

Abstract
Carbonic anhydrase IX (CA IX) is highly expressed in rapidly proliferating and highly glycolytic cells, where it serves to enhance acid-regulatory capacity. Pulmonary microvascular endothelial cells (PMVECs) actively utilize aerobic glycolysis and acidify media, whereas pulmonary arterial endothelial cells (PAECs) primarily rely on oxidative phosphorylation and minimally change media pH. Therefore, we hypothesized that CA IX is critical to PMVEC angiogenesis because of its important role in regulating pH. To test this hypothesis, PMVECs and PAECs were isolated from Sprague-Dawley rats. CA IX knockout PMVECs were generated using the CRISPR-Cas9 technique. During serum-stimulated growth, mild acidosis (pH 6.8) did not affect cell counts of PMVECs, but it decreased PAEC cell number. Severe acidosis (pH 6.2) decreased cell counts of PMVECs and elicited an even more pronounced reduction of PAECs. PMVECs had a higher CA IX expression compared with PAECs. CA activity was higher in PMVECs compared with PAECs, and enzyme activity was dependent on the type IX isoform. Pharmacological inhibition and genetic ablation of CA IX caused profound dysregulation of extra- and intracellular pH in PMVECs. Matrigel assays revealed impaired angiogenesis of CA IX knockout PMVECs in acidosis. Lastly, pharmacological CA IX inhibition caused profound cell death in PMVECs, whereas genetic CA IX ablation had little effect on PMVEC cell death in acidosis. Thus CA IX controls PMVEC pH necessary for angiogenesis during acidosis. CA IX may contribute to lung vascular repair during acute lung injury that is accompanied by acidosis within the microenvironment.
Keywords: heterogeneity, Matrigel, network formation, pulmonary circulation, SLC-0111
INTRODUCTION
Acidosis is common among critically ill patients and is often associated with poor outcomes (30). A study of 9,799 patients in the ICU showed that the mortality rate of patients with metabolic acidosis is nearly twice as high as that of patients without metabolic acidosis (21). Although in vivo and ex vivo studies have shown detrimental effects of acidosis on vascular and cardiac function, clinical data demonstrating direct effects of protons on target cells are lacking because of multiple confounding conditions that often accompany acid base abnormalities in critically ill patients (31). Furthermore, present therapeutic options, including sodium bicarbonate, Tris-hydroxymethyl-aminomethane, carbicarb, and renal replacement therapy, have failed to improve major outcomes (46a, 47). Therefore, in selected clinical situations, such as in acute respiratory distress syndrome when lung-protective ventilation with low tidal volume is critical to prevent hyperinflation, a moderate degree of acidosis is permitted (3). However, it remains uncertain as to whether acidosis plays any role in short- and long-term pulmonary disease pathophysiology. Thus there needs to be a better understanding of the mechanisms of acidosis-induced lung injury, with potential development of effective therapeutic strategies.
Although severe acidosis is generally associated with poor outcomes, there are subpopulations of patients with relatively better prognosis in a context-dependent manner. For example, in a retrospective observational study of 2,156 patients, 77 patients presented with extreme acidosis, as defined by a pH of <7.0. The mortality rate in these patients was 100% in cases of hypoxic cardiac arrest, whereas it was 0% in cases of diabetic ketoacidosis (1). Such variability in acidosis-related consequences is also observed at a cellular level. Disturbance of acid base homeostasis is generally considered to be harmful for normal cellular function (33). In infectious and inflammatory lung conditions, such as pneumonia and acute respiratory distress syndrome (ARDS), which can accompany systemic and microenvironmental acidosis, acidosis exacerbates lung injury by suppressing neutrophil phagocytosis (40) and IL-1-dependent inflammatory responses (54, 55). However, acidosis can be helpful in selected pathological conditions. During ischemia reperfusion, acidosis is protective for recovering myocytes (27) and neuronal cells (16), providing a rationale for the therapeutic strategy referred to as ischemic postconditioning. Acidosis is also helpful for cancer cell growth (18). At an early stage of growth, cancer cells are exposed to a poorly vascularized hypoxic environment. Therefore, cells that develop highly proliferative and glycolytic phenotypes are selected to survive, which in turn induces microenvironmental acidosis resulting from an enhanced metabolic phenotype with a higher degree of acid production (20). This increased acid production is overcome by an enhanced proton-buffering capacity, which enables them to preserve intracellular pH while acidifying the extracellular environment.
The bicarbonate buffer system is an important mechanism that cells utilize to enhance buffering capacity. Carbonic anhydrases (CAs; EC 4.2.1.1.) are a family of ubiquitous enzymes that contribute to the bicarbonate buffer system by catalyzing the reversible conversion of proton and bicarbonate to carbon dioxide and water (29). Among five distinct classes, the α-class is found in humans, and it is further subclassified into 15 isoforms (17). CA IX is the most active isoform catalyzing the carbon dioxide hydration reaction (23) and is highly expressed in rapidly growing and highly glycolytic cells that are exposed to hypoxic and acidic conditions, such as cancer cells (19). Its expression generally correlates with cancer invasiveness, metastasis, and poor disease outcomes (26). Studies are ongoing to test it as a therapeutic target, with selected monoclonal antibody and small molecule inhibitors presently in clinical use or in clinical trials (50). Although CA IX has been extensively studied in cancer cells, no studies have shown whether CA IX has any role in normal acid base regulation or disease pathophysiology in nontransformed pulmonary endothelial cells.
We have previously reported that pulmonary endothelial cells display heterogeneity in their metabolic profiles (43). Typically, pulmonary microvascular endothelial cells (PMVECs) are highly glycolytic and routinely acidify media, whereas pulmonary arterial endothelial cells (PAECs) primarily utilize oxidative phosphorylation and keep their media pH neutral. Endothelial cells of extrapulmonary vascular origin have a similar metabolic pattern to that of PMVECs, and the pattern mimics metabolic characteristics of cancer cells in terms of active aerobic glycolysis (56). It is not known whether acidosis is helpful or harmful for endothelial cell angiogenesis and whether PMVECs may have a mechanism to protect their intracellular pH. Considering the similarity of metabolic patterns between PMVECs and cancer cells, we hypothesized that CA IX, a known critical regulator of pH and angiogenesis of cancer cells, plays an important role in pH regulation and angiogenesis during acidosis of nontransformed PMVECs.
MATERIALS AND METHODS
Isolation of rat lung endothelial cells.
Procedures for isolation of rat endothelial cells were approved by the University of South Alabama Institutional Animal Care and Use Committee. PMVECs and PAECs were isolated from male Sprague-Dawley rats as previously described (52).
Growth curves.
Cells were seeded at a density of 1.0 × 105 cells per well in six-well plates in DMEM (Thermo Fischer, Grand Island, NY), 10% FCS, and 1% penicillin-streptomycin at 37°C in room air, 5% CO2. The next day media was replaced with bicarbonate-free media (Thermo Fischer) with different buffer and pH 30 mM HEPES with pH 7.4 or 30 mM 2-(N-morpholino)ethanesulfonic acid (MES) with pH 6.8, 6.6, or 6.2, and grown for 7 days with daily media change at 37°C in room air, 0% CO2. Cells were photographed, trypsinized, and counted every 24 h.
Media and reagent pH measurement.
The pH was measured in the medium or reagents using a pH meter (Denver Instrument, Bohemia, NY).
Western blot analysis.
Cells were seeded at 2.0 × 105 cells/well on six-well plates on bicarbonate buffered media. Three days after cell seeding, media was changed to either HEPES-buffered pH 7.4, or MES-buffered pH 6.8, 6.6, or 6.2 media. Twenty-four hours later, cells were collected and subjected to immunoblot analysis as previously described (43). Primary antibodies from BioScience Slovakia (Bratislava, Slovakia) and Cell Signaling (Danvers, MA) were used for CA IX (M75, 1: 800 dilution) and GAPDH (1: 1,000 dilution), respectively.
