

Abstract
Evidence is accumulating that exposure to cigarette smoke (CS) increases the risk of developing acute respiratory distress syndrome (ARDS). Streptococcus pneumoniae is the most common cause of bacterial pneumonia, which in turn is the leading cause of ARDS. Chronic smokers have increased rates of pneumococcal colonization and develop more severe pneumococcal pneumonia than nonsmokers; yet mechanistic connections between CS exposure, bacterial pneumonia, and ARDS pathogenesis remain relatively unexplored. We exposed mice to 3 wk of moderate whole body CS or air, followed by intranasal inoculation with an invasive serotype of S. pneumoniae. CS exposure alone caused no detectable lung injury or bronchoalveolar lavage (BAL) inflammation. During pneumococcal infection, CS-exposed mice had greater survival than air-exposed mice, in association with reduced systemic spread of bacteria from the lungs. However, when mice were treated with antibiotics after infection to improve clinical relevance, the survival benefit was lost, and CS-exposed mice had more pulmonary edema, increased numbers of BAL monocytes, and elevated monocyte and lymphocyte chemokines. CS-exposed antibiotic-treated mice also had higher serum surfactant protein D and angiopoietin-2, consistent with more severe lung epithelial and endothelial injury. The results indicate that acute CS exposure enhances the recruitment of immune cells to the lung during bacterial pneumonia, an effect that may provide microbiological benefit but simultaneously exposes the mice to more severe inflammatory lung injury. The inclusion of antibiotic treatment in preclinical studies of acute lung injury in bacterial pneumonia may enhance clinical relevance, particularly for future studies of current or emerging tobacco products.
Keywords: acute lung injury, ARDS, cigarette smoke, pneumococcus, pneumonia
INTRODUCTION
Acute respiratory distress syndrome (ARDS) affects nearly 200,000 patients each year with high associated morbidity and mortality (64). While chronic exposure to cigarette smoke (CS) is a well-established causal factor in chronic obstructive pulmonary disease (COPD) and malignancy, there is increasing evidence of the substantial risks of both active and passive CS exposure on acute pulmonary disease, including ARDS. CS has now been associated with an increased risk of ARDS in the setting of trauma, transfusion, and nonpulmonary sepsis (10, 11, 75), as well as with primary graft dysfunction (pulmonary edema) after lung transplantation (18). In addition, lungs from cigarette smokers that were studied ex vivo have increased edema and reduced alveolar fluid clearance (84). Similarly, our research group recently reported that healthy human smokers exposed to nebulized lipopolysaccharide (LPS) have increased inflammatory cytokines and protein in bronchoalveolar lavage (BAL) compared with nonsmokers (48).
The most common etiology of ARDS is pneumonia, and the most frequent responsible pathogen in bacterial pneumonia is Streptococcus pneumoniae (53). CS exposure increases the incidence of pneumococcal pneumonia in patients (80) and the risk that it will be complicated by septic shock (25) and mortality (49). However, CS has also been shown to increase the risk of pneumococcal nasopharyngeal colonization (8, 13), and chronic CS exposure leading to COPD or emphysema reduces overall health and structural lung defenses against infection. Thus, it remains unclear how CS exposures limited in length and intensity may affect the natural history of pneumococcal pneumonia and ARDS.
Long-term (exceeding 6 mo) heavy CS exposure in mice causes robust inflammation involving both innate and adaptive immune cells and produces alveolar destruction reminiscent of emphysema (38). Much less is known about how shorter-term exposures to more moderate levels of CS affect the severity of lung injury in response to acute infectious insults. Given the rapidly evolving landscape of tobacco products, including e-cigarettes (14), there is a compelling need to develop improved models for testing the impact of both established and novel tobacco products on acute pulmonary complications, including ARDS. We recently reported that intratracheal LPS caused more severe neutrophilic lung injury in CS exposed mice compared with controls without CS exposure (29), analogous to our studies in human volunteers (48). Studies of CS exposure and pneumococcal infection in mice have yielded mixed results, with some researchers reporting that smoke exposure increases illness severity (70) and others reporting the opposite (5). Notably, these discrepant results may have reflected differences in the intensity of CS exposure or strain differences in response to CS itself (90). Importantly, mouse and rat models of cigarette smoke exposure followed by challenge with live bacterial pathogens have lacked antibiotic treatment (5, 7, 19, 26, 27, 36, 42, 52, 59, 70, 73, 79, 81), a cornerstone of the care of patients presenting to medical care with suspected infection (31).
For these studies, our initial objective was to test the effect of a limited and well-tolerated CS exposure on lung injury and mortality in mice during pneumococcal lung infection. We hypothesized that CS-exposed mice would have more severe lung injury and a higher mortality from pneumococcal pneumonia. Contrary to our hypothesis, CS exposed mice had improved survival, primarily related to a reduction in the extrapulmonary dissemination of bacteria from the lungs. Therefore, our second objective was to test the effect of CS in antibiotic-treated mice with pneumococcal pneumonia, reasoning that there was more clinical relevance to include antibiotic therapy in these experiments, particularly since there is an increased emphasis on identifying patients at risk for developing ARDS when they present with pneumonia in the emergency department (45). A final objective of this work was to use our refined model to identify biomarkers that may be useful in evaluating the acute pulmonary toxicity of novel tobacco products.
MATERIALS AND METHODS
Animals.
Adult (8- to 10-wk old) female C57BL/6 mice were ordered from NCI (Frederick, MD), housed in pathogen-free housing, and cared for in accord with National Institutes of Health guidelines by the Laboratory Animal Resource Center of the University of California, San Francisco (UCSF). All experiments were conducted under protocols approved by the UCSF Institutional Animal Care and Use Committee. Group size was determined to ensure adequate statistical power based on our extensive experience with models of acute lung injury (21, 29, 39).
Smoke exposure.
Mice were exposed to smoke generated by a Teague TE-10 smoking machine using 3R4F cigarettes (47). The LPS content of 3R4F Kentucky research cigarettes is in the middle of the range of 11 types of commercially available cigarettes tested at 9 pmol/mg (37). Following 5 days of acclimation to increasing smoke concentrations of 20, 40, 60, 80, and 100 mg/m3 total suspended particulates (TSP) for 2 h a day, mice underwent 12 days of exposure to 100 mg/m3 for 5 h a day, with rest on weekends. Control mice were housed in the same room within the barrier facility but not exposed to smoke. This CS exposure was designed to model the recent initiation of active smoking. In some experiments, mice were exposed for 2 h daily to a lower CS concentration meant to mimic second-hand smoke exposure, TSP 3 mg/m3. This lower CS concentration was achieved by mixing concentrated sidestream smoke and fresh air into an aging chamber using an adjustable air amplifier and continuous monitoring of suspended particulate matter with a Sidepack AM 510 aerosol monitor (TSI, Shoreview, MN). For context, a smoke-filled bar may reach TSP 1–2 mg/m3 (71) whereas mouse models of CS exposure have used levels as high as 980 mg/m3 (27).
Bacterial infection, antibiotic administration, and microbiology.
S. pneumoniae serotype 19F (49619; ATCC, Manassas, VA), was grown in brain-heart broth (237500; Becton-Dickinson, Sparks, MD) and harvested at midlog phase, spun down, and resuspended in PBS at different dilutions. Mice were anesthetized deeply with isoflurane and inoculated intranasally with 50 µl of bacteria. In some experiments, ceftriaxone (150 mg/kg ip) was administered every 12 h beginning 12 h after inoculation. This dose was selected on the basis of known pharmacokinetics and proven efficacy in a mouse model of pneumococcal pneumonia (68). In other experiments, S. pneumoniae was delivered by intraperitoneal injection. Bacterial titers of BAL, blood, and spleen minced in 5 ml of PBS were measured by serial dilution and plaque counting on sheep blood agar plates.
Oxygenation measurements during the experiments.
Pulse oximetry was measured using the MouseOx+ cervical collar system (Starr Life Sciences), as we have done in prior studies (29). The mean SpO2 during 5 min of recording was calculated for each time point.
Lung injury end points.
Mice underwent overdose of ketamine and xylazine, bilateral thoracotomy, and exsanguination by right ventricular puncture. The lungs were removed and homogenized in 1 ml of PBS, and samples of blood, lung homogenate, and homogenate supernatant were weighed before and after desiccation. Systemic hemoglobin and hematocrit were measured with a Hemavet 950 cell counter (Drew Scientific, Waterbury, CT). Another fraction of homogenate was assayed for hemoglobin concentration, and the blood volume of the lung was calculated, permitting assessment of the excess extravascular lung water (i.e., pulmonary edema in the interstitial and air spaces above the level in normal mice of the same size), as in prior work (30, 72). In other animals, after exsanguination the trachea was cannulated, and the lungs were lavaged twice with 250 µl of PBS. BAL cell count was measured with a Coulter counter, cytospin preparations of BAL fluid were made and stained with Hema 3 solution (ThermoFisher Scientific, Waltham, MA), and 400 cells/mouse were analyzed at ×100 magnification and classified as neutrophils, lymphocytes, or monocytic cells. BAL protein was measured with a BCA protein assay (ThermoFisher Scientific). For histology, lungs were fixed by intratracheal installation of 1 ml of 4% paraformaldehyde followed by overnight fixation, dehydration, paraffin embedding, and staining of 4-µm sections with hematoxylin and eosin.
Measurement of protein biomarkers of inflammation and lung injury.
