.1038/nature02488. [DOI] [PubMed] [Google Scholar]
- 13.Hennings JC, Andrini O, Picard N, Paulais M, Huebner AK, Cayuqueo IK, Bignon Y, Keck M, Corniere N, Bohm D, Jentsch TJ, Chambrey R, Teulon J, Hubner CA, Eladari D. The ClC-K2 chloride channel is critical for salt handling in the distal nephron. J Am Soc Nephrol 28: 209–217, 2017. doi: 10.1681/ASN.2016010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim H, Na YR, Kim SY, Yang EG. Protein kinase C isoforms differentially regulate hypoxia-inducible factor-1α accumulation in cancer cells. J Cell Biochem 117: 647–658, 2016. doi: 10.1002/jcb.25314. [DOI] [PubMed] [Google Scholar]
- 15.Kim WY, Jung JH, Park EY, Yang CW, Kim H, Nielsen S, Madsen KM, Kim J. Expression of protein kinase C isoenzymes α, βI, and δ in subtypes of intercalated cells of mouse kidney. Am J Physiol Renal Physiol 291: F1052–F1060, 2006. doi: 10.1152/ajprenal.00016.2006. [DOI] [PubMed] [Google Scholar]
- 16.Knepper MA, Good DW, Burg MB. Ammonia and bicarbonate transport by rat cortical collecting ducts perfused in vitro. Am J Physiol Renal Fluid Electrolyte 249: F870–F877, 1985. doi: 10.1152/ajprenal.1985.249.6.F870. [DOI] [PubMed] [Google Scholar]
- 17.Knepper MA, Good DW, Burg MB. Mechanism of ammonia secretion by cortical collecting ducts of rabbits. Am J Physiol Renal Fluid Electrolyte l 247: F729–F738, 1984. doi: 10.1152/ajprenal.1984.247.5.F729. [DOI] [PubMed] [Google Scholar]
- 18.Leviel F, Hübner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, Hassan H, Parker MD, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, Chambrey R, Eladari D. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 120: 1627–1635, 2010. doi: 10.1172/JCI40145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Q, Chen X, Macdonnell SM, Kranias EG, Lorenz JN, Leitges M, Houser SR, Molkentin JD. Protein kinase Calpha, but not PKCbeta or PKCgamma, regulates contractility and heart failure susceptibility: implications for ruboxistaurin as a novel therapeutic approach. Circ Res 105: 194–200, 2009. doi: 10.1161/CIRCRESAHA.109.195313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang CL, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab 14: 352–364, 2011. doi: 10.1016/j.cmet.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meissner T, Mayatepek E, Kinner M, Santer R. Urinary alpha-ketoglutarate is elevated in patients with hyperinsulinism-hyperammonemia syndrome. Clin Chim Acta 341: 23–26, 2004. doi: 10.1016/j.cccn.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 22.Nanami M, Lazo-Fernandez Y, Pech V, Verlander JW, Agazatian D, Weinstein AM, Bao HF, Eaton DC, Wall SM. ENaC inhibition stimulates HCl secretion in the mouse cortical collecting duct. I. Stilbene-sensitive Cl− secretion. Am J Physiol Renal Physiol 309: F251–F258, 2015. doi: 10.1152/ajprenal.00471.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pinelli L, Nissant A, Edwards A, Lourdel S, Teulon J, Paulais M. Dual regulation of the native ClC-K2 chloride channel in the distal nephron by voltage and pH. J Gen Physiol 148: 213–226, 2016. doi: 10.1085/jgp.201611623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rafiqi FH, Zuber AM, Glover M, Richardson C, Fleming S, Jovanović S, Jovanović A, O’Shaughnessy KM, Alessi DR. Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol Med 2: 63–75, 2010. doi: 10.1002/emmm.200900058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Redling S, Pfaff IL, Leitges M, Vallon V. Immunolocalization of protein kinase C isoenzymes alpha, beta I, beta II, delta, and epsilon in mouse kidney. Am J Physiol Renal Physiol 287: F289–F298, 2004. doi: 10.1152/ajprenal.00273.2003. [DOI] [PubMed] [Google Scholar]