In vitro CA activity.
Cells were grown to confluence in 100-mm dishes, rinsed with PBS, and scraped into bicarbonate-free pH 8.0 DMEM. Cells were loaded on 24-well plates at 5.0 × 105 cells/well, pretreated with DMSO, acetazolamide (AZ) (Sigma, St. Louis, MO), or CA IX-specific inhibitor, SLC-0111 (gift from Dr. Supuran, Italy) for 15 min. Maximally CO2 saturated bicarbonate-free DMEM with pH 5.0 was added at equal amounts per well with continuous pH measurement.
Intracellular pH measurement.
Cells were seeded on glass coverslips and grown to confluence for 3 days. Media was changed to HEPES-buffered pH 7.4 or MES-buffered pH 6.8 media, and cells were treated with DMSO, AZ, or SLC-0111. Two days later, cells were rinsed with HBSS and incubated in 1 µM of BCECF-AM (Thermo Fischer) for 15 min. After two rinses with HBSS, coverslips were transferred into the Attofluor chamber (Thermo Fischer). With the use of a Nikon 1X70 fluorescence microscope system with dual excitation (440 and 490 nm) and single emission (535 nm) wavelengths and Felix fluorescence analysis software (Photon Technology International, Birmingham, NJ), ratiometric measurement of the fluorescence of three to five fields with ~5 cells/field per coverslip was performed. For calibration, 10 µM nigericin sodium (Adipogen, San Diego, CA) with pH 6.0 to 8.0 solution containing 120 mM KCl, 2 mM CaCl2·2H2O, 1 mM MgCl2, and 10 mM glucose in 20 mM HEPES (or MES) was used.
Generation of CA IX-depleted cells.
CA IX knockout (KO) PMVECs were generated using CRISPR-Cas9 gene-editing technology as previously described (4). To knock out the gene that encodes CA IX in PMVECs, two constructs, which express single-guide RNAs (sgRNAs) directed against sequences CGCCTTAGAGGACCTACCGA and GAGAGGCGGATCCACCCGGT, were generated and cotransfected into PMVECs with plasmids pX330-U6-Chimeric_BB-CBh-hSpCas9 (42230; Addgene, Cambridge, MA), pExodus CMV.Trex2 (40210; Addgene), and pEF1aEGFP. Forty-eight hours after transfection, cells were subjected to fluorescence-activated cell sorting, and enhanced green fluorescent protein (EGFP)-positive cells were plated at 500, 1,500, and 3,000 cells/150-mm dish. One hundred forty-four resulting colonies were picked, expanded in 24-well plates, and screened by PCR to identify clones with putative indels. Two primer mixes were used for screening, each consisting of two primers flanking the targeted site (CATGCTCAAGTTTGCCTTCTAC+ CTGTCCACTCCTCAGTTCTATTT) and one primer specific for targeted site (GGAGGATTCCCTCGCCT or GAGGCGGATCCACCCGG). Wild-type clones are expected to produce two PCR products, 1,081 bp and either 349 bp or 293 bp. Putative clones with indels were identified by disappearance of a smaller PCR product. CA IX inactivation in these clones was further verified by Western blotting with M75 antibody (BioScience Slovakia).
Cytotoxicity assay.
Cells were seeded at 5.0 × 105 cells/well on six-well plates on bicarbonate-buffered media. The next day, cells were treated with DMSO (0.15%), AZ (150 µM), or SLC-0111 (150 µM). Three days after the inhibitor treatment, lactate dehydrogenase (LDH) activity was measured on culture media using Cytotoxicity Detection Assay Kit (Roche Diagnostics, Mannheim, Germany) using manufacturer protocols.
Matrigel network formation assay.
Matrigel (356231; Corning, Corning, NY) was thawed overnight at 4°C. On the day of the experiment, Matrigel was loaded (30 µl per well) in 96-well plates, while the plates were kept on top of ice. The plates were incubated at 37°C with 5% CO2-room air for 1 h. Cells were trypsinized from 10-cm dishes when ~70% confluent and seeded at cell-type-specific optimal densities (4.0–8.0 × 104 cells per well), with a total cell solution volume of 80 µl per well. Cells were incubated at 37°C with 5% CO2-room air for 24 h. Pictures were taken at 24 h. Images were analyzed using the “Angiogenesis Analyzer” tool, programmed in ImageJs macro language.
Statistics.
One- and two-way ANOVA, Student’s t-tests, and Bonferroni post hoc were used, as appropriate. Significance was denoted as P < 0.05.
RESULTS
PMVECs are more resistant to acidosis-induced population growth inhibition compared with PAECs.
We have previously demonstrated that PMVECs preferentially utilize aerobic glycolysis and acidify the media, whereas PAECs utilize oxidative phosphorylation and maintain neutral media pH (34, 43). Whether acidosis affects PMVEC growth or whether PMVECs have a resistance mechanism against acidosis is unknown. To address this issue, we performed growth curves on PMVECs and PAECs over a proton concentration range. PMVEC counts were not affected by relatively mild acidosis of pH 6.8 but were decreased by pH 6.6 and 6.2 (Fig. 1, A and B; pH 7.4 vs. 6.6 and 6.2, P < 0.05). PAEC counts were decreased at the mildest degree of acidosis, pH 6.8, with a further concentration-dependent decrease at pH 6.6 and 6.2 (Fig. 1, C and D; pH 7.4 vs. 6.8, 6.6, and 6.2, P < 0.05). In pH 6.2, PMVEC counts slowly increased and plateaued by day 7, whereas PAEC counts decreased starting at day 5; there were only a few cells viable by day 7. These data suggest that acidosis has differential effects on cell population growth of PMVECs and PAECs.
Fig. 1.

Pulmonary microvascular endothelial cells (PMVECs) survive better than do pulmonary arterial endothelial cells (PAECs) in acidosis. PMVECs and PAECs were seeded at 1.0 × 105 cells/well on 6-well plates on bicarbonate-buffered media. 1 day after cell seeding, media was changed to either HEPES-buffered pH 7.4 or 2-(N-morpholino)ethanesulfonic acid (MES)-buffered pH 6.8, 6.6, or 6.2 media. Cells were imaged and counted with daily media change. A and B: PMVECs grew to confluence and stayed healthy for 7 days in pH 7.4, 6.8, and 6.6, but the cells underwent growth arrest in pH 6.2. C and D: PAECs grew to confluence and stayed healthy for 7 days in pH 7.4 but showed visible cell count decrease in pH 6.8, 6.6, and 6.2 in a dose-dependent manner. Data represent means ± SD. 2-way ANOVA and Bonferroni post hoc tests were used to compare between different pH groups. For each cell type and pH group, at least 5 separate experiments were performed. *Significant difference (P < 0.05) in 7.4 vs. 6.2 groups. #Significant difference (P < 0.05) in 7.4 vs. 6.6 groups. ^Significant difference (P < 0.05) in 7.4 vs. 6.8 groups.
PMVECs have a higher CA IX expression compared with PAECs, and acidic media decreases CA IX expression.
We hypothesized that PMVECs have higher expression of critical enzymes for acid base regulation, which confers a population growth benefit in an acid environment. CA IX is an isoform of CA, which is a critical constituent of the bicarbonate buffer system. Although CA IX is highly relevant in pathological conditions, such as cancer and pulmonary arterial hypertension (59), its expression and function in normal pulmonary endothelial cells is unknown. We grew PMVECs and PAECs to confluence and exposed them to different degrees of acidosis, pH 7.4 vs. 6.8 vs. 6.2, for 1 day. Western blot analysis of cell lysates revealed baseline expression of CA IX in PMVECs but not in PAECs (Fig. 2B). Acidic media decreased CA IX expression in a concentration-dependent manner in PMVECs (Fig. 2B; PMVECs pH 7.4 vs. 6.2, P < 0.05). Thus CA IX expression parallels cell counts comparing pH 7.4 vs. 6.2 within PMVEC groups.
