

Abstract
Cisplatin is used to treat many solid cancers, but its dose-limiting side effect is nephrotoxicity, causing acute kidney injury in 30% of patients. Previously, we have developed a mouse model that better recapitulates the cisplatin dosing regimen humans receive and found that repeated dosing of cisplatin induces interstitial renal fibrosis. Chronic kidney disease is progressive and is characterized by chronic inflammation, worsening interstitial fibrosis, development of glomerulosclerosis, and endothelial dysfunction. To determine if damage caused by repeated cisplatin dosing results in bona fide chronic kidney disease, mice were treated with our repeated dosing regimen and then aged for 6 mo. These mice had progressive, chronic inflammation and worsened interstitial fibrosis compared with mice euthanized after day 24. Mice aged for 6 mo developed glomerular pathologies, and endothelial dysfunction was persistent. Mice treated with only two doses of cisplatin had little inflammation or kidney damage. Thus repeated dosing of cisplatin causes long-term effects that are characteristic of chronic kidney disease. This translational mouse model of cisplatin injury may better represent the 70% of patients that do not develop clinical acute kidney injury and can be used to identify both biomarkers for early injury, as well as novel therapeutic targets for the prevention of cisplatin-induced chronic kidney disease.
Keywords: chronic kidney disease, cisplatin, fibrosis, nephrotoxicity
INTRODUCTION
The dose-limiting side effect of cisplatin (CDDP), a potent chemotherapeutic, is nephrotoxicity, leading to acute kidney injury (AKI) in 30% of patients (1, 31, 35). AKI is the rapid loss of kidney function over a period of several days and is often diagnosed by a doubling in serum creatinine (SCr) or blood urea nitrogen (BUN), although clinical definitions of AKI vary greatly (43, 60). Patients with AKI may require dialysis and have a high mortality rate ranging from 20 to 80% (9, 61, 62).
Patients who develop CDDP-induced AKI will have their dosing schedule altered or be switched to a less nephrotoxic chemotherapeutic. Until recently, it was believed that most patients with AKI recover their kidney function without long-term, adverse effects. Recent epidemiological studies indicate that this is not the case for all patients (5–7, 14, 36). Patients may have improper or maladaptive repair of their injury, leading to the development of chronic kidney disease (CKD) (14). Patients with AKI are 10 times more likely to develop CKD (6, 7). As for the 70% of patients who do not meet clinical criteria for CDDP-induced AKI, it is believed that these patients are not at risk for poor long-term outcomes. However, recent meta-analyses have indicated that even a 25% increase in SCr is associated with a 70% increased mortality risk (10). Little is known about the risk of developing CKD in the 70% of patients who do not meet clinical criteria for CDDP-induced AKI.
CKD is characterized by replacement of healthy renal tissue by scar tissue, resulting in a progressive loss of kidney function (58). As cancer survival rates improve, there will be an increase in the population of patients who may be at risk for developing CKD after CDDP treatment (2, 19, 50). The best population for studying the chronic, long-term sequelae in the kidney after CDDP treatment is the pediatric cancer patient. Skinner et al. (50) found that 10 years post-treatment with carboplatin or CDDP, there was a significant decrease in estimated glomerular filtration rate (eGFR) in 11% of patients, and 15% of patients still had nephrotoxicity. These data highlight the long-lasting effects of CDDP.
Thus treatment of CDDP-induced kidney injury (CDDP-KI) and the subsequent development of CKD is an important field of study. Although developing therapeutics for the treatment of AKI has been at the forefront of kidney research, there are currently no clinically approved agents. The field of AKI research relies heavily on mouse models to study the pathophysiology of AKI. However, these mouse models are neither clinically nor physiologically relevant to patients and thus hinder the development of drugs that would be therapeutic in patients. This is especially true for CDDP-KI. The established mouse model of CDDP-KI uses one high dose of CDDP (10–30 mg/kg) that causes severe kidney injury and loss of function. Clinically, patients receive lower, repeated doses of CDDP to maintain therapeutic efficacy while curtailing nephrotoxic side effects. In this mouse model of CDDP-KI, the kidney damage is fatal to mice 3–4 days after the initial dose of CDDP (47, 51). Therefore, long-term outcome studies are not feasible.
To address the limitations of this mouse model of CDDP-KI, our group and others have shown that the repeated administration of low-dose CDDP causes interstitial fibrosis, indicative of CKD (22, 41, 47, 48, 54, 59). However, CKD is a progressive disease with many different pathologies, including glomerulosclerosis, endothelial dysfunction and damage, and chronic inflammation. In addition, there is a lack of knowledge about which mechanisms may contribute to these pathologies in the setting of CDDP-KI. The ability to identify these mechanisms would aid in the development of therapies to target the development and progression of CKD after CDDP treatment.
We have shown that mice develop interstitial fibrosis and are able to survive the entirety of our repeated dosing regimen (7 mg/kg CDDP once per week for 4 wk) (47). We hypothesized that due to the progressive nature of CKD, glomerular pathologies and endothelial damage may only be seen at a later time point. We treated mice and allowed them to age for 6 mo. We also wanted to determine when the development of interstitial fibrosis begins and which processes are involved in the initiation of fibrosis. To this end, we also treated mice with only two doses of CDDP.
After dose 2 of CDDP there was subtle inflammation. After dose 4 of CDDP, there was still no change in kidney function, but a significant increase in inflammation, endothelial dysfunction, and the development of interstitial fibrosis. Inflammation was still present 6 mo post-CDDP treatment, with worsened interstitial fibrosis. Furthermore, mice treated with CDDP and aged for 6 mo developed glomerular pathologies. These data indicate that the nephrotoxic effects of repeated CDDP dosing are long term, leading to the maintenance of chronic inflammation, worsened interstitial fibrosis, and glomerular/endothelial changes contributing to decreased kidney function.
MATERIALS AND METHODS
Animals.
Animal procedures were approved by the IACUC at University of Louisville, and followed guidelines set forth by American Veterinary Medical Association. FVB/n mice (The Jackson Laboratory) were maintained on a 12:12-h light/dark cycle and provided food/water ad libitum. Pharmaceutical-grade CDDP was purchased directly from the hospital pharmacy. Mice were intraperitoneally injected with either saline (vehicle) or CDDP (7 mg/kg) once per week for either 2 wk or 4 wk. At day 10, a cohort of saline and CDDP-treated mice were euthanized. At day 24, a cohort of saline and CDDP-treated mice were euthanized. A third cohort of mice received four doses of saline or CDDP and were euthanized 6 mo later (Fig. 1). Mice were monitored, and samples were prepared as previously described (47).
Fig. 1.