Cytokines were measured by Luminex using a 20-plex kit (Mouse Magnetic 20-Plex, ThermoFisher Scientific) and a custom multiplex kit from R&D Systems [CC-chemokine ligand 7 (CCL7, C-X-C ligand 12 (CXCL12), intercellular adhesion molecule 1 (ICAM-1), matrix metalloproteinase-8 (MMP-8), MMP-9, and tumor necrosis factor receptor 1 (TNFR1)]. In addition, biomarkers of lung endothelial and alveolar epithelial injury were measured with this same kit [angiopoietin-2 (Ang-2) and surfactant protein D (SP-D)].
Statistical analyses.
Comparisons between two groups were done with an unpaired t-test or Mann-Whitney U-test (when data were not normally distributed). Comparisons of more than two groups were made with ANOVA or Kruskal-Wallis. Repeated-measures ANOVA was used for comparisons of multiple groups over more than one time point, and two-way interaction terms were created for treatment group and time. Spearman or Pearson correlations were used depending on the normality of data distribution. Log-rank was used for survival analysis. P < 0.05 was considered to be statistically significant. Statistical analyses were performed with Stata (StataCorp, College Station, TX) and graphs were produced in Prism (GraphPad, La Jolla, CA).
RESULTS
S. pneumoniae produced dose-dependent lung injury.
Mice underwent intranasal inoculation with between 1 × 107 and 1 × 108 colony forming units (cfu) of an invasive serotype of pneumococcus (19F), producing dose-dependent weight loss (Fig. 1A), hypothermia (Fig. 1B), arterial hypoxemia (Fig. 1C), and pulmonary edema as measured by excess extravascular lung water (Fig. 1D). A dose of 1 × 108 cfu resulted in ~60% mortality and severe lobar pneumonia in surviving mice, in contrast to 3 × 107 cfu, which resulted in patchy pneumonia and no mortality (Fig. 1E). Doses of 2 × 108 cfu or greater were associated with severe hypothermia and death within 12–24 h (data not shown).
Fig. 1.

Streptococcus pneumoniae dose response. A–C: mice inoculated intranasally with between 1 × 107 and 1 × 108 cfu of S. pneumoniae developed dose-dependent weight loss, hypothermia, and arterial hypoxemia measured in freely moving mice. Data are means (SD), n = 5 per dosing group; *P < 0.0001 for group, time, and interaction term (group × time) by repeated-measures ANOVA; ^P < 0.0001 for group, P = 0.23 for time, P = 0.0003 for interaction term; #P = 0.0001 for group, P < 0.0001 for time, P = 0.0004 for interaction term. D: severity of lung injury as assessed by pulmonary edema was greatest in the 1 × 108 dose group; % by Mann-Whitney. E: representative low-power photomicrographs showing normal lung, patchy pneumonia 7 days following intranasal inoculation with 3 × 107 cfu S. pneumoniae, and profound lung consolidation with a dense inflammatory infiltrate following inoculation with 1 × 108 cfu S. pneumoniae.
Brief, mild cigarette smoke exposure did not affect pneumococcal lung injury.
Mice were exposed for 2 days to 2 h per day of sidestream cigarette smoke (CS) at 3 mg/m3 total suspended particulate (TSP) to model second-hand smoke exposure (Fig. 2A). Following CS exposure, mice were inoculated with 5 × 107 cfu of S. pneumoniae. No difference was observed in weight loss (Fig. 2B), hypothermia (Fig. 2C), arterial oxygen saturation (Fig. 2D), or excess extravascular lung water (Fig. 2E).
Fig. 2.

Low-dose cigarette smoke (CS) exposure did not affect pneumococcal lung injury. A: schema depicting experimental procedures. Mice were exposed to low-dose sidestream smoke for 2 h/day on subsequent days and then inoculated with 5 × 107 cfu S. pneumoniae. B: weight loss declined over time but was similar in air- and CS-exposed mice. Data are means (SD), n = 10 per group; *P = 0.60 for group, P < 0.0001 for time, P = 0.66 for interaction term (group*time) by repeated measures ANOVA. C: Core body temperature was not different 48 h postinfection, P = 0.11 by Mann-Whitney. D: arterial hypoxemia did not differ between air- and CS-exposed mice; P = 0.1 by unpaired t-test. E: pulmonary edema 48 h postinfection was moderate and did not differ based on air- and CS-exposed mice; P = 0.43 by unpaired t-test.
More intense CS exposure improved survival during severe pneumococcal pneumonia.
To model the recent initiation of active smoking, mice were exposed to 2.5 wk of CS at 100 mg/m3 TSP (Fig. 3A), an exposure that we have previously demonstrated produces no significant inflammation as assessed by histology, BAL cellularity, or elevation in inflammatory cytokines (29). The day following the last CS exposure, mice underwent inoculation with 1 × 108 cfu of S. pneumoniae. We selected a higher bacterial inoculum here (than in Fig. 2) to model more severe pneumonia. Unexpectedly, CS-exposed mice had a significant survival benefit (Fig. 3B). The improved survival in the CS-exposed mice was associated with more weight loss (Fig. 3C), less hypothermia (Fig. 3D), and a similar degree of arterial hypoxemia (Fig. 3E), peripheral leukopenia (Fig. 3F), and pulmonary edema (Fig. 3G) in surviving mice.
Fig. 3.

Moderate-dose CS exposure increases survival in severe pneumococcal pneumonia. A: schema depicting smoke exposure followed by intranasal inoculation with 1 × 108 cfu S. pneumoniae. B: CS-exposed mice had a significant survival advantage through euthanasia at 48 h; *by log-rank test, n = 20 mice per group. C: weight loss was slightly greater over time in surviving CS-exposed mice. Data are means (SD), n = 20 per group; ^P = 0.09 for group, P < 0.0001 for time, P = 0.001 for interaction term (group* × time) by repeated-measures ANOVA. D: hypothermia in surviving mice was less severe in CS-exposed mice over time. Data are means (SD), n = 20 per group; #P = 0.4 for group, P < 0.0001 for time, P = 0.0003 for interaction term by repeated-measures ANOVA. E: arterial hypoxemia in surviving mice worsened over time but did not differ according to prior CS exposure. Data are means (SD), n = 20 per group; %P = 0.67 for group, P < 0.0001 for time, P = 0.48 for interaction term by repeated-measures ANOVA. F: peripheral leukopenia at 48 h among surviving mice was similar; P = 0.44 by Mann-Whitney. G: pulmonary edema in surviving mice was similar 48 h postinfection in CS- and air-exposed mice; P = 0.8 by Mann-Whitney.
The survival benefit of CS exposure did not extend to severe nonpulmonary pneumococcal infection.
To determine whether CS conferred a general protective effect against severe pneumococcal infection, we developed an intraperitoneal (ip) inoculation model. Although primary pneumococcal peritonitis is not nearly as common as pneumonia, it represents ~1% of invasive pneumococcal disease (82). Mice were injected ip with increasing doses of S. pneumoniae, with 50% survival obtained with 1 × 108 cfu (Fig. 4A). Notably, mice either succumbed to this infection or rapidly recovered by 48 h. In the next set of experiments, we exposed mice to 2.5 wk of CS or air (as in Fig. 3A), and the following day, mice were inoculated with 1 × 108 cfu ip of S. pneumoniae. As shown in Fig. 4B, both air- and CS-exposed mice had high mortality with minimal lung injury in surviving mice (Fig. 4C). Although measurement of pulmonary edema could not be accomplished in mice that had died, the gross weight of the lungs did not differ between air and CS exposure, suggesting a similar degree of mild lung injury in both groups in this model of rapidly lethal pneumococcal peritonitis (Fig. 4D).
Fig. 4.

Prior CS exposure does not protect against intraperitoneal S. pneumoniae. A: survival of naïve mice with increasing doses of ip S. pneumoniae, n = 5 per group. B: prior CS exposure had no effect on 24-h survival following ip challenge with 1 × 108 cfu S. pneumoniae. n = 20 per group; P = 0.68 by log-rank test. C: pulmonary edema was minimal in both CS- and air-exposed surviving mice 24 h after ip S. pneumoniae; P = 0.32 by unpaired t-test. D: lungs extracted from dead mice did not differ in weight based on prior CS exposure, suggesting a similar degree of pulmonary edema; P = 0.68 by unpaired t-test.
CS exposure reduces the systemic spread of infection during severe pneumococcal pneumonia.
To determine whether the survival effect of CS in the pneumonia model was related to the severity of lung injury, we repeated the experiment with the moderate smoke exposure and intranasal pneumococcal inoculation (Fig. 5A), focusing on the 24-h time point before the survival curves separated. As shown in Fig. 5B, there was a significant improvement in hypothermia in CS-exposed mice. Interestingly, arterial hypoxemia was significantly worse in CS-exposed mice, opposite the survival benefit (Fig. 5C). However, BAL protein (Fig. 5D) and lung water (Fig. 5E) did not differ significantly with CS exposure, indicating that the difference in hypoxemia might be related to other factors such as differences in ventilation-perfusion matching. Notably, the effect of the modest group temperature difference on oxygen-hemoglobin interactions is likely to be insignificant (61).
Fig. 5.