Fig. 2.
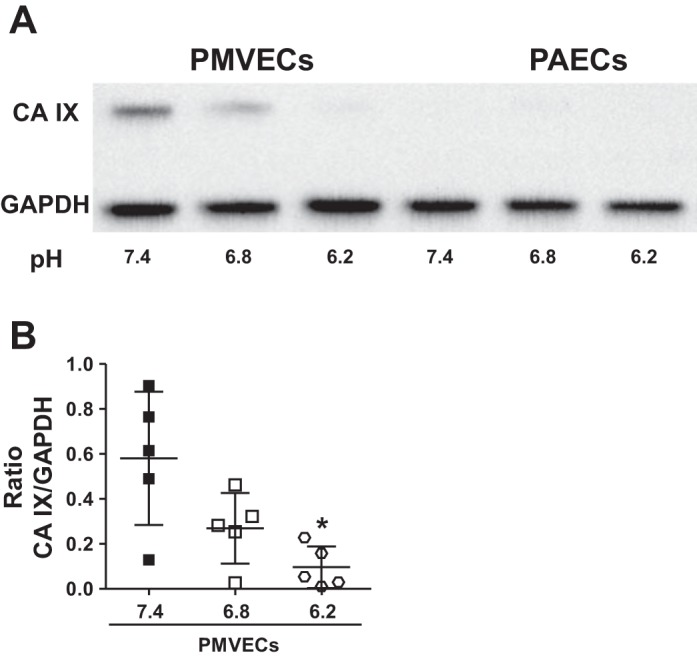
Pulmonary microvascular endothelial cells (PMVECs) have a higher carbonic anhydrase IX (CA IX) expression than do pulmonary arterial endothelial cells (PAECs), and acidic media decreases CA IX expression in both PMVECs and PAECs. PMVECs and PAECs were seeded at 2.0 × 105 cells/well on 6-well plates on bicarbonate-buffered media. 3 days after cell seeding, media was changed to either HEPES-buffered pH 7.4 or 2-(N-morpholino)ethanesulfonic acid (MES)-buffered pH 6.8, 6.6, or 6.2 media. 24 h later, whole cell lysates were collected, and Western blotting (A) was performed to assess protein abundance. CA IX expression was higher in PMVECs compared with PAECs, and it was decreased by acidic media. Data represent means ± SD. 5 separate experiments were performed. 1-way ANOVA was used to compare different pH groups within each cell type (B). *Significant difference (P < 0.05) from pH 7.4 group for each cell type.
CA IX is an important determinant of extracellular pH regulation in PMVECs.
We have demonstrated that CA IX expression parallels cell counts of PMVECs in an acidic environment. To investigate whether CA IX contributes to PMVEC acid tolerance, we first measured extracellular CA activity. PMVECs and PAECs were suspended in minimally buffered bicarbonate-free media and loaded on 24-well plates with equal cell numbers. Maximally saturated CO2 solution was added with continuous pH monitoring of the media. Upon addition of CO2-saturated solution, pH immediately dropped, indicating CO2 conversion to proton. The speed and degree of the drop in pH correlate with CA function of the cells (9). In PMVECs, there was a significant acceleration and augmentation of the pH decrease following CO2 challenge, indicating increased CA function (Fig. 3A; no cells vs. control, P < 0.05). In contrast, in PAECs, there was no change in either the rate or the magnitude of decreased pH (Fig. 3B; no cells vs. control, P = ns). To confirm that these pH changes are mediated by CA activity, cells were pretreated with either AZ (nonspecific CA inhibitor) or SLC-0111 (specific CA IX inhibitor) for 15 min before CO2 challenge. Both AZ and SLC-0111 impaired the PMVEC response to CO2 load (Fig. 3A; no cells vs. AZ vs. SLC-0111, P < 0.05), suggesting that CA IX mediated the higher buffering capacity of PMVECs. Next, to confirm that the inhibitory effect of SLC-0111 on PMVEC extracellular pH regulation is CA IX specific, we generated CA IX KO PMVECs using CRISPR-Cas9 gene-editing technique (Fig. 3C). CA IX deletion was verified by RT-PCR and immunoblot (Fig. 3D). Figure 3E shows a loss of extracellular CA function in five individual CA IX KO PMVEC colonies (wild-type vs. CA IX KO, P < 0.05), confirming that CA IX is critical to extracellular pH regulation in PMVECs.
Fig. 3.

Carbonic anhydrase IX (CA IX) is an important determinant of extracellular pH regulation in pulmonary microvascular endothelial cells (PMVECs). PMVECs (A), pulmonary arterial endothelial cells (PAECs) (B), and wild-type (E) and CA IX knockout (KO) PMVECs were grown to confluence in 100-mm dishes. Cells were scraped in bicarbonate-free media with adjusted pH of 8.0. Total volume of media per dish was proportional to total cell counts. Cell solution with equal cell concentration was loaded on 24-well plates. A and B: PMVECs and PAECs were pretreated with DMSO (0.15%), acetazolamide (AZ) (150 µM), or SLC-0111 (150 µM) for 15 min in room temperature. A, B, and E: maximally saturated CO2 solution was added to each well while pH changes were continuously recorded. For clarity of data, values measured at 20 s are separately presented as bar graphs on the bottom of each graph depicting continuous pH monitoring. CA IX mediated accelerated CO2 conversion to proton in PMVECs. Data represent means + SD. 5 or more separate experiments were performed. 1-way ANOVA was used to compare between groups. *Significant difference (P < 0.05) from No cell group. #Significant difference (P < 0.05) from wild-type PMVECs. C: sgRNAs guide Cas9 nucleases to target sequences to make double-strand breaks and indels in CA IX gene. D: 5 representative wild-type and CA IX KO clones were subjected to PCR and Western blot (WB). Primers target to produce 2 PCR products for wild-type and 1 PCR product for CA IX KO clones. WB of whole cell lysate shows successful deletion of CA IX gene in PMVECs.
CA IX is critical to intracellular pH regulation in PMVECs.
Intracellular pH homeostasis is critical for normal cellular function. Therefore, cells have multiple mechanisms to protect intracellular pH (6). CA IX is extensively studied in cancer cells for its important role in intracellular pH regulation with a subsequent survival benefit (9). Earlier we have shown that PMVECs have higher CA IX expression than do PAECs and that it mediates an enhanced extracellular buffering capacity. To investigate whether CA IX protects intracellular pH, we measured intracellular pH in PMVECs using the pH-sensitive fluorescent dye BCECF-AM. PMVECs were grown to confluence and incubated in different pH media while being treated with CA inhibitors for 3 days. This experiment allows another way of testing cellular buffer capacity by increasing proton load over a relatively longer time period, compared with experiments in Fig. 3, where we challenged cells with an acute and excessive CO2 load. pH 6.8 did not reduce PMVEC intracellular pH (Fig. 4A; pH 7.4 DMSO vs. pH 6.8 DMSO, P = ns). However, upon SLC-0111 treatment, intracellular pH profoundly dropped in both pH 7.4 and 6.8 groups (Fig. 4A; DMSO vs. SLC-0111, P < 0.05). In addition, SLC-0111 treatment decreased extracellular pH in both pH 7.4 and 6.8 groups (Fig. 4B; DMSO vs. SLC-0111, P < 0.05), with an additional significant decrease in the pH 6.8 group compared with the pH 7.4 group (Fig. 4B; pH 7.4 SLC-0111 vs. pH 6.8 SLC-0111, P < 0.05). When CA IX KO PMVECs were challenged with pH 6.2 media for 3 days, intracellular pH dropped more significantly compared with wild-type (Fig. 4C; wild-type vs. CA IX KO, P < 0.05). These findings suggest that CA IX plays a critical role in intra- and extracellular pH regulation in PMVECs.