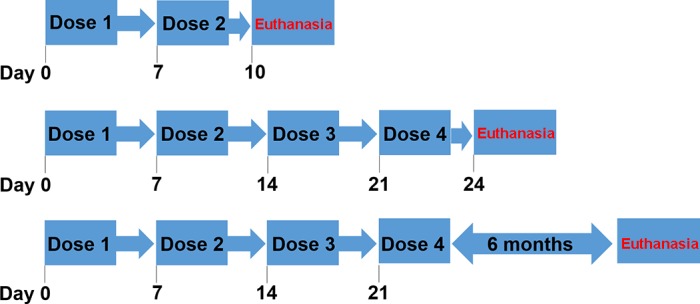
Repeated dosing regimen of cisplatin (CDDP). Eight-week-old male FVB mice were treated with either 2 doses saline vehicle or CDDP (7 mg/kg) and euthanized at day 10 (top). Another group was treated with either 4 doses of saline vehicle or CDDP (7 mg/kg) and euthanized at day 24 (middle) or aged out 6 mo posttreatment and then euthanized (bottom).
BUN determination.
BUN was determined in plasma per manufacturer’s protocol (80146, AMS Diagnostics).
ELISAs.
ELISAs for neutrophil gelatinase associated lipocalin (NGAL; DY1857, R&D Systems) and albuminuria (1011, Exocell) were performed on urine per manufacturers’ protocols.
Gene expression.
RNA was isolated using EZNA Total RNA Kit (R6834-02, Omega) per manufacturer’s protocol. cDNA was made from 1 μg RNA with High-Capacity Reverse Transcriptase (4368814, Life Technologies) per manufacturer’s protocol. Gene-specific cDNA was quantified with real-time qRT-PCR using either predesigned TAQman assays or self-designed SYBR assays. The following TAQman assays (Life Technologies) were used: Tnfα (Mm00443258_m1), Il-6 (Mm00446190_m1), Cxcl1 (Mm04207460_m1), Mcp-1 (Mm00441242_m1), Cdkn1a (Mm01303209_m1), Ctgf (Mm01192932_m1), Col1α1 (Mm00801666_m1), Arg-1 (Mm00475991_m1), and B2m (Mm00437762_m1). The following primers were self-designed: Inos, Cdkn2a, Endothelin-1, Vcam-1, Icam-1, Timp-1, Tgfβ1, and Fn1. Real-time qRT-PCR was done with either iTaq Universal Probes Supermix (172-5134, Bio-Rad) or iTaq Universal Sybr Green Supermix (172–5124, Bio-Rad).
Immunohistochemistry.
αSMA immunohistochemistry (IHC) was performed as previously published (47). For F4/80 IHC, kidney sections were rehydrated with Histoclear and an ethanol gradient. Target retrieval solution (S1699, Dako) was diluted 1:10 in ddH2O, and slides were incubated in solution at 72°C overnight. The next morning, slides were washed in 1× Tris-buffered saline (TBS) for 5 min. Slides were blocked with hydrogen peroxide, avidin, and biotin as described for αSMA. Slides were washed with TBS-0.01% Triton (TBS-T) for 5 min and blocked with 10% goat serum (31873, Invitrogen) in TBS-T for 30 min. Primary F4/80 antibody (ab16911, Abcam) was added at a 1:50 dilution in 10% goat serum. Slides were coverslipped and incubated at room temperature for 2 h. Slides were washed with TBST 2× for 5 min. Biotinylated rat 2° antibody (BA-9401, Vector) was added and incubated for 15 min. Slides were rinsed with TBS 2× for 5 min, and ABC reagent (PK-6104, Vectastain) was added for 30 min, followed by two 5-min TBS washes. DAB substrate solution (E885-200 ml, Amresco) was added to slides at room temperature for ~10 min. Slides were counterstained and mounted as described previously (47). For CD31 IHC, kidney sections were rehydrated as described above. Sodium citrate buffer (pH 6.0) was used for antigen retrieval for 40 min at 95°C. Slides were serum blocked with goat serum for 2 h at room temperature (PK-7100, Vector Laboratories) and washed twice with TBS 0.01% Triton X-100 for 5 min. Slides were then blocked for avidin and biotin as described above. CD31 antibody (ab124431, Abcam) was added to slides at 1:500 dilution in TBS with 1% BSA. TBS with 1% BSA was added to negative controls. Slides were coverslipped and incubated overnight at 4°C. The following morning, slides were washed twice with TBS 0.01% Triton X-100 for 5 min. Endogenous peroxidases were blocked with 3% hydrogen peroxide for 30 min, and slides were washed with TBS 0.01% Triton X-100. Biotinylated goat anti-rabbit IgG antibody in TBS with 1% BSA [(1:25,000), PK-7100, Vector Laboratories] was added to slides and incubated for 30 min at room temperature. Slides were then rinsed 2 times for 5 min before Vector ABC reagent was added (PK-7100, Vector Laboratories) for 30 min at room temperature. Slides were then rinsed two times for 5 min with TBS 0.01% Triton X-100. NovaRed peroxidase substrate solution (SK-4800, Vector Laboratories) was added to slides for 5 min, and then slides were rinsed in dH2O for 5 min. Slides were counterstained with Modified Mayer’s hematoxylin and processed as described above.
Sirius red/Fast green staining for total collagen.
Sirius red/Fast green (SR/FG) staining was performed as previously described (47). Optical density of Sirius red was quantified using Fiji-ImageJ and the following equation: OD = log (maximum intensity/mean intensity).
Statistical analysis.
Data are expressed as means ± SE for all experiments. Multiple comparisons were analyzed by two-way ANOVA, and group means were compared using Tukey posttests. The criteria for statistical differences were: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
RESULTS
Changes in kidney function and injury occur with repeated CDDP treatment and after 6-mo age out.
Eight-week-old male FVB mice were treated every week for either two doses or four doses of saline or CDDP (7 mg/kg) and euthanized 72 h after the final dose. Another group of mice was treated with four doses of saline or CDDP (7 mg/kg) and aged for 6 mo. BUN and albuminuria are common measures of kidney function, and NGAL is a FDA-approved urine biomarker for kidney injury (13, 33, 43, 60). BUN did not significantly increase after dose 2 (D2) or dose 4 (D4). BUN was significantly increased 6 mo post-CDDP treatment (6 mo post-CDDP) (Fig. 2A). Albuminuria was elevated at D2 and D4 of CDDP, but returned to baseline 6 mo post-CDDP (Fig. 2B). NGAL was slightly elevated after D2, but significantly increased after D4. NGAL levels were not elevated 6 mo post-CDDP (Fig. 2C). Taken together, these data indicate that mice develop albuminuria after D2, but albuminuria was no longer detectable 6 mo post-CDDP. In comparison, significant changes in BUN do not occur until 6 mo post-CDDP, indicating a progressive and permanent loss of kidney function even when injury, as measured by NGAL and albuminuria, was no longer present.