Prior CS exposure reduces bacteremia during pneumococcal pneumonia. A: schema depicting smoke exposure and infection. B: CS-exposed mice were less hypothermic than air-exposed mice, *by unpaired t-test. C: arterial hypoxemia was more severe in CS-exposed mice, in contrast to the survival benefit, *by unpaired t-test. D: bronchoalveolar lavage (BAL) protein, a gross measure of the permeability of the alveolar-capillary barrier, did not differ with regard to prior CS exposure 24 h following pneumococcal inoculation; P = 0.62 by unpaired t-test. E: pulmonary edema was not different in CS- and air-exposed mice at 24 h; P = 0.7 by unpaired t-test. F: prior CS exposure reduced recoverable pneumococci in blood by several orders of magnitude, *by Mann-Whitney. G: splenic pneumococci were also reduced by prior CS exposure, *by Mann-Whitney.
Because mice respond to overwhelming infection with hypothermia rather than fever (62), we suspected the survival difference might be due to a difference in systemic infection; therefore, we measured bacterial loads in the blood (Fig. 5F) and spleen (Fig. 5G) at 24 h. CS-exposed mice had reduced systemic bacterial burden in pneumococcal pneumonia by several orders of magnitude. Notably, body temperature at 24 h was inversely correlated with systemic bacterial load (log of blood cfu), Pearson r = −0.78, P = 0.0004. To determine whether differences in systemic bacterial burden were due to a CS-induced reduction in airspace bacteria, we performed an additional experiment, identical to the protocol depicted in Fig. 5A except with a euthanasia time of 16 rather than 24 h postinfection. As shown in Fig. 6A, CS-exposed mice again had significantly higher body temperature than air-exposed mice at this earlier time point. However, BAL bacterial loads were not different with regard to prior smoke exposure (Fig. 6B), indicating that both groups of mice had very high levels of pneumococcal airspace burden early after infection. Interestingly, BAL myeloperoxidase (MPO) activity was significantly higher in CS-exposed mice (Fig. 6C), consistent with a more vigorous innate immune response within the airspaces.
Fig. 6.
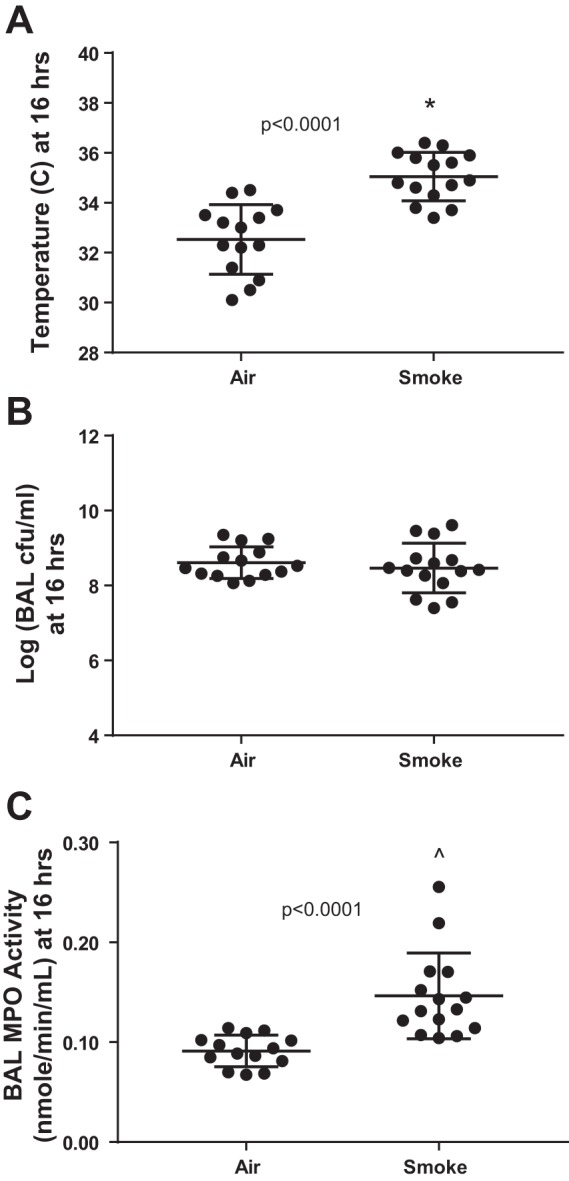
The CS-associated reduction in bacteremia is not due to reduced pneumococcal burden within the airspaces. A: CS-exposed mice were less hypothermic than air-exposed mice at 16 h postinfection, *by unpaired t-test. B: airspace pneumococcal burden at 16 h postinfection was similar in air- and CS-exposed mice. C: myeloperoxidase (MPO) activity within BAL was significantly higher in CS-exposed mice, ^by Mann-Whitney.
A model of severe pneumococcal pneumonia treated with antibiotics.
Because patients presenting with pneumonia and sepsis are uniformly treated with potent antipneumococcal antibiotics, we decided to enhance the clinical relevance of this model by treating infected mice with ceftriaxone, a third-generation cephalosporin with favorable pharmacokinetics and potent antipneumococcal activity. In preliminary experiments, we observed that a delay of 4 h between infection and the first dose of antibiotics resulted in minimal lung injury and 100% survival, whereas a delay of more than 24 h frequently resulted in severe and progressive lung injury and high mortality (data not shown). Therefore, we selected a ceftriaxone dose of 150 mg/kg and dosing frequency of 12 h, based on prior work in mice showing a favorable pharmacokinetic profile and efficacy against several strains of the pneumococcus (68). Mice were inoculated with 1 × 108 S. pneumonia and treated with ceftriaxone beginning 12 h postinfection for 3 doses as shown in Fig. 7A. Treated mice had more weight loss (Fig. 7B) and a significant improvement in hypothermia (Fig. 7C). Thus (as in Fig. 3C), weight loss and hypothermia, commonly assessed clinical parameters, seemed discordant as regards the health of the animals. We therefore tested whether these parameters might be related in a counterintuitive manner. Interestingly, across both antibiotic-treated and untreated mice, temperature was directly correlated with weight loss (%change from baseline, Spearman r = 0.68, P = 0.007), consistent with hypothermia-reducing activity and/or metabolic rate. By 48 h postinfection, antibiotic-treated mice had greatly reduced bacterial burden in BAL (Fig. 7D) and reduced MPO activity (Fig. 7E), indicating decreased degranulation of neutrophils and monocytes/macrophages. Histological analysis confirmed a major reduction in tissue neutrophils 48 h postinfection in antibiotic-treated mice (Fig. 7F).
Fig. 7.

Model of pneumococcal pneumonia treated with antibiotics. A: schema depicting experimental protocol. Mice were inoculated with S. pneumoniae and then injected with saline or ceftriaxone 150 mg/kg ip at 12, 24, and 36 h postinfection, followed by euthanasia at 48 h. B and C: mice treated with antibiotics had greater weight loss and less hypothermia. Data are means (SD), n = 7–8 per group; *P = 0.05 for group, P < 0.0001 for time, P = 0.17 for interaction term by repeated-measures ANOVA; ^P = 0.0001 for group, P < 0.0001 for time, P = 0.24 for interaction term. D: recoverable pneumococci in BAL were greatly reduced by 48 h with antibiotic treatment, #by Mann-Whitney. E: BAL myeloperoxidase activity was significantly reduced 48 h postinfection in antibiotic-treated mice, %P = 0.007 vs. No Abx, P = 0.006 vs. Uninfected by Mann-Whitney. F: representative high-power photomicrographs showing neutrophil-predominant inflammation 48 h post-untreated infection, reduced in mice treated with antibiotics but still clearly present relative to uninfected mice.
Prior moderate CS exposure increases lung injury in antibiotic-treated pneumococcal pneumonia.
We next repeated the CS exposure shown earlier to have a survival benefit in untreated pneumococcal infection, this time treating all mice with ceftriaxone beginning 12 h after bacterial inoculation (Fig. 8A). As shown in Fig. 8B, nearly all mice in both groups survived to 48 h (25/25 CS exposed vs. 22/25 air exposed). CS-exposed mice had greater weight loss than air-exposed mice (Fig. 8C) and were less hypothermic (Fig. 8D). However, CS-exposed mice had more pulmonary edema, as indicated by increased extravascular lung water (Fig. 8E). Histological analysis revealed moderate alveolar septal thickening in both groups, with a shift from neutrophilic to monocytic inflammation in CS-exposed mice (Fig. 8, F and G). Importantly, both air- and CS-exposed antibiotic-treated mice had less severe lung injury than air- and CS-exposed non-antibiotic-treated mice (compare excess lung water in Figs. 3G and 8E). Thus, antibiotics, rather than worsening lung injury, differentially reduced injury severity with regard to CS exposure.
Fig. 8.

CS exposure increases lung injury in pneumococcal pneumonia treated with antibiotics. A: schema depicting experimental procedures. Mice were exposed to moderate CS or air and then infected with 1 × 108 cfu S. pneumoniae and treated with ceftriaxone 150 mg/kg ip at 12, 24, and 36 h postinfection, followed by euthanasia at 48 h. B: survival did not differ between CS- and air-exposed mice, n = 25 mice per group; P = 0.08 by log-rank test. C: weight loss was greater in CS-exposed mice over time. Data are means (SD), n = 25 mice per group; *P < 0.0001 for group and time, P = 0.004 for interaction term (group × time) by repeated measures ANOVA. D: CS-exposed mice were less hypothermic, but this difference decreased with time as air-exposed mice gained body temperature during antibiotic treatment. Data are means (SD), n = 25 mice per group; ^P < 0.0001 for group, time, and interaction term (group × time) by repeated-measures ANOVA. E: pulmonary edema was significantly greater in mice previously exposed to CS, #by Mann-Whitney. F: representative high-power photomicrograph of a hematoxylin and eosin (H&E)-stained section from an air-exposed mouse 48 h postinfection showing an inflammatory infiltrate composed mostly of neutrophils (dotted arrows) and monocytes/macrophages (solid arrow). G: CS-exposed mice had a subtle increase in septal thickening and greater numbers of monocytes/macrophages (solid arrows) relative to neutrophils (dotted arrow).