Fig. 4.
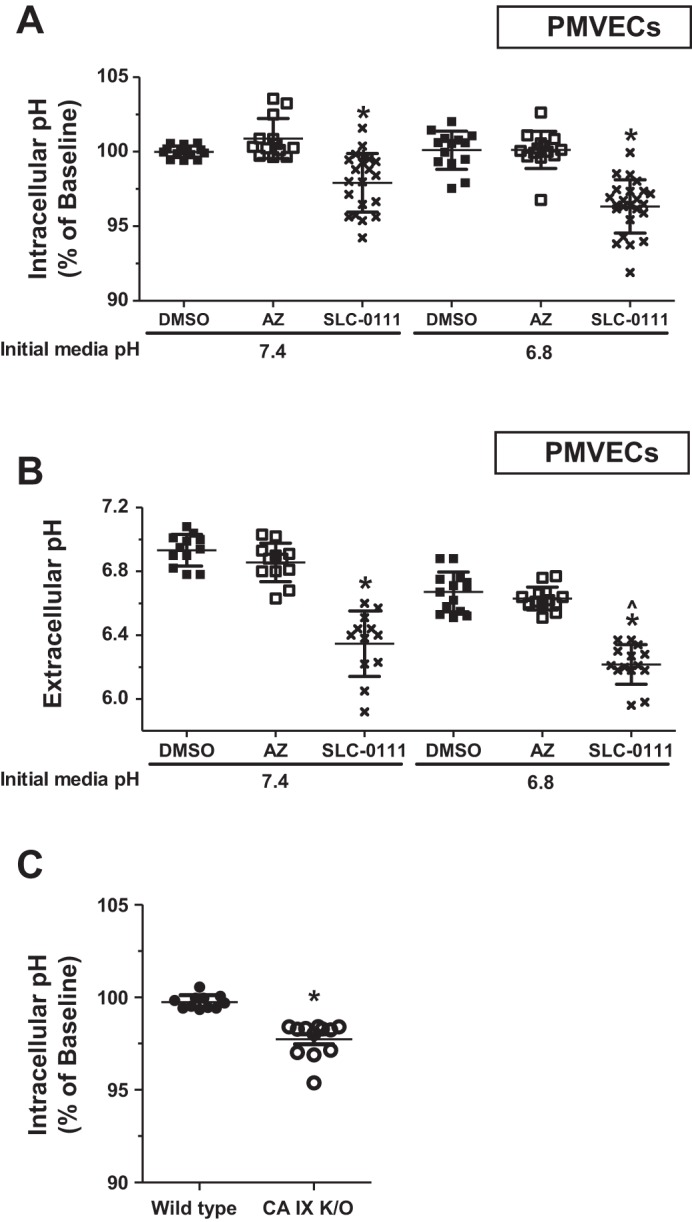
Carbonic anhydrase IX (CA IX) is critical to intracellular pH regulation in pulmonary microvascular endothelial cells (PMVECs). A and B: PMVECs were seeded at 2.0 × 105 cells/well on 6-well plates with glass coverslips on bicarbonate-buffered media and grown to confluence for 3 days. Media was changed to HEPES-buffered pH 7.4 or 2-(N-morpholino)ethanesulfonic acid (MES)-buffered pH 6.8 media, and cells were treated with DMSO (0.15%), acetazolamide (AZ) (150 µM), or SLC-0111 (150 µM). 2 days after inhibitor treatment, intracellular (A) and extracellular (B) pH was measured by pH-sensitive fluorescent dye BCECF-AM and pH meter, respectively. C: wild-type and CA IX knockout (KO) PMVECs were grown to confluence for 3 days as described above, and media was changed to MES-buffered pH 6.2 media. 3 days after media change, intracellular pH was measured by BCECF-AM. Intracellular pH is reported as percentage of baseline [pH 7.4 DMSO (A) and wild-type (C)]. Either chemical or genetic CA IX inhibition decreased intracellular pH in PMVECs. Data represent means ± SD. 5 or more separate experiments were performed. 1-way ANOVA was used to compare between groups. *Significant difference (P < 0.05) from DMSO (A and B) or wild-type (C) groups. ^Significant difference (P < 0.05) from pH 7.4 SLC-0111 group.
CA IX activity controls PMVEC angiogenesis.
Our group has previously demonstrated that PMVECs preferentially utilize aerobic glycolysis, which causes extracellular acidosis (44). Data so far suggest that acidosis is detrimental to pulmonary endothelial cell population growth and that PMVECs utilize CA IX as a protective mechanism to maintain intra- and extracellular pH homeostasis. To further investigate whether CA IX and its pH regulatory function contribute to angiogenesis in acidic conditions, we performed a Matrigel network formation assay with pH 6.2 media. Compared with our previous study where we assessed angiogenic capacity of PMVECs in pH 7.4 media (34), the number of junctions and branches were lower in both wild-type and KO PMVECs in pH 6.2 media (Fig. 5A). Compared with wild-type, CA IX KO PMVECs formed significantly fewer junctions and branches in pH 6.2 media (Fig. 5A; wild-type vs. CA IX KO, P < 0.05), suggesting a critical role of CA IX in angiogenesis during acidosis.
Fig. 5.

Carbonic anhydrase IX (CA IX) activity controls pulmonary microvascular endothelial cell (PMVEC) angiogenesis. A: wild-type and CA IX knockout (KO) PMVECs were seeded on Matrigel-coated 96-well plates at 4.0 × 104 cells/well in pH 6.2 media. Images were obtained 24 h after seeding at ×10 magnification. Representative images are shown for each group. Networks were quantified by ImageJ software. Mean values of wild-type PMVEC network quantification in pH 7.4 media from our previous study (34) are depicted with dashed lines for reference. Compared with the reference data, acidic media decreased network formation in PMVECs and more so in CA IX KO. B–E: PMVECs and pulmonary arterial endothelial cells (PAECs) were seeded at 5.0 × 105 cells/well on 6-well plates on bicarbonate-buffered media at pH 7.4. The next day, cells were treated with DMSO (0.15%), acetazolamide (AZ) (150 µM), or SLC-0111 (150 µM). 3 days after the inhibitor treatment, images were obtained, cells were counted, and lactate dehydrogenase (LDH) release and extracellular pH were measured. SLC-0111 causes significant PMVEC (B) cytotoxicity, whereas it has minimal effect on PAECs. SLC-0111 decreased cell counts (C), increased LDH release (D), and decreased extracellular pH in PMVECs (E), whereas it had no effect on PAECs. Data represent means ± SD. 5 or more separate experiments were performed. Unpaired 2-tailed t-test was used to compare wild-type vs. CA IX KO. 1-way ANOVA was used to compare DMSO vs. AZ vs. SLC-0111 groups. ^Significant difference (P < 0.05) from wild-type PMVECs. *Significant difference (P < 0.05) from PMVEC DMSO group.