Fig. 2.

Markers of kidney function and injury during and after repeated cisplatin (CDDP) treatment. Eight-week-old FVB mice were treated with either saline vehicle or CDDP (7 mg/kg) once per week for 2 wk and euthanized 3 days after dose 2. Another group was treated with either saline vehicle or CDDP (7 mg/kg) once per week for 4 wk and euthanized 3 days after dose 4, or allowed to age for 6 mo following treatment (6 mo post-CDDP). A: blood urea nitrogen (BUN) measured in serum. B: albuminuria measured in urine. C: neutrophil gelatinase associated lipocalin (NGAL) measured in urine. Data are expressed as means ± SE; n = 5–10. Statistical significance was determined by 2-way ANOVA followed by Tukey posttest. *P < 0.05, ****P < 0.0001.
Tubular injury occurs with repeated CDDP treatment and after 6-mo age out.
CDDP-induced AKI results proximal tubule cell damage (1, 31, 34, 35). Tubular dilatation occurred after D4, and tubules remained dilatated 6 mo post-CDDP (Fig. 3A). There was a slight loss of brush borders after D2, but the severity increased after D4 and remained elevated 6 mo post-CDDP (Fig. 3B). Tubular degeneration occurred after D4 and was still present 6 mo post-CDDP (Fig. 3C). Severity of tubular cast formation was high at D4 and 6 mo post CDDP (Fig. 3D). Interestingly, there was no tubular necrosis with CDDP treatment (Fig. 3E). Whereas there was a slight increase in inflammatory cell infiltration after both D2 and D4, there was a significant increase in inflammatory cell infiltration 6 mo post-CDDP (Fig. 3F). There was also a significant increase in distal tubule damage 6 mo post-CDDP (Fig. 3G). Taken together, these data indicate that severe tubular injury does not occur until after D4 of CDDP, and that inflammation and distal damage are progressive.
Fig. 3.

Tubular injury during and after repeated cisplatin (CDDP) treatment. Eight-week-old FVB mice were treated with either saline vehicle or CDDP (7 mg/kg) once per week for 2 wk and euthanized 3 days after dose 2. Another group was treated with either saline vehicle or CDDP (7 mg/kg) once per week for 4 wk and euthanized 3 days after dose 4, or allowed to age for 6 mo following treatment (6 mo post-CDDP). Severity of tubular dilatation (A), loss of brush border (B), tubular degeneration (C), tubular casts (D), tubular necrosis (E), inflammation (F), and distal damage (G) was determined by a renal pathologist. Data are expressed as means ± SE; n = 5–7. Statistical significance was determined by 2-way ANOVA followed by Tukey posttest. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Inflammatory cytokine and chemokine levels are altered with repeated CDDP treatment and after 6-mo age out.
Inflammation is one of the hallmarks of AKI, and chronic inflammation contributes to renal fibrosis (14, 15, 21, 30, 31, 34, 35). Thus we wanted to determine levels of disease-related inflammatory cytokines and chemokines. Tnfα is the major mediator of inflammation in CDDP-induced AKI (39, 40). Tnfα mRNA levels were elevated twofold after D2, and levels were significantly increased at D4 (Fig. 4A). Tnfα levels remained significantly elevated 6 mo post-CDDP (Fig. 4A). Il6 increased at D2 and remained elevated after D4 (Fig. 4B). Six months post-CDDP, Il6 levels increased significantly compared with D2 and D4 (Fig. 4B). Mcp-1, a chemokine for monocyte recruitment, increased slightly after D2, and was elevated threefold at D4 and 6 mo post-CDDP (Fig. 4C). Cxcl1, a chemokine for neutrophil recruitment, increased fivefold after D2, and was significantly increased at D4 (Fig. 4D). Six months post-CDDP, Cxcl1 levels were elevated sevenfold (Fig. 4D). These data indicate that inflammatory cytokine and chemokine levels increase with each dose of CDDP examined in this study and remain elevated after CDDP treatment, indicating chronic inflammation.
Fig. 4.

Inflammatory cytokine and chemokine levels during and after repeated cisplatin (CDDP) treatment. Eight-week-old FVB mice were treated with either saline vehicle or CDDP (7 mg/kg) once per week for 2 wk and euthanized 3 days after dose 2. Another group was treated with either saline vehicle or CDDP (7 mg/kg) once per week for 4 wk and euthanized 3 days after dose 4, or allowed to age for 6 mo following treatment (6 mo post-CDDP). mRNA levels of tumor necrosis factor-α (Tnfα) (A), interleukin 6 (Il6) (B), monocyte chemoattractant protein 1 (Mcp-1) (C), and c-x-c motif ligand 1 (Cxcl1) (D) measured in the kidney cortex via qRT-PCR. Data are expressed as means ± SE; n = 5–10. Statistical significance was determined by 2-way ANOVA followed by Tukey posttest. *P < 0.05, **P < 0.01.
Macrophages are present in kidneys with repeated CDDP treatment and after 6-mo age out.
Macrophages mediate CDDP-induced kidney injury (14, 34, 37, 42). With AKI, M1 macrophages are proinflammatory, whereas M2 macrophages are anti-inflammatory, although the roles of these macrophage subtypes are less clearly defined in fibrogenesis and CKD (14, 23, 24, 37, 42). We wanted to determine the presence and subtypes of macrophages with repeated CDDP administration. F4/80 IHC for macrophages indicated that there was no change in number of macrophages after D2, but after D4 there was an increase in macrophages (Fig. 5A). Six months post-CDDP, more macrophages were present (Fig. 5A). Inos is a marker of M1 macrophages (27, 49). Levels of Inos did not change with CDDP treatment (Fig. 5B). Arg-1, a marker of M2 macrophages, increased after D4 (Fig. 5C) (27, 49). Six months post-CDDP treatment, Arg-1 was still elevated (Fig. 5C). These data indicate an increase in macrophages after D4 and 6 mo post-CDDP, and there are elevated levels of Arg-1-expressing macrophages after D4 and 6 mo post-CDDP.
Fig. 5.

Total macrophages and macrophage subtypes during and after repeated cisplatin (CDDP) treatment. Eight-week-old FVB mice were treated with either saline vehicle or CDDP (7 mg/kg) once per week for 2 wk and euthanized 3 days after dose 2 (D2). Another group was treated with either saline vehicle or CDDP (7 mg/kg) once per week for 4 wk and euthanized 3 days after dose 4 (D4), or allowed to age for 6 mo following treatment (6 mo post-CDDP). F4/80 IHC staining for total macrophages (A). mRNA levels of inducible nitric oxide synthase (iNos) (B) and arginase-1 (Arg-1) (C) measured in the kidney cortex via qRT-PCR. Data are expressed as means ± SE; n = 5–10.