Prior CS exposure changes the composition of inflammatory cells and cytokines in airspaces.
At 48 h postinfection, CS-exposed mice had a lower percentage of neutrophils and a higher percentage of monocytic cells in BAL, with no change in the percentage of lymphocytes (Fig. 9A). Because overall BAL cell number trended higher in CS-exposed mice (mean 413 vs. 309, P = 0.29 by Mann-Whitney), the absolute numbers of neutrophils in BAL were similar in CS- and air-exposed mice, whereas monocytic cells were significantly increased and lymphocytes trended higher relative to air-exposed mice (Fig. 9B). Notably, the CS exposure alone (without infection) did not result in any change in BAL cell number or composition (data not shown). We next measured the concentration of key chemokines and cytokines in BAL (Fig. 9C). KC (murine homolog of IL-8, a potent neutrophil chemoattractant) was detected at relatively low levels in both groups. In contrast, the monocyte chemokine macrophage inflammatory protein (MIP)-1α (CCL3) and the lymphocyte chemokine CXCL9 were both significantly increased in BAL of CS-exposed mice relative to nonsmoked mice (Fig. 9C). BAL levels of IL-6, MCP-1, MCP-2, MCP-3, and CXCL12 were consistent with a shift toward increased monocyte and lymphocyte chemokines in CS-exposed mice (Table 1), mirroring the cellular infiltrate in BAL and histology observed 48 h postinfection.
Fig. 9.

CS exposure before pneumococcal pneumonia changes the cellular composition of airspace inflammation. A: CS exposure increases the percentage of monocytes/macrophages in BAL at the expense of neutrophils, whereas the percentage of lymphocytes is unchanged, *by Mann-Whitney. B: given the trend toward higher BAL cell counts in CS-exposed mice, the total number of BAL neutrophils was unchanged, whereas total BAL monocytes/macrophages were significantly increased, and total BAL lymphocytes trended higher, ^by unpaired t-test. C: the concentration of key neutrophil, monocyte/macrophage, and lymphocyte chemokines was measured in BAL and corrected for total protein. KC (murine homolog of IL-8) was detected at very low levels and not different with regard to CS exposure. However, macrophage inflammatory protein-1α (MIP-1α) and C-X-C ligand 9 (CXCL9) were significantly increased in CS-exposed mice, *by Mann-Whitney.
Table 1.
CS exposure increases monocyte and lymphocyte chemokines measured in BAL
| Ligand | Cell* | Air (n = 8) | Smoke (n = 9–10) | Ratio† (S/A) | P‡ |
|---|---|---|---|---|---|
| KC | N | 25 | 53 | 2.1 | 0.61 |
| IL-6 | N | 198 | 180 | 0.9 | 0.24 |
| MIP-1α | M | 1,159 | 3,103 | 2.7 | 0.001 |
| MCP-1 | M | 837 | 1,340 | 2.4 | 0.008 |
| MCP-2 | M | 747 | 1,159 | 1.6 | 0.04 |
| MCP-3 | M | 1,155 | 2,400 | 2.1 | 0.21 |
| CXCL9 | L | 814 | 2,013 | 2.5 | 0.02 |
| CXCL12 | L | 38,072 | 64,603 | 1.7 | 0.009 |
Median values at 48 h postinfection are expressed in pg/mg bronchoalveolar lavage (BAL) protein. CS, cigarette smoke; KC, murine homolog of IL-8; IL-6, interleukin-6; MIP, macrophage inflammatory protein; MCP, monocyte chemoattractant protein; CXCL, C-X-C ligand.
Predominant target cell for each cytokine. N, neutrophil; M, monocyte/macrophage; L, lymphocyte.
Smoke-to-air ratio, bolded values significant by P < 0.05;
uncorrected P value.
Cellular mediators of lung injury.
To determine possible mediators of lung injury, we measured lung neutrophil elastase (NE), MPO, and granzyme B. Notably, MPO is present in both neutrophils and monocytes (32, 50). Because inhibitory substances in lung homogenate precluded its use in the elastase and MPO enzymatic assays, we used cell-free BAL for these experiments. Although BAL NE did not differ between CS-exposed and air-exposed mice (data not shown), BAL MPO was significantly higher in CS-exposed mice (Fig. 10A), similar to the nonantibiotic pneumococcal model (Fig. 6C). Granzyme B is a serine protease contained in the cytotoxic granules of lymphocytes (2). As shown in Fig. 10B, lung homogenate levels of granzyme B trended higher in CS-exposed mice. Interestingly, the concentration of granzyme B was unrelated to the extent of pulmonary edema (excess extravascular lung water) in air-exposed mice (Fig. 10C) but was significantly associated with the extent of pulmonary edema in CS-exposed mice (Fig. 10D).
Fig. 10.

Prior CS exposure increases inflammatory cell cytotoxic proteins during pneumococcal pneumonia. A: CS-exposed mice had greater myeloperoxidase activity than air-exposed mice, *by Mann-Whitney. B: lung levels of granzyme B, a serine protease contained in cytotoxic lymphocyte granules, were not significantly increased by prior CS exposure, P = 0.14 by Mann-Whitney. C: granzyme B levels were unrelated to the level of pulmonary edema in air-exposed mice, Spearman r = −0.07, P = 0.81. D: in contrast, granzyme B levels predicted the extent of pulmonary edema in CS-exposed mice, Spearman r = 0.58, P = 0.04.
Antibiotic treatment causes major changes in BAL cytokines in CS-exposed mice.
Given that the effect of CS exposure on outcomes was so different in the untreated and antibiotic-treated models of pneumococcal pneumonia, we analyzed these model differences further by comparing the BAL cytokine profile of CS-exposed mice with and without antibiotic treatment. As show in Table 2, antibiotic treatment in CS-exposed mice was associated with significant reductions in the potent inflammatory molecules IL-1α, IL-17, TNFα, and IL-1β, a marker of inflammasome activation. Interestingly, the greatest differences between antibiotic-treated and untreated mice were neutrophil-associated KC (70-fold higher in untreated mice), and IL-6 (9-fold higher in untreated mice). In contrast, most monocyte (except MIP-1α) and lymphocyte chemokines were unchanged or trended higher with antibiotic treatment.
Table 2.
Antibiotic treatment in CS-exposed mice induces significant changes in BAL cytokine profile
| Ligand | −Abx (n = 3–5) | +Abx (n = 8–9) | Ratio* −/+ | P† |
|---|---|---|---|---|
| IL-1α | 442 | 152 | 2.9 | 0.004 |
| IL-1β | 196 | 129 | 1.5 | 0.001 |
| TNFα | 1,924 | 438 | 4.4 | 0.001 |
| IL-17 | 5.3 | 1.5 | 3.5 | 0.03 |
| KC | 6,940 | 98 | 70.8 | 0.001 |
| IL-6 | 5,439 | 599 | 9.1 | 0.02 |
| MIP-1α | 44,925 | 11,737 | 3.8 | 0.03 |
| MCP-1 | 5,564 | 6,036 | 0.9 | >0.99 |
| MCP-2 | 6,260 | 5,378 | 1.2 | 0.71 |
| MCP-3 | 2,275 | 3,603 | 0.6 | 0.10 |
| CXCL9 | 9,475 | 8,769 | 1.1 | 0.44 |
| CXCL12 | 148,956 | 254,114 | 0.6 | 0.09 |
Median values at 48 h postinfection are expressed in pg/ml.
Ratio of −Abx to +Abx, bolded values significant by P < 0.05;
uncorrected P value.
CS exposure increases lung epithelial and endothelial injury.
SP-D, a product of alveolar epithelial type II cells that is released into the circulation during lung epithelial injury (57), is increased in the blood of patients with ARDS (33), and it predicts worse outcomes in patients with ARDS (20, 83). Serum SP-D has also been shown to be increased during acute lung injury in rodents induced by nebulized LPS (28), bleomycin (24, 57), and hydrochloric acid (57). As shown in Fig. 11A, blood levels of SP-D in antibiotic-treated pneumococcal pneumonia (including both air- and CS-exposed mice) were highly correlated with the degree of pulmonary edema (Spearman r = 0.71, P < 0.0001), consistent with its established role as a biomarker of alveolar epithelial injury. SP-D was significantly elevated in mice previously exposed to CS (Fig. 11B). Ang-2 is released by vascular endothelium by a variety of inflammatory insults and interferes with Ang-1 signaling through Tie-2, increasing vascular permeability (63). Levels of Ang-2 in the blood of patients are associated with poor outcomes in sepsis-associated lung injury (9) and have been shown to predict the development of ARDS (3). As shown in Fig. 11C, CS-exposed mice had significantly higher blood levels of Ang-2, consistent with increased endothelial injury and permeability. Using different methods, other investigators have reported that CS exposure increases lung endothelial injury (40). Notably, CS exposure alone did not increase either SP-D or Ang-2 in uninjured mice (data not shown).