To investigate whether CA IX is critical to PMVEC survival, we assessed PMVEC and PAEC counts and LDH release with and without CA inhibition in bicarbonate-buffered media. Three days after inhibitor treatment, AZ had no effect on cell survival, but SLC-0111 caused significant PMVEC monolayer disruption (Fig. 5B), decreased cell counts (Fig. 5C; DMSO vs. SLC-0111, P < 0.05), and increased LDH release (Fig. 5D; DMSO vs. SLC-0111, P < 0.05). Note the heterogeneity of response, (e.g., cell number and LDH) in PMVECs treated with SLC-0111. In the PMVEC SLC-0111 group, four individual data points are distinguishable from others in the same group by relatively higher cell counts and low LDH (Fig. 5D). The variability reflects cytotoxicity progression; given more time, all cells within the well died. In contrast, PAEC growth was not affected by either AZ or SLC-0111 (Fig. 5, B–D). Consistent with our earlier findings, there was a significant decrease in extracellular pH following SLC-0111 treatment in PMVECs (Fig. 5E; DMSO vs. SLC-0111, P < 0.05), which also parallels cell death. Although chemical inhibition of CA IX by SLC-0111 caused profound cell death in wild-type PMVECs, there was no statistical difference in cell death between wild-type and CA IX KO PMVECs when they were grown in acidic media for 3 days. Therefore, our findings suggest that CA IX is critical to PMVEC angiogenesis during acidosis and may contribute to survival in a context-dependent manner (Fig. 6).
Fig. 6.
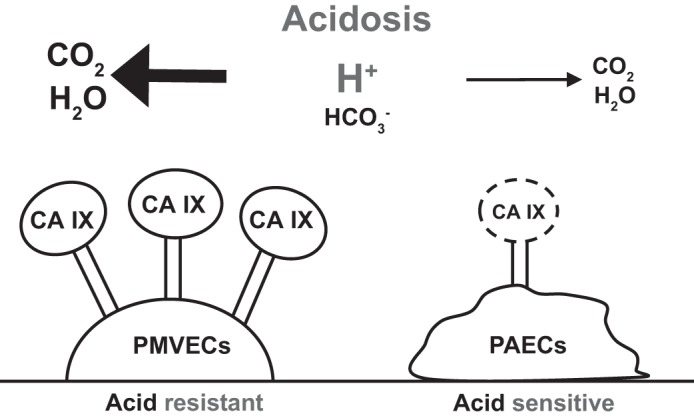
Pulmonary microvascular endothelial cells (PMVECs) are more acid resistant than are pulmonary arterial endothelial cells (PAECs) because of higher carbonic anhydrase IX (CA IX) expression and enhanced buffering capacity. PMVECs have higher CA IX expression than do PAECs; thus they more efficiently convert protons to carbon dioxide, relieving acid load. Our study suggests that CA IX is a potential mechanism of PMVEC acid resistance. Dashed lines represent low or no expression of CA IX.
DISCUSSION
In this study, we have investigated mechanisms involved in heterogeneity of pulmonary endothelial cell population growth in an acidic environment. We have demonstrated that PMVECs are more resistant to acidosis-induced growth inhibition compared with PAECs. CA IX expression is higher in PMVECs compared with PAECs, and acidosis decreases CA IX expression in a concentration-dependent manner. CA IX expression also parallels PMVEC cell counts in acidosis. CA IX inhibition in PMVECs resulted in dysregulation of extra- and intracellular pH. CA IX KO impaired angiogenic capacity in an acidic environment, and chemical inhibition of CA IX caused profound cell death in PMVECs. Our data suggest that CA IX regulates PMVEC pH necessary for angiogenesis during acidosis.
Acidosis has context-dependent effects on cellular function. Infection and inflammation often results in microenvironmental acidosis, which impairs bacterial clearance, enhances proinflammatory responses, and causes apoptosis. For example, in the porcine cystic fibrosis lung, acidic airway surface pH decreases bacterial killing (45), and, in mouse glial cells, acidosis drives damage-associated molecular pattern-induced interleukin-1β secretion independently of the inflammasome (14). Acidosis induces phosphatidylinositol 3-kinase/Akt and ERK pathway-mediated human neutrophil activation (37), and it upregulates NF-κB important for activation of inducible nitric oxide synthase in macrophages contained within inflammatory lesions (5). Acidosis induces activation of cardiac myocyte Bcl-2 family protein BNIP3 (33), neural cell calcium-permeable acid-sensing ion channels (58), and human vascular endothelial cell endoplasmic reticulum stress pathways (12), which all result in apoptosis. Our data show that acidosis inhibits growth of both PMVECs and PAECs in a dose-dependent manner, but the pH threshold responsible for this significant inhibitory effect is lower in PMVECs. In addition, in pH 6.2 media where growth of both cell types was suppressed, PMVEC counts continued to rise until day 7, but PAEC counts peaked at day 3 and dropped thereafter. To understand the mechanisms involved in the higher acid resistance of PMVECs compared with PAECs, we investigated the function of CA IX.
CA IX was originally identified in HeLa cells in 1986 (41) as a transmembrane zinc-containing metalloenzyme with an extracellular active catalytic site. The active enzyme is a dimeric structure stabilized by disulfide bonds (2). CA IX KO mice develop normally early in their lives, except for the presence of gastric epithelial hyperplasia and cysts (22). By 8 to 10 mo, these mice develop vacuolar degenerative changes in the brain (42). In normal tissue, CA IX is most highly expressed in the epithelium of the gastrointestinal tract, where cells are routinely exposed to dynamic pH changes (36). Hypoxia upregulates CA IX expression (35) along with other genes related to pH regulation, angiogenesis, and glucose and iron metabolism (17). High cell density (32) and DNA methylation (39) are other factors that increase CA IX expression. Studies have reported variable degrees of CA IX expression in normal lung tissue, possibly related to the various antibodies used and methods of detection. Simi et al. (51) reported positive baseline CA IX mRNA and protein expression in normal human lung tissue using M75, a mouse monoclonal antibody directed against the proteoglycan-like domain of CA IX. We have used the same antibody and detected abundant CA IX expression in rat PMVECs but not in PAECs. This finding led us to hypothesize that CA IX contributes to the acid tolerance of PMVECs. Indeed, CA functional assays supported the ideas that CA IX regulates pH in PMVECs. Unlike reported data showing an inverse correlation between pH and CA IX expression levels in HeLa cells (25), glioblastoma cells (46), and B cell lymphoma cell line models (8), our PMVECs showed a dose-dependent positive correlation between pH and CA IX expression (R2 = 0.54 ± 0.2, P = 0.02). This correlation may reflect the gradually saturating and failing global buffering system of PMVECs, considering their subsequent growth arrest occurring at pH 6.2, which seems to be below their compensation threshold.
Intracellular pH is tightly regulated by multiple mechanisms because small changes in intracellular pH can cause significant alterations in enzymatic activity, membrane excitability, metabolism, proliferation, and other cellular functions (6). The bicarbonate buffer system catalyzed by CAs is an important determinant of buffering capacity that prevents rapid fluctuation of pH (48). Although the buffering system is highly efficient, it is finite when the sources of extra acid or base load persist. Therefore, various transporters are utilized for sustained regulatory effects, including alkali cation-H+ exchangers such as Na+-H+ exchangers and Na+-H+ antiporters, lactate-H+ cotransporters such as monocarboxylate transporters, bicarbonate transporters such as Na+-coupled transporters (NBCs), and acid-loading transporters such as Cl− exchange anion exchangers (AEs) (6). CA IX physically and functionally interacts with many of these transporters, including NBCs and AEs, forming a transport metabolon, a membrane protein complex involved in regulation of bicarbonate metabolism and transport (38). In our study, CA IX inhibition resulted in profound dysregulation of extra- and intracellular pH and impaired network formation in acidic environments in PMVECs. These results strongly suggest that CA IX is critical to PMVEC angiogenesis during acidosis by regulating acid base homeostasis. Interestingly, nonspecific inhibition of CA with AZ had little effect on intracellular pH or cell survival, but its inhibitory effect on extracellular pH regulation was still significant. We speculate three potential mechanisms: 1) the inhibitory effect of AZ is not as potent as SLC-0111, 2) balance between extra- and intracellular CA isoforms is a more important determinant of global pH regulation than absolute quantity of each isoform, and 3) a CA IX-specific proteoglycan-like domain that is known to play an important role in maintaining enzymatic function under extreme acidosis (11) may be more preferentially inhibited by SLC-0111 than by AZ.