Profibrotic growth factor production occurs with repeated CDDP treatment and after 6-mo age out.
Profibrotic factors like Tgfβ and Ctgf contribute to the development and maintenance of fibrosis (14, 20, 63). Tgfβ levels significantly increased after D4 and remained elevated 6 mo post-CDDP (Fig. 6A). Ctgf increased twofold after D4 and remained elevated 6 mo post-CDDP (Fig. 6B). These data indicate that there is continued production of TGFβ and CTGF even after repeated CDDP treatment.
Fig. 6.

Profibrotic growth factor production during and after repeated cisplatin (CDDP) treatment. Eight-week-old FVB mice were treated with either saline vehicle or CDDP (7 mg/kg) once per week for 2 wk and euthanized 3 days after dose 2. Another group was treated with either saline vehicle or CDDP (7 mg/kg) once per week for 4 wk and euthanized 3 days after dose 4, or allowed to age for 6 mo following treatment (6 mo post-CDDP). mRNA levels of transforming growth factor β (Tgfβ) (A) and connective tissue growth factor (Ctgf) (B) were measured in the kidney cortex via qRT-PCR. Data are expressed as means ± SE; n = 5–10. Statistical significance was determined by 2-way ANOVA followed by Tukey posttest. *P < 0.05, **P < 0.01.
Interstitial fibrosis develops with repeated CDDP treatment and is worsened with 6-mo age out.
Development of interstitial fibrosis is a hallmark of CKD (14, 58). After D4, interstitial fibrosis was present and severity increased 6 mo post-CDDP (Fig. 7A). Fibrosis is marked by an increase in both collagen and fibronectin, and myofibroblasts, which deposit collagen, can be detected by αSMA expression (57). IHC indicated an increase in αSma-positive staining after D4 (Fig. 7B). Six months post-CDDP, there was more αSma-positive staining (Fig. 7B). Sirius red/Fast green (SR/FG) staining for total collagen (red stain) indicated no change in total collagen levels after D2, but there was a significant increase after D4 (Fig. 7C). Levels of total collagen remained elevated 6 mo post-CDDP (Fig. 7C). Col1a1 levels were not elevated after D2 of CDDP, but increased threefold after D4 and remained elevated 6 mo post-CDDP (Fig. 7D). Fibronectin levels increased only after D4, and remained elevated twofold 6 mo post-CDDP (Fig. 7D). Thus data indicate that fibrosis develops only after D4, and may be worsened 6 mo post-CDDP.
Fig. 7.

Development of interstitial fibrosis during and after repeated cisplatin (CDDP) treatment. Eight-week-old FVB mice were treated with either saline vehicle or CDDP (7 mg/kg) once per week for 2 wk and euthanized 3 days after dose 2. Another group was treated with either saline vehicle or CDDP (7 mg/kg) once per week for 4 wk and euthanized 3 days after dose 4, or allowed to age for 6 mo following treatment (6 mo post-CDDP). A: interstitial fibrosis levels determined by a renal pathologist. B: presence of myofibroblasts determined by α-smooth muscle actin (α-SMA) IHC. C: levels of total collagen determined by Sirius red/Fast green (SR/FG) staining and optical density quantification of Sirius red staining. Levels of collagen 1 type 1a (Col1a1) (D) and fibronectin (Fn1) (E) measured in kidney cortex via qRT-PCR. Data are expressed as means ± SE; n = 5–10. Statistical significance was determined by 2-way ANOVA followed by Tukey posttest. *P < 0.05, **P < 0.01.
Glomerular pathologies occur after 6-mo age out in CDDP-treated mice.
CKD is associated with damage to glomeruli, ultimately resulting in decreased glomerular filtration rate (GFR) (16, 25, 58). Renal pathology indicated there was mesangial hypercellularity after D2 and D4 of CDDP (Fig. 8A). Interestingly, both saline- and CDDP-treated mice showed increased mesangial hypercellularity 6 mo post-CDDP (Fig. 8A). Six months post-CDDP, there was a significant increase in periglomerular fibrosis and thickening of the basement membrane (Fig. 8, B and C). Taken together, these data indicate that most glomerular changes occur 6 mo post-CDDP treatment, which may contribute to loss of renal function.
Fig. 8.

Indexes of glomerular damage during and after repeated cisplatin (CDDP) treatment. Eight-week-old FVB mice were treated with either saline vehicle or CDDP (7 mg/kg) once per week for 2 wk and euthanized 3 days after dose 2. Another group was treated with either saline vehicle or CDDP (7 mg/kg) once per week for 4 wk and euthanized 3 days after dose 4 or allowed to age for 6 mo following treatment (6 mo post-CDDP). Severity of mesangial hypercellularity (A), periglomerular fibrosis (B), and thickening of basement membrane (C) were determined by a renal pathologist. Data are expressed as means ± SE; n = 5–7. Statistical significance was determined by 2-way ANOVA followed by Tukey posttest. **P < 0.01, ***P < 0.001, ****P < 0.0001.
Endothelial dysfunction occurs with repeated CDDP treatment, and damage is present with 6-mo age out.
Endothelial dysfunction and subsequent damage is a major component of CKD. Endothelial dysfunction is marked by an increased production of vasoconstrictors and improper regulation of inflammatory processes (3, 12). Endothelin-1 is a potent vasoconstrictor that is involved in the regulation of endothelial cells and is increased when endothelial dysfunction occurs (3, 12, 46, 53). Endothelin-1 significantly increased after D4, and remained elevated 6 mo post-CDDP (Fig. 9A). During endothelial dysfunction, adhesion molecule expression is elevated, which contribute to inflammation by promoting adhesion of leukocytes to the surface of endothelium (12). Vcam-1 and Icam-1 levels were slightly elevated after D2, but significantly increased after D4 (Fig. 9, B and C). Levels of Vcam-1 and Icam-1 remained slightly elevated 6 mo post CDDP (Fig. 9, B and C). In addition, Timp-1 levels increased after D4 and remained elevated 6 mo post CDDP (Fig. 9D). Finally, CD31 is highly expressed in endothelial cells and is often used as a marker of endothelial damage (28). CD31 IHC staining indicated that there were no detectable changes in CD31 in the cortex (Fig. 9E). However, 6 mo post-CDDP, there was an increase in CD31 expression in the medulla (Fig. 9E). These data indicate that endothelial dysfunction occurs after D2 of CDDP, worsens after D4, and persists 6 mo post-CDDP.
Fig. 9.