Fig. 11.

Prior CS exposure increases markers of alveolar epithelial and endothelial injury in the blood during pneumococcal pneumonia. A: plasma surfactant protein D (SP-D) was highly correlated with the severity of lung injury (extravascular lung water) across CS- and air-exposed mice, Spearman r = 0.71, P < 0.0001. B: serum SP-D was significantly higher 48 h post-injury in CS-exposed mice, *by Mann-Whitney. C: serum angiopoietin-2 (Ang-2), a marker of endothelial injury, was significantly higher in CS-exposed mice, *by Mann-Whitney.
Biomarkers of CS-associated infection-related lung injury.
A major goal of these studies was to identify potential biomarkers of smoking-related lung injury to be tested in future work with blood samples collected prospectively from a cohort of critically ill patients. Therefore, we measured several cytokines and molecules with well-established roles in inflammatory tissue injury in mouse serum samples 48 h postinfection in the antibiotic-treated pneumococcal pneumonia model. As shown in Table 3, MMP-8 and -9 were significantly increased in CS-exposed mice, along with the lymphocyte chemokine CXCL9 and the monocyte chemokine MIP-1α.
Table 3.
Blood biomarkers of CS exposure-related lung injury in antibiotic-treated pneumonia
| Ligand | Air (n = 8) | Smoke (n = 9–10) | Ratio* (S/A) | P† |
|---|---|---|---|---|
| MMP-9 | 13,008 | 49,834 | 3.8 | 0.0085 |
| MIP-1α | 73.2 | 138.2 | 1.9 | 0.03 |
| VEGF | 1.6 | 3.0 | 1.9 | 0.007 |
| SP-D | 68,692 | 12,6446 | 1.8 | 0.04 |
| GM-CSF | 5.5 | 8.5 | 1.5 | 0.12 |
| CXCL9 | 130.4 | 200.3 | 1.5 | 0.045 |
| Ang-2 | 22,314 | 34,147 | 1.5 | 0.0003 |
| IL-17 | 7.8 | 10.5 | 1.4 | 0.02 |
| MMP-8 | 309,065 | 445,069 | 1.4 | 0.009 |
| FGF-2 | 124 | 159.7 | 1.3 | 0.006 |
| IL-6 | 31.2 | 41.6 | 1.3 | 0.20 |
| IFNγ | 19.4 | 23.2 | 1.2 | 0.50 |
| IL-1β | 94.7 | 105.3 | 1.1 | 0.15 |
| IL-2 | 6.5 | 7.1 | 1.1 | 0.47 |
| MCP-3 | 1,209 | 1,299 | 1.1 | 0.76 |
| IL-5 | 14.9 | 15.0 | 1.0 | >0.99 |
| ICAM-1 | 54,245 | 54,956 | 1.0 | 0.68 |
| CXCL12 | 13,254 | 12,845 | 1.0 | 0.67 |
| IL-12 | 28.2 | 24.3 | 0.9 | 0.09 |
| MCP-1 | 72.7 | 63.7 | 0.9 | 0.85 |
| TNFR1 | 1,394 | 1,259 | 0.9 | 0.46 |
| TNFα | 13.1 | 10.8 | 0.8 | 0.85 |
| CXCL10 | 306.7 | 238.5 | 0.8 | 0.57 |
| KC | 1,328 | 732.2 | 0.6 | 0.24 |
| IL-10 | Too low to measure | |||
| IL-13 | Too low to measure | |||
| IL-4 | Too low to measure | |||
Median serum values at 48 h postinfection are expressed in pg/ml. MMP, matrix metalloproteinase; VEGF, vascular endothelial growth factor; SP-D, surfactant protein D; GM-CSF, granulocyte-macrophage colony-stimulating factor; Ang-2, angiopoietin-2; FGF, fibroblast growth factor; IFN, interferon; ICAM, intercellular adhesion molecule; TNF, tumor necrosis factor; TNFR, TNF receptor.
Ratio of CS exposed to air exposed, bolded values significant by P < 0.05;
uncorrected P value.
DISCUSSION
The main findings of these experiments can be summarized as follows. First, several weeks of CS exposure improved survival during subsequent challenge with pneumococcal pneumonia in mice. Second, this survival benefit was likely due to reduced dissemination of bacteria from the lungs into the systemic circulation and did not generalize to extrapulmonary pneumococcal sepsis. Third, when antibiotic treatment was introduced into the model of acute bacterial pneumonia, the survival benefit of CS exposure was lost, and CS-exposed mice instead suffered more severe lung injury relative to air-exposed control mice, including evidence of lung endothelial and alveolar epithelial damage. Fourth, CS-exacerbated lung injury was associated with increased accumulation of alveolar monocytes and monocyte-related airspace chemokines.
CS exposure is known to increase the risk of ARDS in trauma and in nonpulmonary sepsis (10, 11). Our group previously reported that healthy human smokers (compared with nonsmokers) have increased BAL protein after inhaling nebulized LPS, a model of gram-negative pneumonia (48). Similarly, we (29) recently reported that short-term moderate CS exposure increases lung injury in response to intratracheal LPS in mice. Other investigators have reported similar results with LPS after short-term CS exposure in mice (40, 67). Although well suited to experimental models, LPS lacks many characteristics of live bacteria and even at high doses causes only mild lung injury in mice that are naturally resistant to endotoxin (22).
To the best of our knowledge, we here report for the first time that CS exposure improves survival in a mouse model of pneumonia employing live bacteria in the absence of antibiotics. Our CS exposure of 100 mg/m3 for ~3 wk causes no obvious BAL or histological inflammation or increase in inflammatory cytokines (29), making it moderate by comparison to studies employing exposures of 250 mg/m3 or greater, which have consistently demonstrated significant inflammation from CS itself (26, 27, 42, 52). CS-exposed mice had no difference in lung injury or airspace bacterial burden but were less hypothermic and had decreased bacteremia by several orders of magnitude. Notably, CS exposure provided no protection against death from pneumococcal peritonitis. These results are consistent with moderate CS exposure inducing an enhanced, localized, innate immune response in the lung to invading lung pathogens that decreases translocation into the blood.
There are at least 13 published reports in which mice and rats have been exposed to CS followed by bacterial challenge for which detailed methods are available (5, 7, 19, 26, 27, 36, 42, 52, 59, 70, 73, 79, 81). CS exposures (TSP) in these studies have ranged between 15 and 980 mg/m3 with total exposure durations from 4 days to 9 mo. Several groups have reported that prior CS exposure increases bacterial loads following challenge with intratracheal S. pneumoniae (42) and Pseudomonas aeruginosa (19, 73). However, other researchers have reported that CS-exposed mice had either no change (36) or decreased lung bacteria following challenge with Haemophilus influenza (26, 27, 52), P. aeruginosa (5), and S. pneumoniae (5). Several methodological differences have been cited to explain these different results, including intensity and duration of CS exposure, size of the bacterial inoculum, and time points and tissues examined. Mouse strain, in particular, may be especially important, with well-characterized strain differences in physiological responses to hypoxia and hypercapnea (1), CS-induced inflammation (90), and recently reported strain-dependent susceptibility to CS priming with endotoxin-induced lung injury (67). However, no study of bacterial pneumonia and CS in rodents has employed antibiotics, to our knowledge.
We are interested in the mechanisms by which CS predisposes patients to develop ARDS during critical illness (10, 11, 75). Recognizing that the survival results we obtained in mice were highly discordant from human studies demonstrating that smokers are at increased risk of invasive pneumococcal disease (74) and death from pneumococcal pneumonia (6), we sought to improve the clinical relevance of our model. Patients with pneumonia are uniformly treated with broad-spectrum antibiotics within 1–2 h of presenting for medical care (31). Notably, treatment of serious pneumococcal infections with effective antibiotics releases large quantities of bacterial cell wall products over a short time and has been shown to produce a wave of inflammation that can worsen organ injury (43, 76, 77). This phenomenon is well described in patients with pneumococcal meningitis and is the basis for coadministration of antibiotics and systemic glucocorticoids. In addition, all indications from our experiments without antibiotics were that the mice were dying of systemic infection, not due to the severity of the pneumonia, making it difficult to assess the effects of CS exposure on the degree of acute lung injury, which was our primary objective.
In our work developing the antibiotic-treated model of pneumococcal pneumonia, we found that three doses of ceftriaxone beginning 12 h after infection nearly sterilized the airspaces by 48 h, improved hypothermia, and significantly reduced lung neutrophils in naïve mice, as well as MPO levels in cell-free BAL, suggesting reduced degranulation of neutrophils and/or monocytes. The timing of the first dose of antibiotics was critical, with early initiation (under 6 h) resulting in minimal lung injury and later initiation (after 24 h) resulting in progressive hypothermia, hypoxemia, and death. The progressive organ injury phenotype observed with the later initiation of antibiotics is reminiscent of multiorgan failure that frequently develops in patients with septic shock despite the administration of effective antimicrobial therapy (31).
Applying antibiotic treatment to our moderate-smoking model, we found that CS no longer significantly improved survival but instead caused greater lung injury in association with elevated numbers of monocytes and a trend toward increased lymphocytes. MPO levels were higher in the BAL of CS-exposed mice, suggesting either greater degranulation of neutrophils (which would be consistent with reduced percentage of polymorphonuclear leukocytes in BAL at 48 h), or a predominantly monocytic source.