Acidosis upregulates vascular endothelial growth factor (VEGF) in various human cancer cells and is thought to contribute to angiogenesis (49). Studies assessing whether CA IX plays any role in nontransformed endothelial cell angiogenesis are lacking. Here, acidosis and CA IX deletion additively impaired PMVEC network formation. To our knowledge, this is first evidence that CA IX is a critical determinant of angiogenic potential in PMVECs during acidosis. In cancer cells and renal epithelial cells, CA IX increases cell migration by interacting with bicarbonate transporters in lamellipodia (53). In human umbilical vein endothelial cells (HUVECs), acidosis abolishes VEGF-induced AKT pathway activation, enhanced proliferation, and survival (15). Acidosis also activates G protein-coupled receptor 4 (GPR4) and enhances HUVEC-monocyte adhesion through the cAMP/Epac pathway (7). Angiostatin is an interesting molecule that enhances intracellular acidosis and anoikis in normal endothelial cells under acidic conditions (57). These studies offer relevant insights on our future studies to elucidate mechanisms involved in CA IX-mediated PMVEC angiogenesis during acidosis.
Whether or not CA IX contributes to PMVEC survival remains uncertain. Although SLC-0111 induced dose-dependent cell death in PMVECs, genetic deletion of CA IX did not similarly cause cytotoxicity. We consider two possibilities for an increased cell death by chemical CA IX inhibition compared with genetic inhibition: 1) SLC-0111 has off-target effects on other CA isoforms, and 2) SLC-0111-induced accelerated glycolysis (data not shown), in conjunction with rapid loss of CA IX (faster rate of CA IX level change compared with KO), generates a more acute acid imbalance than does the global genetic deletion. Further studies are needed to rigorously elucidate these mechanisms.
Evidence is abundant that organs undergoing infection or inflammation develop local acidosis even before overwhelming systemic acidosis ensues (45). When measured by microelectrode or noninvasive imaging approaches, local tissue pH commonly ranges from 6.0 to 7.0 in the microenvironment of ischemic and inflammatory tissues (12, 24). The cause of local acidosis is thought to be dysregulated cell metabolism with enhanced glycolysis and/or defective blood perfusion to remove acidic metabolic byproducts (13). GPR4 mediates acidosis-induced endoplasmic reticulum stress and cell death in human vascular endothelial cells (12), but detailed effects of acidosis on endothelial cells and the mechanisms of their response to acidosis are still poorly understood. CA IX expression is increased in pulmonary arterial hypertension, but whether it is involved in disease pathophysiology has never been reported (10). Our data suggest that CA IX may play an important role in endothelial cell function and survival in normal and variable pathological conditions that accompany microenvironmental acidosis and hypoxia.
In summary, we report differential acid sensitivity among PMVECs and PAECs. CA IX expression is higher in PMVECs and decreases with extracellular acidosis, which correlates with cell population growth. CA IX is critical to PMVECs angiogenesis during acidosis by regulating extra- and intracellular pH. These findings may be especially relevant to understanding the pathophysiology of acidosis-related lung injury, such as in infection and inflammation, which often accompany microenvironmental acidosis.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-66299 and HL-60024 (T. Stevens).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.Y.L., M.A., and T.S. conceived and designed research; J.Y.L., M.A., N.K., V.P., and R.W. performed experiments; J.Y.L., M.A., and T.S. analyzed data; J.Y.L., M.A., and T.S. interpreted results of experiments; J.Y.L. and T.S. prepared figures; J.Y.L., M.A., and T.S. drafted manuscript; J.Y.L., M.A., and T.S. edited and revised manuscript; J.Y.L. and T.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Drs. Sarah Sayner and Chung-Sik Choi for assistance with intracellular pH measurement and Drs. Claudiu T. Supuran and Fabrizio Carta, University of Florence, for kindly offering SLC-0111.
REFERENCES
- 1.Allyn J, Vandroux D, Jabot J, Brulliard C, Galliot R, Tabatchnik X, Combe P, Martinet O, Allou N. Prognosis of patients presenting extreme acidosis (pH <7) on admission to intensive care unit. J Crit Care 31: 243–248, 2016. doi: 10.1016/j.jcrc.2015.09.025. [DOI] [PubMed] [Google Scholar]
- 2.Alterio V, Hilvo M, Di Fiore A, Supuran CT, Pan P, Parkkila S, Scaloni A, Pastorek J, Pastorekova S, Pedone C, Scozzafava A, Monti SM, De Simone G. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc Natl Acad Sci USA 106: 16233–16238, 2009. doi: 10.1073/pnas.0908301106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amato MBP, Barbas CSV, Medeiros DM, Magaldi RB, Schettino GP, Lorenzi-Filho G, Kairalla RA, Deheinzelin D, Munoz C, Oliveira R, Takagaki TY, Carvalho CR. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med 338: 347–354, 1998. doi: 10.1056/NEJM199802053380602. [DOI] [PubMed] [Google Scholar]
- 4.Balczon R, Morrow KA, Zhou C, Edmonds B, Alexeyev M, Pittet JF, Wagener BM, Moser SA, Leavesley S, Zha X, Frank DW, Stevens T. Pseudomonas aeruginosa infection liberates transmissible, cytotoxic prion amyloids. FASEB J 31: 2785–2796, 2017. doi: 10.1096/fj.201601042RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellocq A, Suberville S, Philippe C, Bertrand F, Perez J, Fouqueray B, Cherqui G, Baud L. Low environmental pH is responsible for the induction of nitric-oxide synthase in macrophages. Evidence for involvement of nuclear factor-kappaB activation. J Biol Chem 273: 5086–5092, 1998. doi: 10.1074/jbc.273.9.5086. [DOI] [PubMed] [Google Scholar]
- 6.Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol 11: 50–61, 2010. doi: 10.1038/nrm2820. [DOI] [PubMed] [Google Scholar]
- 7.Chen A, Dong L, Leffler NR, Asch AS, Witte ON, Yang LV. Activation of GPR4 by acidosis increases endothelial cell adhesion through the cAMP/Epac pathway. PLoS One 6: e27586, 2011. doi: 10.1371/journal.pone.0027586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen LQ, Howison CM, Spier C, Stopeck AT, Malm SW, Pagel MD, Baker AF. Assessment of carbonic anhydrase IX expression and extracellular pH in B-cell lymphoma cell line models. Leuk Lymphoma 56: 1432–1439, 2015. doi: 10.3109/10428194.2014.933218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiche J, Ilc K, Laferrière J, Trottier E, Dayan F, Mazure NM, Brahimi-Horn MC, Pouysségur J. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res 69: 358–368, 2009. doi: 10.1158/0008-5472.CAN-08-2470. [DOI] [PubMed] [Google Scholar]
- 10.Cotroneo E, Ashek A, Wang L, Wharton J, Dubois OD, Bozorgi SS, Busbridge M, Alavian KN, Wilkins MR, and Zhao L. Iron homeostasis and pulmonary hypertension: iron deficiency leads to pulmonary vascular remodeling in the rat. Circ Res 116: 1680–1690, 2015. doi: 10.1161/CIRCRESAHA.116.305265. [DOI] [PubMed] [Google Scholar]