- 26.Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, Green ED. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci USA 98: 4221–4226, 2001. doi: 10.1073/pnas.071516798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwiebert EM, Karlson KH, Friedman PA, Dietl P, Spielman WS, Stanton BA. Adenosine regulates a chloride channel via protein kinase C and a G protein in a rabbit cortical collecting duct cell line. J Clin Invest 89: 834–841, 1992. doi: 10.1172/JCI115662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi PP, Cao XR, Sweezer EM, Kinney TS, Williams NR, Husted RF, Nair R, Weiss RM, Williamson RA, Sigmund CD, Snyder PM, Staub O, Stokes JB, Yang B. Salt-sensitive hypertension and cardiac hypertrophy in mice deficient in the ubiquitin ligase Nedd4-2. Am J Physiol Renal Physiol 295: F462–F470, 2008. doi: 10.1152/ajprenal.90300.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terada Y, Knepper MA. Thiazide-sensitive NaCl absorption in rat cortical collecting duct. Am J Physiol Renal Fluid Electrolyte 259: F519–F528, 1990. doi: 10.1152/ajprenal.1990.259.3.F519. [DOI] [PubMed] [Google Scholar]
- 30.Tokonami N, Morla L, Centeno G, Mordasini D, Ramakrishnan SK, Nikolaeva S, Wagner CA, Bonny O, Houillier P, Doucet A, Firsov D. α-Ketoglutarate regulates acid-base balance through an intrarenal paracrine mechanism. J Clin Invest 123: 3166–3171, 2013. doi: 10.1172/JCI67562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uwai Y, Kawasaki T, Nabekura T. D-Malate decreases renal content of α-ketoglutarate, a driving force of organic anion transporters OAT1 and OAT3, resulting in inhibited tubular secretion of phenolsulfonphthalein, in rats. Biopharm Drug Dispos 38: 479–485, 2017. doi: 10.1002/bdd.2089. [DOI] [PubMed] [Google Scholar]
- 32.Verlander JW, Hassell KA, Royaux IE, Glapion DM, Wang ME, Everett LA, Green ED, Wall SM. Deoxycorticosterone upregulates PDS (Slc26a4) in mouse kidney: role of pendrin in mineralocorticoid-induced hypertension. Hypertension 42: 356–362, 2003. doi: 10.1161/01.HYP.0000088321.67254.B7. [DOI] [PubMed] [Google Scholar]
- 33.Wall SM. augments net acid secretion by a ouabain-sensitive mechanism in isolated perfused inner medullary collecting ducts. Am J Physiol Renal Fluid Electrolyte 270: F432–F439, 1996. doi: 10.1152/ajprenal.1996.270.3.F432. [DOI] [PubMed] [Google Scholar]
- 34.Wall SM, Kim YH, Stanley L, Glapion DM, Everett LA, Green ED, Verlander JW. NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: role in Cl- conservation. Hypertension 44: 982–987, 2004. doi: 10.1161/01.HYP.0000145863.96091.89. [DOI] [PubMed] [Google Scholar]
- 35.Wall SM, Weinstein AM. Cortical distal nephron Cl− transport in volume homeostasis and blood pressure regulation. Am J Physiol Renal Physiol 305: F427–F438, 2013. doi: 10.1152/ajprenal.00022.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu X, Jin T. The novel functions of the PLC/PKC/PKD signaling axis in G protein-coupled receptor-mediated chemotaxis of neutrophils. J Immunol Res 2015: 817604, 2015. doi: 10.1155/2015/817604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang SS, Lo YF, Wu CC, Lin SW, Yeh CJ, Chu P, Sytwu HK, Uchida S, Sasaki S, Lin SH. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol 21: 1868–1877, 2010. doi: 10.1681/ASN.2009121295. [DOI] [PMC free article] [PubMed] [Google Scholar]