We found that blood levels of SP-D were strongly correlated with the severity of lung injury (extravascular lung water) in mice during antibiotic-treated pneumococcal pneumonia. This finding is consistent with prior reports in patients that SP-D is an important prognostic biomarker in ARDS and an indicator of the degree of alveolar epithelial injury (20, 83). CS exposure was associated with elevated serum SP-D, consistent with greater lung epithelial injury. Additionally, elevated serum Ang-2 suggests that CS-exposed mice suffered greater endothelial injury, similar to what has been reported by others in CS-exposed mice following challenge with endotoxin (7, 40) and P. aeruginosa (7). The combination of lung endothelial and alveolar epithelial injury is a well-established mechanism that leads to protein-rich pulmonary edema in experimental models (89) and in clinical studies (85, 86).
The pattern of BAL chemokines that we observed in the antibiotic-treated model is consistent with increased mobilization of monocytes and lymphocytes into the airspaces of CS-exposed mice during severe bacterial infection. The role of macrophages in CS-related lung inflammation is well established. Macrophages have been shown to be activated by CS to release chemokines for monocytes, neutrophils, and lymphocytes, generate reactive oxygen species, and release elastolytic enzymes such as the matrix metalloproteinases. High-intensity CS exposure in mice recruits monocytes into the lung within several days (12). Basilico et al. (5) recently reported that a CS exposure (100 mg/m3 TSP for 6 wk) similar to ours resulted in a reduced lung burden of S. pneumoniae and P. aeruginosa in association with increased bone marrow release of inflammatory Ly-6Chi monocytes. These authors also reported that neutropenic mice (which, as expected, suffered very high bacterial burdens compared with wild type), had bacterial loads reduced to wild-type levels by CS exposure. The increased numbers of monocytes and macrophages that we observed in the lungs of CS-exposed mice are consistent with these data and suggest that these cells may play an important role in the confinement of the infection to the lung.
Interestingly, we found that the severity of lung injury in CS-exposed (but not air-exposed) mice correlated with tissue levels of the lymphocyte serine protease granzyme B (2), suggesting that recruited lymphocytes may differentially impact acute bacterial inflammation in the setting of prior CS exposure. CD8+ T cells have long been associated with chronic smoking-related lung disease in patients (54, 65), and mice deficient in CD8+ T cells are protected against emphysema resulting from chronic CS exposure (44). BAL levels of granzyme B are increased in smokers and correlate with bronchial epithelial cell apoptosis (34). Similarly, NK cells isolated from the sputum of COPD patients have increased granzyme B expression, cytotoxicity, and expression of CXCR3 (78), a major T cell chemokine receptor (17). Although most studies have focused on chronic CS exposure, during intense CS exposure in mice, CD8+ T cells are recruited to the lung within only 3 days (51). Interestingly, in one study, CXCR3 knockout mice were protected against both acute CS-induced T cell recruitment and lung injury (51). In our experiments, CS-exposed infected mice had significant elevations in BAL CXCL9, one of the major CXCR3 ligands and lymphocyte chemoattractants, previously shown to be increased in the sputum of patients with COPD (15).
In contrast, we observed a reduced percentage of neutrophils in BAL and low levels (<100 pg/ml) of the neutrophil chemokine KC (murine homolog of IL-8/CXLC8) at 48 h in the antibiotic-treated model. BAL KC in CS-exposed mice was reduced over 70-fold with antibiotic treatment (relative to no antibiotics), IL-6 was reduced nearly 10-fold, TNFα was reduced over fourfold, and IL-1β, a marker of inflammasome activation, was also significantly reduced. Meanwhile, levels of monocyte and lymphocyte chemokines generally remained similar or even trended higher compared with non-antibiotic-treated mice. The results indicate that ongoing bacterial presence in the lungs perpetuates intense neutrophil-dominated inflammation. The omission of antibiotic treatment in animal models of severe pneumonia may thus limit their applicability to the clinical setting, in which progressive organ dysfunction including ARDS frequently occurs despite effective treatment of the causative pathogen (31) and reductions in inflammatory cytokines such as IL-6 and IL-8 over time (58).
A major objective of this work was to identify biomarkers of CS-related acute lung injury. As discussed above, SP-D and Ang-2 are established ARDS prognostic biomarkers reflecting lung epithelial and endothelial injury, and we found that both biomarkers were elevated in the blood of CS-exposed mice with bacterial pneumonia. Other investigators have emphasized the role of CS in causing lung endothelial injury (7, 40, 41, 66). MMP-9 (gelatinase B) was increased in the blood of CS-exposed mice nearly fourfold. MMP-9 is a collagenase expressed by many types of cells, including neutrophils (55), monocytes (88), and lymphocytes (87), with complex roles in lung inflammation and remodeling (4). MMP-8, another collagenase expressed by neutrophils (56) and monocytes (16), was also significantly elevated in the blood of CS-exposed infected mice. MMPs are known to be activated by CS (69), and sputum MMP-8 distinguishes early-stage COPD patients from active asymptomatic smokers and nonsmokers (35). Although MMP-8 and -9 have both been shown to be elevated in the airspaces of patients with ARDS (23, 60), whether CS exposure differentially affects MMP levels in smokers with ARDS is not yet known. Finally, the lymphocyte chemokine CXCL9 and the monocyte chemokine MIP-1α were elevated in the serum of CS-exposed infected mice. We (29) recently reported CS-associated increases in blood CXCL9 following injury with intratracheal endotoxin, suggesting that diverse inflammatory stimuli may elicit common biomarker signatures following CS exposure.
There are some limitations to these studies. Although the CS exposure is moderate in duration, it does have clinical relevance to our published clinical studies showing an association between ARDS and CS exposure (10, 11, 75). We have not determined the mechanism by which CS exposure reduces bacterial dissemination, but in light of the CS-associated increase in BAL MPO activity, we hypothesize that it may involve a more robust innate immune response from macrophages, recruited monocytes, and possibly lymphocytes. Also, we have not identified all of the potential mechanisms that account for the greater degree of pneumococcal lung injury in the antibiotic-treated mice with CS exposure, although the cell and chemokine data indicate a major role for monocytes and monocyte-derived chemokines in mediating the increase in lung endothelial and alveolar epithelial injury. Future experiments with broadly immunosuppressive agents such as corticosteroids and specific inhibition of lymphocyte and monocyte subsets using genetic manipulation may be helpful in elucidating these mechanistic pathways. We propose that this model of bacterial pneumonia and lung injury that develops in antibiotic-treated mice has considerable clinical relevance to patients who often progress to develop ARDS despite appropriate antibiotic treatment (46) and should be valuable to other investigators who test novel therapeutics in preclinical models of ARDS.
In conclusion, compared with controls, moderate CS exposure in mice over a 3-wk period resulted in improved survival following bacterial pneumonia with S. pneumoniae in the absence of antibiotics, primarily explained by reduced bacteremia. However, when CS exposed mice with pneumococcal pneumonia were treated with antibiotics, as would usually be the case in the clinical setting, the degree of acute lung injury was greater in the CS-exposed mice, with evidence of more pulmonary edema and higher elevations of markers of alveolar epithelial injury (SP-D) and lung endothelial injury (Ang-2). The mechanisms for this greater lung injury in the antibiotic-treated CS-exposed mice may be explained, in part, by a higher concentration of monocyte-derived chemokines and monocytes. The antibiotic-treated S. pneumoniae model may be useful for future studies of the acute pulmonary impact of current and emerging tobacco products, including the identification of biomarkers reflecting tobacco product-related lung injury.
GRANTS
This work was supported by Grant no. 1P50 CA-180890 from the National Cancer Institute and Food and Drug Administration Center for Tobacco Products. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health (NIH) or the Food and Drug Administration. Additional support was from National Heart, Lung, and Blood Institute Grants R01-HL-51854 and R37-HL-51856, and 24RT-0020 University of California Tobacco-Related Disease Research Program.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.G., C.C., S.N., and M.M. conceived of and designed the research. J.G., L.C., J.A., X.F., and N.T. performed the experiments. M.S. and S.S. conceived of, designed, and built the low concentration cigarette smoke generation and exposure system. J.G., S.N., C.C., and M.M analyzed the data, interpreted the results, prepared the figures, and drafted and edited the manuscript. All authors approved the final version of the manuscript.
ACKNOWLEDGMENTS
We are grateful to Hanjing Zhuo for assistance with statistical analysis.