- 11.De Simone G, Supuran CT. Carbonic anhydrase IX: biochemical and crystallographic characterization of a novel antitumor target. Biochim Biophys Acta 1804: 404–409, 2010. doi: 10.1016/j.bbapap.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 12.Dong L, Krewson EA, Yang LV. Acidosis activates endoplasmic reticulum stress pathways through GPR4 in human vascular endothelial cells. Int J Mol Sci 18: 278, 2017. doi: 10.3390/ijms18020278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong L, Li Z, Leffler NR, Asch AS, Chi J-T, Yang LV. Acidosis activation of the proton-sensing GPR4 receptor stimulates vascular endothelial cell inflammatory responses revealed by transcriptome analysis. PLoS One 8: e61991, 2013. doi: 10.1371/journal.pone.0061991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edye ME, Lopez-Castejon G, Allan SM, Brough D. Acidosis drives damage-associated molecular pattern (DAMP)-induced interleukin-1 secretion via a caspase-1-independent pathway. J Biol Chem 288: 30485–30494, 2013. doi: 10.1074/jbc.M113.478941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faes S, Uldry E, Planche A, Santoro T, Pythoud C, Demartines N, Dormond O. Acidic pH reduces VEGF-mediated endothelial cell responses by downregulation of VEGFR-2; relevance for anti-angiogenic therapies. Oncotarget 7: 86026–86038, 2016. doi: 10.18632/oncotarget.13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan Y-Y, Shen Z, He P, Jiang L, Hou WW, Shen Y, Zhang X-N, Hu W-W, Chen Z. A novel neuroprotective strategy for ischemic stroke: transient mild acidosis treatment by CO2 inhalation at reperfusion. J Cereb Blood Flow Metab 34: 275–283, 2014. doi: 10.1038/jcbfm.2013.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frost SC, McKenna R. Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications. Berlin, Germany: Springer Science & Business Media, 2013. [Google Scholar]
- 18.Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer 8: 56–61, 2008. doi: 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- 19.Giatromanolaki A, Koukourakis MI, Sivridis E, Pastorek J, Wykoff CC, Gatter KC, Harris AL. Expression of hypoxia-inducible carbonic anhydrase-9 relates to angiogenic pathways and independently to poor outcome in non-small cell lung cancer. Cancer Res 61: 7992–7998, 2001. [PubMed] [Google Scholar]
- 20.Gillies RJ, Gatenby RA. Metabolism and its sequelae in cancer evolution and therapy. Cancer J 21: 88–96, 2015. doi: 10.1097/PPO.0000000000000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gunnerson KJ, Saul M, He S, Kellum JA. Lactate versus non-lactate metabolic acidosis: a retrospective outcome evaluation of critically ill patients. Crit Care 10: R22, 2006. doi: 10.1186/cc3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gut MO, Parkkila S, Vernerová Z, Rohde E, Závada J, Höcker M, Pastorek J, Karttunen T, Gibadulinová A, Závadová Z, Knobeloch KP, Wiedenmann B, Svoboda J, Horak I, Pastoreková S. Gastric hyperplasia in mice with targeted disruption of the carbonic anhydrase gene Car9. Gastroenterology 123: 1889–1903, 2002. doi: 10.1053/gast.2002.37052. [DOI] [PubMed] [Google Scholar]
- 23.Hilvo M, Baranauskiene L, Salzano AM, Scaloni A, Matulis D, Innocenti A, Scozzafava A, Monti SM, Di Fiore A, De Simone G, Lindfors M, Jänis J, Valjakka J, Pastoreková S, Pastorek J, Kulomaa MS, Nordlund HR, Supuran CT, Parkkila S. Biochemical characterization of CA IX, one of the most active carbonic anhydrase isozymes. J Biol Chem 283: 27799–27809, 2008. doi: 10.1074/jbc.M800938200. [DOI] [PubMed] [Google Scholar]
- 24.Huang Y, McNamara JO. Ischemic stroke: “acidotoxicity” is a perpetrator. Cell 118: 665–666, 2004. doi: 10.1016/j.cell.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Ihnatko R, Kubes M, Takacova M, Sedlakova O, Sedlak J, Pastorek J, Kopacek J, Pastorekova S. Extracellular acidosis elevates carbonic anhydrase IX in human glioblastoma cells via transcriptional modulation that does not depend on hypoxia. Int J Oncol 29: 1025–1033, 2006. [PubMed] [Google Scholar]
- 26.Ilie M, Mazure NM, Hofman V, Ammadi RE, Ortholan C, Bonnetaud C, Havet K, Venissac N, Mograbi B, Mouroux J, Pouysségur J, Hofman P. High levels of carbonic anhydrase IX in tumour tissue and plasma are biomarkers of poor prognostic in patients with non-small cell lung cancer. Br J Cancer 102: 1627–1635, 2010. doi: 10.1038/sj.bjc.6605690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inserte J, Ruiz-Meana M, Rodríguez-Sinovas A, Barba I, Garcia-Dorado D. Contribution of delayed intracellular pH recovery to ischemic postconditioning protection. Antioxid Redox Signal 14: 923–939, 2011. doi: 10.1089/ars.2010.3312. [DOI] [PubMed] [Google Scholar]
- 29.Kaluz S, Kaluzová M, Liao SY, Lerman M, Stanbridge EJ. Transcriptional control of the tumor-and hypoxia-marker carbonic anhydrase 9: a one transcription factor (HIF-1) show? Biochim Biophys Acta 1795: 162–172, 2009. doi: 10.1016/j.bbcan.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kellum JA. Metabolic acidosis in patients with sepsis: epiphenomenon or part of the pathophysiology? Crit Care Res 6: 197–203, 2004. [PubMed] [Google Scholar]
- 31.Kimmoun A, Novy E, Auchet T, Ducrocq N, Levy B. Hemodynamic consequences of severe lactic acidosis in shock states: from bench to bedside. Crit Care 19: 175, 2015. doi: 10.1186/s13054-015-0896-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kopacek J, Barathova M, Dequiedt F, Sepelakova J, Kettmann R, Pastorek J, Pastorekova S. MAPK pathway contributes to density-and hypoxia-induced expression of the tumor-associated carbonic anhydrase IX. Biochim Biophys Acta 1729: 41–49, 2005. doi: 10.1016/j.bbaexp.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 33.Kubasiak LA, Hernandez OM, Bishopric NH, Webster KA. Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc Natl Acad Sci USA 99: 12825–12830, 2002. doi: 10.1073/pnas.202474099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JY, McMurtry SA, Stevens T. Single cell cloning generates lung endothelial colonies with conserved growth, angiogenic, and bioenergetic characteristics. Pulm Circ 7: 777–792, 2017. doi: 10.1177/2045893217731295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loncaster JA, Harris AL, Davidson SE, Logue JP, Hunter RD, Wycoff CC, Pastorek J, Ratcliffe PJ, Stratford IJ, West CM. Carbonic anhydrase (CA IX) expression, a potential new intrinsic marker of hypoxia: correlations with tumor oxygen measurements and prognosis in locally advanced carcinoma of the cervix. Cancer Res 61: 6394–6399, 2001. [PubMed] [Google Scholar]
- 36.Luong-Player A, Liu H, Wang HL, Lin F. Immunohistochemical reevaluation of carbonic anhydrase IX (CA IX) expression in tumors and normal tissues. Am J Clin Pathol 141: 219–225, 2014. doi: 10.1309/AJCPVJDS28KNYZLD. [DOI] [PubMed] [Google Scholar]