Markers of endothelial dysfunction and damage during and after repeated cisplatin (CDDP) treatment. Eight-week-old FVB mice were treated with either saline vehicle or CDDP (7 mg/kg) once per week for 2 wk and euthanized 3 days after dose 2. Another group was treated with either saline vehicle or CDDP (7 mg/kg) once per week for 4 wk and euthanized 3 days after dose 4 or allowed to age for 6 mo following treatment (6 mo post-CDDP). mRNA levels of Endothelin-1, (A), vascular adhesion protein 1 (Vcam-1) (B), intercellular adhesion molecule 1 (Icam-1) (C), and TIMP metallopeptidase inhibitor 1 (Timp-1) (D) were measured in kidney via qRT-PCR. E: CD31 IHC stain for endothelial cell damage in cortex and medulla. Data are expressed as means ± SE; n = 5–10. Data are expressed as means ± SE; n = 5–10. Statistical significance was determined by 2-way ANOVA followed by Tukey posttest. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
DISCUSSION
By 2020, it is estimated that there will be 15 million new cases of cancer each year (18). Approximately one-fifth of these patients will receive CDDP as part of their therapeutic regimen. Although it is established that patients with AKI are at greater risk for developing CKD, the novel finding of our study is that mild injury caused by repeated, low doses of CDDP that does not meet the clinical criteria for AKI can progress to CKD. Clinically, there is no stringent definition for AKI, but RIFLE (risk, injury, failure, loss of kidney function, and end-stage kidney disease) criteria define it as a doubling in SCr, and 30% of patients that receive cisplatin will develop AKI according to this definition (4, 11, 43). However, little is known as to whether the 70% who do not develop clinical AKI are also at risk for poor, long-term outcomes such as increased mortality risk or development of CKD (6, 8, 10, 19). In meta-analyses by Chertow et al. (8) and Coca et al. (10), some cohorts included in these studies defined AKI as a 25% increase in SCr. Even with this mildest form of AKI, there was a 70% increase in mortality risk (10, 26).
SCr is a poor marker for defining AKI and does not accurately reflect true renal function. Measures of SCr are affected by age, sex, protein intake, and drug metabolism (56). Furthermore, changes in SCr do not occur until there is 50% or greater loss of nephrons (52). Even without changes in kidney function, we began to see pathologies that were indicative of acute tubular injury, although the severity of these pathologies was not significant. However, there was a 26% increase in BUN after D4 of CDDP, and tubular pathologies were more severe, indicating a mild AKI phenotype. We believe our model presents the novel opportunity to identify pathologies and their contributing mechanisms that occur early on with CDDP-induced kidney injury, and in doing so, will aid in the identification of more reliable and accurate biomarkers for both AKI and CKD.
Furthermore, our study is the first to look at long-term effects post-CDDP treatment and to show that bona fide CKD can result from repeated CDDP administration. Six-month-age out of FVB/n mice treated with CDDP (7 mg/kg once per week for 4 wk) indicated worsened chronic inflammation of the kidney and progressive interstitial fibrosis (Fig. 10). These mice had endothelial dysfunction and glomerular pathologies secondary to tubular injury. This ultimately culminated in a significant decline in kidney function, indicative of CKD.
Fig. 10.

Overview of pathological changes that occur during and after repeated cisplatin (CDDP) treatment. Mice treated with 2 doses of CDDP developed albuminuria, mild tubular pathologies, slight inflammation, and mild endothelial dysfunction. There was also increased level of NGAL in urine. Mice treated with 4 doses of CDDP and euthanized at day 24 still had albuminuria. In these mice, NGAL levels peaked and there were more severe tubular pathologies, as well as endothelial dysfunction. In addition, mice treated with 4 doses of CDDP and euthanized at day 24 developed interstitial fibrosis, which coincided with an increase in total macrophages and increase in profibrotic growth factors. After 6-mo age out, mice treated with 4 doses of CDDP no longer had detectable levels of albuminuria or NGAL, but inflammation and interstitial fibrosis worsened. In addition, these mice began to develop periglomerular fibrosis.
Although kidney injury 6 mo post-CDDP treatment was not detected by NGAL, inflammation persisted. Of note, Il6 was significantly increased 6 mo post-CDDP treatment. IL6 enhances the response of injured tubular cells to profibrotic stimuli, and high circulating levels of IL6 are commonly found in patients with end-stage renal disease (ESRD) (38). Zhang et al. (64) have also shown that IL6 levels increase in the kidneys of patients with CKD and that IL6 activation led to the expression of fibrotic genes and endothelin-1, indicating the role of IL6 as a major driver of CKD. In our model, Il6 mRNA levels in the kidney were highest in mice 6 mo post-CDDP. Thus the robust increase in Il6 seen at this time point may be indicative of progressive CKD. This provides evidence that IL6 may be a potential therapeutic target for preventing the development and progression of CKD after CDDP-induced kidney injury, as inhibition or depletion would affect a number of contributing pathways. Indeed, in a model of acute peritoneal inflammation, IL6−/− mice did not develop fibrosis (15).
The continual production of profibrotic growth factors also contributes to worsening interstitial fibrosis. TGF-β and CTGF are derived in part from senescent cells that have undergone cell cycle arrest with injury (14, 20, 63). These profibrotic growth factors can stimulate proliferation and differentiation of fibroblasts to myofibroblasts (14). Indeed, αSMA-positive myofibroblasts increased after D4 and remained present 6 mo post-CDDP. Myofibroblasts contribute to accumulation of collagen as evidenced by SR/FG staining, indicative of interstitial fibrosis. Concurrently, periglomerular fibrosis developed 6 mo post-CDDP, contributing to the overall presence of fibrosis, and indicating development of glomerular pathologies with this model.
The development of interstitial fibrosis and glomerular pathologies after CDDP treatment recapitulates outcomes in a porcine model of CDDP treatment (45). Robbins et al. found that 24 wk after an intravenous injection of low-dose CDDP, mature large white female pigs developed interstitial fibrosis in the deep cortex and medulla and that glomeruli had thickened Bowman’s capsules and capillary changes (44, 45). We saw a similar interstitial fibrosis pattern in the kidneys of mice treated with repeated CDDP treatment and aged out for 6 mo, as well as glomerular changes at this time point. Porcine kidneys are similar to human kidneys in structure and have the ability to develop renal fibrosis and glomerular changes that are similar to what is seen with human CKD progression (17, 55). However, renal studies with pigs are still relatively new, and mechanistic studies are difficult because genetically manipulated porcine models are still limited (55). Since we are able to recapitulate a similar fibrotic phenotype with our mouse model, this further suggests that our repeated dosing regimen is a clinically relevant model of CDDP nephrotoxicity. In addition, because this model utilizes mice, mechanistic studies are easier and more cost effective.