REFERENCES
- 1.Adachi T, Ogawa H, Okabe S, Kitamuro T, Kikuchi Y, Shibahara S, Shirato K, Hida W. Mice with blunted hypoxic ventilatory response are susceptible to respiratory disturbance during hypoxia. Tohoku J Exp Med 209: 125–134, 2006. doi: 10.1620/tjem.209.125. [DOI] [PubMed] [Google Scholar]
- 2.Afonina IS, Cullen SP, Martin SJ. Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme B. Immunol Rev 235: 105–116, 2010. doi: 10.1111/j.0105-2896.2010.00908.x. [DOI] [PubMed] [Google Scholar]
- 3.Agrawal A, Matthay MA, Kangelaris KN, Stein J, Chu JC, Imp BM, Cortez A, Abbott J, Liu KD, Calfee CS. Plasma angiopoietin-2 predicts the onset of acute lung injury in critically ill patients. Am J Respir Crit Care Med 187: 736–742, 2013. doi: 10.1164/rccm.201208-1460OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atkinson JJ, Senior RM. Matrix metalloproteinase-9 in lung remodeling. Am J Respir Cell Mol Biol 28: 12–24, 2003. doi: 10.1165/rcmb.2002-0166TR. [DOI] [PubMed] [Google Scholar]
- 5.Basilico P, Cremona TP, Oevermann A, Piersigilli A, Benarafa C. Increased myeloid cell production and lung bacterial clearance in mice exposed to cigarette smoke. Am J Respir Cell Mol Biol 54: 424–435, 2016. doi: 10.1165/rcmb.2015-0017OC. [DOI] [PubMed] [Google Scholar]
- 6.Bello S, Menéndez R, Antoni T, Reyes S, Zalacain R, Capelastegui A, Aspa J, Borderías L, Martin-Villasclaras JJ, Alfageme I, Rodríguez de Castro F, Rello J, Luis M, Ruiz-Manzano J. Tobacco smoking increases the risk for death from pneumococcal pneumonia. Chest 146: 1029–1037, 2014. doi: 10.1378/chest.13-2853. [DOI] [PubMed] [Google Scholar]
- 7.Borgas D, Chambers E, Newton J, Ko J, Rivera S, Rounds S, Lu Q. Cigarette smoke disrupted lung endothelial barrier integrity and increased susceptibility to acute lung injury via histone deacetylase 6. Am J Respir Cell Mol Biol 54: 683–696, 2016. doi: 10.1165/rcmb.2015-0149OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brook I, Gober AE. Recovery of potential pathogens and interfering bacteria in the nasopharynx of smokers and nonsmokers. Chest 127: 2072–2075, 2005. doi: 10.1378/chest.127.6.2072. [DOI] [PubMed] [Google Scholar]
- 9.Calfee CS, Gallagher D, Abbott J, Thompson BT, Matthay MA; NHLBI ARDS Network . Plasma angiopoietin-2 in clinical acute lung injury: prognostic and pathogenetic significance. Crit Care Med 40: 1731–1737, 2012. doi: 10.1097/CCM.0b013e3182451c87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calfee CS, Matthay MA, Eisner MD, Benowitz N, Call M, Pittet J-F, Cohen MJ. Active and passive cigarette smoking and acute lung injury after severe blunt trauma. Am J Respir Crit Care Med 183: 1660–1665, 2011. doi: 10.1164/rccm.201011-1802OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calfee CS, Matthay MA, Kangelaris KN, Siew ED, Janz DR, Bernard GR, May AK, Jacob P, Havel C, Benowitz NL, Ware LB. Cigarette smoke exposure and the acute respiratory distress syndrome. Crit Care Med 43: 1790–1797, 2015. doi: 10.1097/CCM.0000000000001089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campos KKD, Manso RG, Gonçalves EG, Silva ME, de Lima WG, Menezes CAS, Bezerra FS. Temporal analysis of oxidative effects on the pulmonary inflammatory response in mice exposed to cigarette smoke. Cell Immunol 284: 29–36, 2013. doi: 10.1016/j.cellimm.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Cardozo DM, Nascimento-Carvalho CM, Andrade A-LSS, Silvany-Neto AM, Daltro CHC, Brandão M-AS, Brandão AP, Brandileone M-CC. Prevalence and risk factors for nasopharyngeal carriage of Streptococcus pneumoniae among adolescents. J Med Microbiol 57: 185–189, 2008. doi: 10.1099/jmm.0.47470-0. [DOI] [PubMed] [Google Scholar]
- 14.Chun LF, Moazed F, Calfee CS, Matthay MA, Gotts JE. Pulmonary toxicity of e-cigarettes. Am J Physiol Lung Cell Mol Physiol 313: L193–L206, 2017. doi: 10.1152/ajplung.00071.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Costa C, Rufino R, Traves SL, Lapa E Silva JR, Barnes PJ, Donnelly LE. CXCR3 and CCR5 chemokines in induced sputum from patients with COPD. Chest 133: 26–33, 2008. doi: 10.1378/chest.07-0393. [DOI] [PubMed] [Google Scholar]
- 16.Craig VJ, Polverino F, Laucho-Contreras ME, Shi Y, Liu Y, Osorio JC, Tesfaigzi Y, Pinto-Plata V, Gochuico BR, Rosas IO, Owen CA. Mononuclear phagocytes and airway epithelial cells: novel sources of matrix metalloproteinase-8 (MMP-8) in patients with idiopathic pulmonary fibrosis. PLoS One 9: e97485, 2014. doi: 10.1371/journal.pone.0097485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.D’Ambrosio D, Mariani M, Panina-Bordignon P, Sinigaglia F. Chemokines and their receptors guiding T lymphocyte recruitment in lung inflammation. Am J Respir Crit Care Med 164: 1266–1275, 2001. doi: 10.1164/ajrccm.164.7.2103011. [DOI] [PubMed] [Google Scholar]
- 18.Diamond JM, Lee JC, Kawut SM, Shah RJ, Localio AR, Bellamy SL, Lederer DJ, Cantu E, Kohl BA, Lama VN, Bhorade SM, Crespo M, Demissie E, Sonett J, Wille K, Orens J, Shah AS, Weinacker A, Arcasoy S, Shah PD, Wilkes DS, Ware LB, Palmer SM, Christie JD; Lung Transplant Outcomes Group . Clinical risk factors for primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med 187: 527–534, 2013. doi: 10.1164/rccm.201210-1865OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drannik AG, Pouladi MA, Robbins CS, Goncharova SI, Kianpour S, Stämpfli MR. Impact of cigarette smoke on clearance and inflammation after Pseudomonas aeruginosa infection. Am J Respir Crit Care Med 170: 1164–1171, 2004. doi: 10.1164/rccm.200311-1521OC. [DOI] [PubMed] [Google Scholar]
- 20.Eisner MD, Parsons P, Matthay MA, Ware L, Greene K; Acute Respiratory Distress Syndrome Network . Plasma surfactant protein levels and clinical outcomes in patients with acute lung injury. Thorax 58: 983–988, 2003. doi: 10.1136/thorax.58.11.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fang X, Abbott J, Cheng L, Colby JK, Lee JW, Levy BD, Matthay MA. Human mesenchymal stem (stromal) cells promote the resolution of acute lung injury in part through lipoxin A4. J Immunol 195: 875–881, 2015. doi: 10.4049/jimmunol.1500244. [DOI] [PubMed] [Google Scholar]
- 22.Fink MP. Animal models of sepsis. Virulence 5: 143–153, 2014. doi: 10.4161/viru.26083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fligiel SEG, Standiford T, Fligiel HM, Tashkin D, Strieter RM, Warner RL, Johnson KJ, Varani J. Matrix metalloproteinases and matrix metalloproteinase inhibitors in acute lung injury. Hum Pathol 37: 422–430, 2006. doi: 10.1016/j.humpath.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 24.Fujita M, Shannon JM, Ouchi H, Voelker DR, Nakanishi Y, Mason RJ. Serum surfactant protein D is increased in acute and chronic inflammation in mice. Cytokine 31: 25–33, 2005. doi: 10.1016/j.cyto.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Vidal C, Ardanuy C, Tubau F, Viasus D, Dorca J, Liñares J, Gudiol F, Carratalà J. Pneumococcal pneumonia presenting with septic shock: host- and pathogen-related factors and outcomes. Thorax 65: 77–81, 2010. doi: 10.1136/thx.2009.123612. [DOI] [PubMed] [Google Scholar]
- 26.Gaschler GJ, Skrtic M, Zavitz CCJ, Lindahl M, Onnervik P-O, Murphy TF, Sethi S, Stämpfli MR. Bacteria challenge in smoke-exposed mice exacerbates inflammation and skews the inflammatory profile. Am J Respir Crit Care Med 179: 666–675, 2009. doi: 10.1164/rccm.200808-1306OC. [DOI] [PubMed] [Google Scholar]
- 27.Gaschler GJ, Zavitz CCJ, Bauer CMT, Stämpfli MR. Mechanisms of clearance of nontypeable Haemophilus influenzae from cigarette smoke-exposed mouse lungs. Eur Respir J 36: 1131–1142, 2010. doi: 10.1183/09031936.00113909. [DOI] [PubMed] [Google Scholar]
- 28.Gaunsbaek MQ, Rasmussen KJ, Beers MF, Atochina-Vasserman EN, Hansen S. Lung surfactant protein D (SP-D) response and regulation during acute and chronic lung injury. Hai 191: 295–303, 2013. doi: 10.1007/s00408-013-9452-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gotts JE, Abbott J, Fang X, Yanagisawa H, Takasaka N, Nishimura SL, Calfee CS, Matthay MA. Cigarette smoke exposure worsens endotoxin-induced lung injury and pulmonary edema in mice. Nicotine Tob Res 19: 1033–1039, 2017. doi: 10.1093/ntr/ntx062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gotts JE, Abbott J, Matthay MA. Influenza causes prolonged disruption of the alveolar-capillary barrier in mice unresponsive to mesenchymal stem cell therapy. Am J Physiol Lung Cell Mol Physiol 307: L395–L406, 2014. doi: 10.1152/ajplung.00110.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ 353: i1585, 2016. doi: 10.1136/bmj.i1585. [DOI] [PubMed] [Google Scholar]