- 37.Martínez D, Vermeulen M, Trevani A, Ceballos A, Sabatté J, Gamberale R, Álvarez ME, Salamone G, Tanos T, Coso OA, Geffner J. Extracellular acidosis induces neutrophil activation by a mechanism dependent on activation of phosphatidylinositol 3-kinase/Akt and ERK pathways. J Immunol 176: 1163–1171, 2006. doi: 10.4049/jimmunol.176.2.1163. [DOI] [PubMed] [Google Scholar]
- 38.Morgan PE, Pastoreková S, Stuart-Tilley AK, Alper SL, Casey JR. Interactions of transmembrane carbonic anhydrase, CAIX, with bicarbonate transporters. Am J Physiol Cell Physiol 293: C738–C748, 2007. doi: 10.1152/ajpcell.00157.2007. [DOI] [PubMed] [Google Scholar]
- 39.Nakamura J, Kitajima Y, Kai K, Hashiguchi K, Hiraki M, Noshiro H, Miyazaki K. Expression of hypoxic marker CA IX is regulated by site-specific DNA methylation and is associated with the histology of gastric cancer. Am J Pathol 178: 515–524, 2011. doi: 10.1016/j.ajpath.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Croinin DF, Nichol AD, Hopkins N, Boylan J, O’Brien S, O’Connor C, Laffey JG, McLoughlin P. Sustained hypercapnic acidosis during pulmonary infection increases bacterial load and worsens lung injury. Crit Care Med 36: 2128–2135, 2008. doi: 10.1097/CCM.0b013e31817d1b59. [DOI] [PubMed] [Google Scholar]
- 41.Oosterwijk E, Ruiter DJ, Hoedemaeker PJ, Pauwels EK, Jonas U, Zwartendijk J, Warnaar SO. Monoclonal antibody G 250 recognizes a determinant present in renal-cell carcinoma and absent from normal kidney. Int J Cancer 38: 489–494, 1986. doi: 10.1002/ijc.2910380406. [DOI] [PubMed] [Google Scholar]
- 42.Pan PW, Parkkila AK, Autio S, Hilvo M, Sormunen R, Pastorekova S, Pastorek J, Haapasalo H, Parkkila S. Brain phenotype of carbonic anhydrase IX-deficient mice. Transgenic Res 21: 163–176, 2012. doi: 10.1007/s11248-011-9520-z. [DOI] [PubMed] [Google Scholar]
- 43.Parra-Bonilla G, Alvarez DF, Al-Mehdi AB, Alexeyev M, Stevens T. Critical role for lactate dehydrogenase A in aerobic glycolysis that sustains pulmonary microvascular endothelial cell proliferation. Am J Physiol Lung Cell Mol Physiol 299: L513–L522, 2010. doi: 10.1152/ajplung.00274.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parra-Bonilla G, Alvarez DF, Alexeyev M, Vasauskas A, Stevens T. Lactate dehydrogenase expression is necessary to sustain rapid angiogenesis of pulmonary microvascular endothelium. PLoS One 8: e75984, 2013. doi: 10.1371/journal.pone.0075984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pezzulo AA, Tang XX, Hoegger MJ, Abou Alaiwa MH, Ramachandran S, Moninger TO, Karp PH, Wohlford-Lenane CL, Haagsman HP, van Eijk M, Bánfi B, Horswill AR, Stoltz DA, McCray PB Jr, Welsh MJ, Zabner J. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 487: 109–113, 2012. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rafajová M, Zatovicová M, Kettmann R, Pastorek J, Pastoreková S. Induction by hypoxia combined with low glucose or low bicarbonate and high posttranslational stability upon reoxygenation contribute to carbonic anhydrase IX expression in cancer cells. Int J Oncol 24: 995–1004, 2004. [PubMed] [Google Scholar]
- 46a.RENAL Replacement Therapy Study Investigators; Bellomo R, Cass A, Cole L, Finfer S, Gallagher M, Lo S, McArthur C, McGuinness S, Myburgh J, Norton R, Scheinkestel C, Su S. Intensity of continuous renal-replacement therapy in critically ill patients. N Engl J Med 361: 1627–1638, 2009. doi: 10.1056/NEJMoa0902413. [DOI] [PubMed] [Google Scholar]
- 47.Rhee KH, Toro LO, McDonald GG, Nunnally RL, Levin DL. Carbicarb, sodium bicarbonate, and sodium chloride in hypoxic lactic acidosis. Effect on arterial blood gases, lactate concentrations, hemodynamic variables, and myocardial intracellular pH. Chest 104: 913–918, 1993. doi: 10.1378/chest.104.3.913. [DOI] [PubMed] [Google Scholar]
- 48.Roos A, Boron WF. Intracellular pH. Physiol Rev 61: 296–434, 1981. doi: 10.1152/physrev.1981.61.2.296. [DOI] [PubMed] [Google Scholar]
- 49.Shi Q, Le X, Wang B, Abbruzzese JL, Xiong Q, He Y, Xie K. Regulation of vascular endothelial growth factor expression by acidosis in human cancer cells. Oncogene 20: 3751–3756, 2001. doi: 10.1038/sj.onc.1204500. [DOI] [PubMed] [Google Scholar]
- 50.Siebels M, Rohrmann K, Oberneder R, Stahler M, Haseke N, Beck J, Hofmann R, Kindler M, Kloepfer P, Stief C. A clinical phase I/II trial with the monoclonal antibody cG250 (RENCAREX®) and interferon-alpha-2a in metastatic renal cell carcinoma patients. World J Urol 29: 121–126, 2011. doi: 10.1007/s00345-010-0570-2. [DOI] [PubMed] [Google Scholar]
- 51.Simi L, Venturini G, Malentacchi F, Gelmini S, Andreani M, Janni A, Pastorekova S, Supuran CT, Pazzagli M, Orlando C. Quantitative analysis of carbonic anhydrase IX mRNA in human non-small cell lung cancer. Lung Cancer 52: 59–66, 2006. doi: 10.1016/j.lungcan.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 52.Stevens T, Creighton J, Thompson WJ. Control of cAMP in lung endothelial cell phenotypes. Implications for control of barrier function. Am J Physiol Lung Physiol 277: L119–L126, 1999. [DOI] [PubMed] [Google Scholar]
- 53.Svastova E, Witarski W, Csaderova L, Kosik I, Skvarkova L, Hulikova A, Zatovicova M, Barathova M, Kopacek J, Pastorek J, Pastorekova S. Carbonic anhydrase IX interacts with bicarbonate transporters in lamellipodia and increases cell migration via its catalytic domain. J Biol Chem 287: 3392–3402, 2012. doi: 10.1074/jbc.M111.286062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Torres IM, Demirdjian S, Vargas J, Goodale BC, Berwin B. Acidosis increases the susceptibility of respiratory epithelial cells to Pseudomonas aeruginosa-induced cytotoxicity. Am J Physiol Lung Cell Mol Physiol 313: L126–L137, 2017. doi: 10.1152/ajplung.00524.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Torres IM, Patankar YR, Berwin B. Acidosis exacerbates in vivo IL-1-dependent inflammatory responses and neutrophil recruitment during pulmonary Pseudomonas aeruginosa infection. Am J Physiol Lung Cell Mol Physiol 314: L225–L235, 2018. doi: 10.1152/ajplung.00338.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Verdegem D, Moens S, Stapor P, Carmeliet P. Endothelial cell metabolism: parallels and divergences with cancer cell metabolism. Cancer Metab 2: 19, 2014. doi: 10.1186/2049-3002-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wahl ML, Owen CS, Grant DS. Angiostatin induces intracellular acidosis and anoikis in endothelial cells at a tumor-like low pH. Endothelium 9: 205–216, 2002. doi: 10.1080/10623320213633. [DOI] [PubMed] [Google Scholar]
- 58.Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell 118: 687–698, 2004. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 59.Zhao L, Oliver E, Maratou K, Atanur SS, Dubois OD, Cotroneo E, Chen CN, Wang L, Arce C, Chabosseau PL, Ponsa-Cobas J, Frid MG, Moyon B, Webster Z, Aldashev A, Ferrer J, Rutter GA, Stenmark KR, Aitman TJ, Wilkins MR. The zinc transporter ZIP12 regulates the pulmonary vascular response to chronic hypoxia. Nature 524: 356–360, 2015. doi: 10.1038/nature14620. [DOI] [PMC free article] [PubMed] [Google Scholar]