An important finding of this study was that the total macrophage population increased with worsening renal fibrosis. Macrophages are key mediators of AKI, and the prevalence of M1 or M2 subtype is an indicator of adaptive or maladaptive repair (14, 24, 37, 42). We found that iNos mRNA levels (M1 macrophages) did not change with CDDP treatment. There was a peak in Arg-1 mRNA levels (M2 macrophages) after D4, and Arg-1 was still elevated 6 mo post-CDDP. This indicates that most of the macrophages present in the kidney after injury are M2 in nature. This is poignant as the kidney has little to no Arg-1 expressed normally, and increases in Arg-1 can be traced to M2 macrophages (29, 32).
Previous studies have indicated that M2 macrophages may only be reparative when there is short-term inflammation. With our model chronic, progressive inflammation occurs, and M2 macrophages may contribute to fibrosis. This finding is not surprising as macrophages secrete chemokines that recruit fibroblasts and activate both resident and recruited myofibroblasts at the site of injury (23). In an ischemia-reperfusion (I/R) study by Kim et al. (23), there was a significant increase in F4/80-positive macrophages during the recovery phase after I/R injury, but total number of macrophages further increased even after the recovery phase. Furthermore, there was a shift from M1 to M2 subtypes after the recovery phase, highlighting the contribution of M2 subtype macrophages in fibrosis (23). Adoptive transfer studies to validate the role of M2 macrophages and their contribution to fibrosis showed that the fibrotic phenotype was only recapitulated with M2 macrophages (23). Our findings also indicate that M2 macrophages are contributing to the progression of fibrosis.
The recruitment of macrophages and leukocytes into the kidney after injury is a process often mediated by endothelial dysfunction (3, 12). Early endothelial dysfunction is marked by an increase in vasoconstrictors and adhesion molecules (12). Endothelin-1 is a sensitive marker of endothelial perturbations, and increases in this vasoconstrictor can contribute to the development of atherosclerosis (3, 12, 28). ICAM-1 and VCAM-1 promote inflammation by allowing leukocytes to adhere to endothelium (12). With our model, slight changes in endothelin-1, VCAM-1, and ICAM-1 can be seen after D2 with our repeated CDDP dosing regimen, and these levels are further elevated after D4, and remain slightly elevated 6 mo post-CDDP, indicating persistent endothelial dysfunction. Loss of CD31 expression at the intercellular junctions of endothelial cells is often indicative of damage, but we saw little to no change in CD31 expression in the cortex (28). However, there was increased CD31 expression in the medulla 6 mo post-CDDP. Cell types other than endothelial cells can also express CD31, particularly leukocytes, and increased expression of CD31 in these cell types may be indicative of early atherosclerosis, although further validation of this would need to be done in our model (28).
In conclusion, our data indicate the effects of repeated treatment with CDDP, which better mimics the clinical dosing regimen, leads to adverse long-term effects without overt AKI. Specifically, these effects include chronic inflammation, worsened interstitial fibrosis, endothelial dysfunction, declined kidney function, and glomerular pathologies associated with CKD. This model is ideal for both identifying biomarkers for early injury detection, as well as potential targets to halt the development of CKD after CDDP treatment. This model also provides a clinically relevant model for testing current, potential kidney injury therapeutics that are being developed.
GRANTS
Support for this work was provided by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-093462 (to L. J. Siskind).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.N.S., L.J.B., and L.J.S. conceived and designed research; C.N.S. performed experiments; C.N.S., M.A.D., J.M., and G.B.O. analyzed data; C.N.S., M.A.D., and G.B.O. interpreted results of experiments; C.N.S. prepared figures; C.N.S. drafted manuscript; C.N.S., M.A.D., J.M., L.J.B., and L.J.S. edited and revised manuscript; C.N.S., L.J.B., and L.J.S. approved final version of manuscript.
REFERENCES
- 1.Arany I, Safirstein RL. Cisplatin nephrotoxicity. Semin Nephrol 23: 460–464, 2003. doi: 10.1016/S0270-9295(03)00089-5. [DOI] [PubMed] [Google Scholar]
- 2.Arga M, Oguz A, Pinarli FG, Karadeniz C, Citak EC, Emeksiz HC, Duran EA, Soylemezoglu O. Risk factors for cisplatin-induced long-term nephrotoxicity in pediatric cancer survivors. Pediatr Int 57: 406–413, 2015. doi: 10.1111/ped.12542. [DOI] [PubMed] [Google Scholar]
- 3.Basile DP. The endothelial cell in ischemic acute kidney injury: implications for acute and chronic function. Kidney Int 72: 151–156, 2007. doi: 10.1038/sj.ki.5002312. [DOI] [PubMed] [Google Scholar]
- 4.Bellomo R, Kellum JA, Ronco C. Defining and classifying acute renal failure: from advocacy to consensus and validation of the RIFLE criteria. Intensive Care Med 33: 409–413, 2007. doi: 10.1007/s00134-006-0478-x. [DOI] [PubMed] [Google Scholar]
- 5.Chawla LS. Acute kidney injury leading to chronic kidney disease and long-term outcomes of acute kidney injury: the best opportunity to mitigate acute kidney injury? Contrib Nephrol 174: 182–190, 2011. doi: 10.1159/000329396. [DOI] [PubMed] [Google Scholar]
- 6.Chawla LS, Amdur RL, Amodeo S, Kimmel PL, Palant CE. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int 79: 1361–1369, 2011. doi: 10.1038/ki.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med 371: 58–66, 2014. doi: 10.1056/NEJMra1214243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol 16: 3365–3370, 2005. doi: 10.1681/ASN.2004090740. [DOI] [PubMed] [Google Scholar]
- 9.Chertow GM, Levy EM, Hammermeister KE, Grover F, Daley J. Independent association between acute renal failure and mortality following cardiac surgery. Am J Med 104: 343–348, 1998. doi: 10.1016/S0002-9343(98)00058-8. [DOI] [PubMed] [Google Scholar]
- 10.Coca SG, Yusuf B, Shlipak MG, Garg AX, Parikh CR. Long-term risk of mortality and other adverse outcomes after acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis 53: 961–973, 2009. doi: 10.1053/j.ajkd.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cruz DN, Ricci Z, Ronco C. Clinical review: RIFLE and AKIN–time for reappraisal. Crit Care 13: 211, 2009. doi: 10.1186/cc7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation 115: 1285–1295, 2007. doi: 10.1161/CIRCULATIONAHA.106.652859. [DOI] [PubMed] [Google Scholar]
- 13.Devarajan P. Neutrophil gelatinase-associated lipocalin: a promising biomarker for human acute kidney injury. Biomarkers Med 4: 265–280, 2010. doi: 10.2217/bmm.10.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol 11: 264–276, 2015. doi: 10.1038/nrneph.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fielding CA, Jones GW, McLoughlin RM, McLeod L, Hammond VJ, Uceda J, Williams AS, Lambie M, Foster TL, Liao CT, Rice CM, Greenhill CJ, Colmont CS, Hams E, Coles B, Kift-Morgan A, Newton Z, Craig KJ, Williams JD, Williams GT, Davies SJ, Humphreys IR, O’Donnell VB, Taylor PR, Jenkins BJ, Topley N, Jones SA. Interleukin-6 signaling drives fibrosis in unresolved inflammation. Immunity 40: 40–50, 2014. doi: 10.1016/j.immuni.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fogo AB. Mechanisms of progression of chronic kidney disease. Pediatr Nephrol 22: 2011–2022, 2007. doi: 10.1007/s00467-007-0524-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forster R, Ancian P, Fredholm M, Simianer H, Whitelaw B; Steering Group of the RETHINK Project . The minipig as a platform for new technologies in toxicology. J Pharmacol Toxicol Methods 62: 227–235, 2010. doi: 10.1016/j.vascn.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 18.Frankish H. 15 million new cancer cases per year by 2020, says WHO. Lancet 361: 1278, 2003. doi: 10.1016/S0140-6736(03)13038-3. [DOI] [PubMed] [Google Scholar]
- 19.Greenberg JH, Coca S, Parikh CR. Long-term risk of chronic kidney disease and mortality in children after acute kidney injury: a systematic review. BMC Nephrol 15: 184, 2014. doi: 10.1186/1471-2369-15-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grgic I, Campanholle G, Bijol V, Wang C, Sabbisetti VS, Ichimura T, Humphreys BD, Bonventre JV. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int 82: 172–183, 2012. doi: 10.1038/ki.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta J, Mitra N, Kanetsky PA, Devaney J, Wing MR, Reilly M, Shah VO, Balakrishnan VS, Guzman NJ, Girndt M, Periera BG, Feldman HI, Kusek JW, Joffe MM, Raj DS; CRIC Study Investigators . Association between albuminuria, kidney function, and inflammatory biomarker profile in CKD in CRIC. Clin J Am Soc Nephrol 7: 1938–1946, 2012. doi: 10.2215/CJN.03500412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katagiri D, Hamasaki Y, Doi K, Negishi K, Sugaya T, Nangaku M, Noiri E. Interstitial renal fibrosis due to multiple cisplatin treatments is ameliorated by semicarbazide-sensitive amine oxidase inhibition. Kidney Int 89: 374–385, 2016. doi: 10.1038/ki.2015.327. [DOI] [PubMed] [Google Scholar]
- 23.Kim MG, Kim SC, Ko YS, Lee HY, Jo SK, Cho W. The role of M2 macrophages in the progression of chronic kidney disease following acute kidney injury. PLoS One 10: e0143961, 2015. doi: 10.1371/journal.pone.0143961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lech M, Gröbmayr R, Ryu M, Lorenz G, Hartter I, Mulay SR, Susanti HE, Kobayashi KS, Flavell RA, Anders HJ. Macrophage phenotype controls long-term AKI outcomes–kidney regeneration versus atrophy. J Am Soc Nephrol 25: 292–304, 2014. doi: 10.1681/ASN.2013020152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levey AS, Coresh J. Chronic kidney disease. Lancet 379: 165–180, 2012. doi: 10.1016/S0140-6736(11)60178-5. [DOI] [PubMed] [Google Scholar]
- 26.Linder A, Fjell C, Levin A, Walley KR, Russell JA, Boyd JH. Small acute increases in serum creatinine are associated with decreased long-term survival in the critically ill. Am J Respir Crit Care Med 189: 1075–1081, 2014. doi: 10.1164/rccm.201311-2097OC. [DOI] [PubMed] [Google Scholar]
- 27.Lisi L, Ciotti GM, Braun D, Kalinin S, Currò D, Dello Russo C, Coli A, Mangiola A, Anile C, Feinstein DL, Navarra P. Expression of iNOS, CD163 and ARG-1 taken as M1 and M2 markers of microglial polarization in human glioblastoma and the surrounding normal parenchyma. Neurosci Lett 645: 106–112, 2017. doi: 10.1016/j.neulet.2017.02.076. [DOI] [PubMed] [Google Scholar]
- 28.Liu L, Shi GP. CD31: beyond a marker for endothelial cells. Cardiovasc Res 94: 3–5, 2012. doi: 10.1093/cvr/cvs108. [DOI] [PubMed] [Google Scholar]
- 29.Marini JC, Keller B, Didelija IC, Castillo L, Lee B. Enteral arginase II provides ornithine for citrulline synthesis. Am J Physiol Endocrinol Metab 300: E188–E194, 2011. doi: 10.1152/ajpendo.00413.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meng XM, Nikolic-Paterson DJ, Lan HY. Inflammatory processes in renal fibrosis. Nat Rev Nephrol 10: 493–503, 2014. doi: 10.1038/nrneph.2014.114. [DOI] [PubMed] [Google Scholar]
- 31.Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of cisplatin nephrotoxicity. Toxins (Basel) 2: 2490–2518, 2010. doi: 10.3390/toxins2112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris SM Jr, Bhamidipati D, Kepka-Lenhart D. Human type II arginase: sequence analysis and tissue-specific expression. Gene 193: 157–161, 1997. doi: 10.1016/S0378-1119(97)00099-1. [DOI] [PubMed] [Google Scholar]
- 33.Noto A, Cibecchini F, Fanos V, Mussap M. NGAL and metabolomics: the single biomarker to reveal the metabolome alterations in kidney injury. BioMed Res Int 2013: 612032, 2013. doi: 10.1155/2013/612032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. BioMed Res Int 2014: 967826, 2014. doi: 10.1155/2014/967826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 73: 994–1007, 2008. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- 36.Palant CE, Amdur RL, Chawla LS. The acute kidney injury to chronic kidney disease transition: a potential opportunity to improve care in acute kidney injury. Contrib Nephrol 187: 55–72, 2016. doi: 10.1159/000442365. [DOI] [PubMed] [Google Scholar]
- 37.Pan B, Liu G, Jiang Z, Zheng D. Regulation of renal fibrosis by macrophage polarization. Cell Physiol Biochem 35: 1062–1069, 2015. doi: 10.1159/000373932. [DOI] [PubMed] [Google Scholar]
- 38.Pecoits-Filho R, Lindholm B, Axelsson J, Stenvinkel P. Update on interleukin-6 and its role in chronic renal failure. Nephrol Dial Transplant 18: 1042–1045, 2003. doi: 10.1093/ndt/gfg111. [DOI] [PubMed] [Google Scholar]
- 39.Ramesh G, Kimball SR, Jefferson LS, Reeves WB. Endotoxin and cisplatin synergistically stimulate TNF-alpha production by renal epithelial cells. Am J Physiol Renal Physiol 292: F812–F819, 2007. doi: 10.1152/ajprenal.00277.2006. [DOI] [PubMed] [Google Scholar]
- 40.Ramesh G, Reeves WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest 110: 835–842, 2002. doi: 10.1172/JCI200215606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ravichandran K, Wang Q, Ozkok A, Jani A, Li H, He Z, Ljubanovic D, Weiser-Evans MC, Nemenoff RA, Edelstein CL. CD4 T cell knockout does not protect against kidney injury and worsens cancer. J Mol Med (Berl) 94: 443–455, 2016. doi: 10.1007/s00109-015-1366-z. [DOI] [PubMed] [Google Scholar]
- 42.Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest 118: 3522–3530, 2008. doi: 10.1172/JCI36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ricci Z, Cruz D, Ronco C. The RIFLE classification for acute kidney injury definition. Am J Surg 198: 152–153, 2009. doi: 10.1016/j.amjsurg.2008.06.033. [DOI] [PubMed] [Google Scholar]
- 44.Robbins ME. Single injection techniques in determining age-related changes in porcine renal function. Int J Appl Radiat Isot 35: 85–91, 1984. doi: 10.1016/0020-708X(84)90189-3. [DOI] [PubMed] [Google Scholar]
- 45.Robbins ME, Bywaters TB, Jaenke RS, Hopewell JW, Matheson LM, Tothill P, Whitehouse E. Long-term studies of cisplatin-induced reductions in porcine renal functional reserve. Cancer Chemother Pharmacol 29: 309–315, 1992. doi: 10.1007/BF00685950. [DOI] [PubMed] [Google Scholar]
- 46.Schiffrin EL. Role of endothelin-1 in hypertension and vascular disease. Am J Hypertens 14: 83S–89S, 2001. doi: 10.1016/S0895-7061(01)02074-X. [DOI] [PubMed] [Google Scholar]
- 47.Sharp CN, Doll MA, Dupre TV, Shah PP, Subathra M, Siow D, Arteel GE, Megyesi J, Beverly LJ, Siskind LJ. Repeated administration of low-dose cisplatin in mice induces fibrosis. Am J Physiol Renal Physiol 310: F560–F568, 2016. doi: 10.1152/ajprenal.00512.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharp CN, Siskind LJ. Developing better mouse models to study cisplatin-induced kidney injury. Am J Physiol Renal Physiol 313: F835–F841, 2017. doi: 10.1152/ajprenal.00285.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H, Hainzl A, Schatz S, Qi Y, Schlecht A, Weiss JM, Wlaschek M, Sunderkötter C, Scharffetter-Kochanek K. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest 121: 985–997, 2011. doi: 10.1172/JCI44490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Skinner R, Parry A, Price L, Cole M, Craft AW, Pearson AD. Persistent nephrotoxicity during 10-year follow-up after cisplatin or carboplatin treatment in childhood: relevance of age and dose as risk factors. Eur J Cancer 45: 3213–3219, 2009. doi: 10.1016/j.ejca.2009.06.032. [DOI] [PubMed] [Google Scholar]
- 51.Skrypnyk NI, Siskind LJ, Faubel S, de Caestecker MP. Bridging translation for acute kidney injury with better preclinical modeling of human disease. Am J Physiol Renal Physiol 310: F972–F984, 2016. doi: 10.1152/ajprenal.00552.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Slocum JL, Heung M, Pennathur S. Marking renal injury: can we move beyond serum creatinine? Transl Res 159: 277–289, 2012. doi: 10.1016/j.trsl.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Su H, Lei CT, Zhang C. Interleukin-6 signaling pathway and its role in kidney disease: an update. Front Immunol 8: 405, 2017. doi: 10.3389/fimmu.2017.00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sugimoto H, Grahovac G, Zeisberg M, Kalluri R. Renal fibrosis and glomerulosclerosis in a new mouse model of diabetic nephropathy and its regression by bone morphogenic protein-7 and advanced glycation end product inhibitors. Diabetes 56: 1825–1833, 2007. doi: 10.2337/db06-1226. [DOI] [PubMed] [Google Scholar]
- 55.Swindle MM, Moody DC, Phillips LD. Swine As Models in Biomedical Research. Ames, IA: Iowa State University Press, 1992. [Google Scholar]
- 56.Tan HL, Yap JQ, Qian Q. Acute kidney injury: tubular markers and risk for chronic kidney disease and end-stage kidney failure. Blood Purif 41: 144–150, 2016. doi: 10.1159/000441269. [DOI] [PubMed] [Google Scholar]
- 57.Tang J, Liu N, Tolbert E, Ponnusamy M, Ma L, Gong R, Bayliss G, Yan H, Zhuang S. Sustained activation of EGFR triggers renal fibrogenesis after acute kidney injury. Am J Pathol 183: 160–172, 2013. doi: 10.1016/j.ajpath.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thakar CV, Christianson A, Himmelfarb J, Leonard AC. Acute kidney injury episodes and chronic kidney disease risk in diabetes mellitus. Clin J Am Soc Nephrol 6: 2567–2572, 2011. doi: 10.2215/CJN.01120211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Torres R, Velazquez H, Chang JJ, Levene MJ, Moeckel G, Desir GV, Safirstein R. Three-dimensional morphology by multiphoton microscopy with clearing in a model of cisplatin-induced CKD. J Am Soc Nephrol 27: 1102–1112, 2016. doi: 10.1681/ASN.2015010079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Van Biesen W, Vanholder R, Lameire N. Defining acute renal failure: RIFLE and beyond. Clin J Am Soc Nephrol 1: 1314–1319, 2006. doi: 10.2215/CJN.02070606. [DOI] [PubMed] [Google Scholar]
- 61.Wald R, Quinn RR, Adhikari NK, Burns KE, Friedrich JO, Garg AX, Harel Z, Hladunewich MA, Luo J, Mamdani M, Perl J, Ray JG; University of Toronto Acute Kidney Injury Research Group . Risk of chronic dialysis and death following acute kidney injury. Am J Med 125: 585–593, 2012. doi: 10.1016/j.amjmed.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 62.Wald R, Quinn RR, Luo J, Li P, Scales DC, Mamdani MM, Ray JG; University of Toronto Acute Kidney Injury Research Group . Chronic dialysis and death among survivors of acute kidney injury requiring dialysis. JAMA 302: 1179–1185, 2009. doi: 10.1001/jama.2009.1322. [DOI] [PubMed] [Google Scholar]
- 63.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543, 2010. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang W, Wang W, Yu H, Zhang Y, Dai Y, Ning C, Tao L, Sun H, Kellems RE, Blackburn MR, Xia Y. Interleukin 6 underlies angiotensin II-induced hypertension and chronic renal damage. Hypertension 59: 136–144, 2012. doi: 10.1161/HYPERTENSIONAHA.111.173328. [DOI] [PMC free article] [PubMed] [Google Scholar]