- 32.Granger V, Faille D, Marani V, Noël B, Gallais Y, Szely N, Flament H, Pallardy M, Chollet-Martin S, de Chaisemartin L. Human blood monocytes are able to form extracellular traps. J Leukoc Biol 102: 775–781, 2017. doi: 10.1189/jlb.3MA0916-411R. [DOI] [PubMed] [Google Scholar]
- 33.Greene KE, Wright JR, Steinberg KP, Ruzinski JT, Caldwell E, Wong WB, Hull W, Whitsett JA, Akino T, Kuroki Y, Nagae H, Hudson LD, Martin TR. Serial changes in surfactant-associated proteins in lung and serum before and after onset of ARDS. Am J Respir Crit Care Med 160: 1843–1850, 1999. doi: 10.1164/ajrccm.160.6.9901117. [DOI] [PubMed] [Google Scholar]
- 34.Hodge S, Hodge G, Nairn J, Holmes M, Reynolds PN. Increased airway granzyme b and perforin in current and ex-smoking COPD subjects. COPD 3: 179–187, 2006. doi: 10.1080/15412550600976868. [DOI] [PubMed] [Google Scholar]
- 35.Ilumets H, Rytilä P, Demedts I, Brusselle GG, Sovijärvi A, Myllärniemi M, Sorsa T, Kinnula VL. Matrix metalloproteinases -8, -9 and -12 in smokers and patients with stage 0 COPD. Int J Chron Obstruct Pulmon Dis 2: 369–379, 2007. [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar S, Smith-Norowitz TA, Kohlhoff S, Apfalter P, Roblin P, Kutlin A, Harkema J, Ng SP, Doherty-Lyons S, Zelikoff JT, Hammerschlag MR. Exposure to cigarette smoke and Chlamydia pneumoniae infection in mice: Effect on infectious burden, systemic dissemination and cytokine responses: A pilot study. J Immunotoxicol 13: 77–83, 2016. doi: 10.3109/1547691X.2015.1006346. [DOI] [PubMed] [Google Scholar]
- 37.Larsson L, Pehrson C, Dechen T, Crane-Godreau M. Microbiological components in mainstream and sidestream cigarette smoke. Tob Induc Dis 10: 13, 2012. doi: 10.1186/1617-9625-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leberl M, Kratzer A, Taraseviciene-Stewart L. Tobacco smoke induced COPD/emphysema in the animal model-are we all on the same page? Front Physiol 4: 91, 2013. doi: 10.3389/fphys.2013.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JS, Su X, Rackley C, Matthay MA, Gupta N. Priming with endotoxin increases acute lung injury in mice by enhancing the severity of lung endothelial injury. Anat Rec (Hoboken) 294: 165–172, 2011. doi: 10.1002/ar.21244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu Q, Sakhatskyy P, Grinnell K, Newton J, Ortiz M, Wang Y, Sanchez-Esteban J, Harrington EO, Rounds S. Cigarette smoke causes lung vascular barrier dysfunction via oxidative stress-mediated inhibition of RhoA and focal adhesion kinase. Am J Physiol Lung Cell Mol Physiol 301: L847–L857, 2011. doi: 10.1152/ajplung.00178.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu Q, Sakhatskyy P, Newton J, Shamirian P, Hsiao V, Curren S, Gabino Miranda GA, Pedroza M, Blackburn MR, Rounds S. Sustained adenosine exposure causes lung endothelial apoptosis: a possible contributor to cigarette smoke-induced endothelial apoptosis and lung injury. Am J Physiol Lung Cell Mol Physiol 304: L361–L370, 2013. doi: 10.1152/ajplung.00161.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lugade AA, Bogner PN, Thatcher TH, Sime PJ, Phipps RP, Thanavala Y. Cigarette smoke exposure exacerbates lung inflammation and compromises immunity to bacterial infection. J Immunol 192: 5226–5235, 2014. doi: 10.4049/jimmunol.1302584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lutsar I, Friedland IR, Jafri HS, Wubbel L, Ahmed A, Trujillo M, McCoig CC, McCracken GH Jr. Factors influencing the anti-inflammatory effect of dexamethasone therapy in experimental pneumococcal meningitis. J Antimicrob Chemother 52: 651–655, 2003. doi: 10.1093/jac/dkg417. [DOI] [PubMed] [Google Scholar]
- 44.Maeno T, Houghton AM, Quintero PA, Grumelli S, Owen CA, Shapiro SD. CD8+ T Cells are required for inflammation and destruction in cigarette smoke-induced emphysema in mice. J Immunol 178: 8090–8096, 2007. doi: 10.4049/jimmunol.178.12.8090. [DOI] [PubMed] [Google Scholar]
- 45.Matthay MA, McAuley DF, Ware LB. Clinical trials in acute respiratory distress syndrome: challenges and opportunities. Lancet Respir Med 5: 524–534, 2017. doi: 10.1016/S2213-2600(17)30188-1. [DOI] [PubMed] [Google Scholar]
- 46.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest 122: 2731–2740, 2012. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Minagawa S, Lou J, Seed RI, Cormier A, Wu S, Cheng Y, Murray L, Tsui P, Connor J, Herbst R, Govaerts C, Barker T, Cambier S, Yanagisawa H, Goodsell A, Hashimoto M, Brand OJ, Cheng R, Ma R, McKnelly KJ, Wen W, Hill A, Jablons D, Wolters P, Kitamura H, Araya J, Barczak AJ, Erle DJ, Reichardt LF, Marks JD, Baron JL, Nishimura SL. Selective targeting of TGF-β activation to treat fibroinflammatory airway disease. Sci Transl Med 6: 241ra79, 2014. doi: 10.1126/scitranslmed.3008074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moazed F, Burnham EL, Vandivier RW, O’Kane CM, Shyamsundar M, Hamid U, Abbott J, Thickett DR, Matthay MA, McAuley DF, Calfee CS. Cigarette smokers have exaggerated alveolar barrier disruption in response to lipopolysaccharide inhalation. Thorax 71: 1130–1136, 2016. doi: 10.1136/thoraxjnl-2015-207886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Naucler P, Darenberg J, Morfeldt E, Ortqvist A, Henriques Normark B. Contribution of host, bacterial factors and antibiotic treatment to mortality in adult patients with bacteraemic pneumococcal pneumonia. Thorax 68: 571–579, 2013. doi: 10.1136/thoraxjnl-2012-203106. [DOI] [PubMed] [Google Scholar]
- 50.Nauseef WM. Insights into myeloperoxidase biosynthesis from its inherited deficiency. J Mol Med (Berl) 76: 661–668, 1998. doi: 10.1007/s001090050265. [DOI] [PubMed] [Google Scholar]
- 51.Nie L, Xiang R, Zhou W, Lu B, Cheng D, Gao J. Attenuation of acute lung inflammation induced by cigarette smoke in CXCR3 knockout mice. Respir Res 9: 82, 2008. doi: 10.1186/1465-9921-9-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nikota JK, Shen P, Morissette MC, Fernandes K, Roos A, Chu DK, Barra NG, Iwakura Y, Kolbeck R, Humbles AA, Stampfli MR. Cigarette smoke primes the pulmonary environment to IL-1α/CXCR-2-dependent nontypeable Haemophilus influenzae-exacerbated neutrophilia in mice. J Immunol 1950: 3134–3145, 2014. [DOI] [PubMed] [Google Scholar]
- 53.Örtqvist A, Hedlund J, Kalin M. Streptococcus pneumoniae: epidemiology, risk factors, and clinical features. Semin Respir Crit Care Med 26: 563–574, 2005. doi: 10.1055/s-2005-925523. [DOI] [PubMed] [Google Scholar]
- 54.O’Shaughnessy TC, Ansari TW, Barnes NC, Jeffery PK. Inflammation in bronchial biopsies of subjects with chronic bronchitis: inverse relationship of CD8+ T lymphocytes with FEV1. Am J Respir Crit Care Med 155: 852–857, 1997. doi: 10.1164/ajrccm.155.3.9117016. [DOI] [PubMed] [Google Scholar]
- 55.Owen CA, Hu Z, Barrick B, Shapiro SD. Inducible expression of tissue inhibitor of metalloproteinases-resistant matrix metalloproteinase-9 on the cell surface of neutrophils. Am J Respir Cell Mol Biol 29: 283–294, 2003. doi: 10.1165/rcmb.2003-0034OC. [DOI] [PubMed] [Google Scholar]
- 56.Owen CA, Hu Z, Lopez-Otin C, Shapiro SD. Membrane-bound matrix metalloproteinase-8 on activated polymorphonuclear cells is a potent, tissue inhibitor of metalloproteinase-resistant collagenase and serpinase. J Immunol 172: 7791–7803, 2004. doi: 10.4049/jimmunol.172.12.7791. [DOI] [PubMed] [Google Scholar]
- 57.Pan T, Nielsen LD, Allen MJ, Shannon KM, Shannon JM, Selman M, Mason RJ. Serum SP-D is a marker of lung injury in rats. Am J Physiol Lung Cell Mol Physiol 282: L824–L832, 2002. doi: 10.1152/ajplung.00421.2000. [DOI] [PubMed] [Google Scholar]
- 58.Parsons PE, Eisner MD, Thompson BT, Matthay MA, Ancukiewicz M, Bernard GR, Wheeler AP; NHLBI Acute Respiratory Distress Syndrome Clinical Trials Network . Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Crit Care Med 33: 1–6, 2005. doi: 10.1097/01.CCM.0000149854.61192.DC. [DOI] [PubMed] [Google Scholar]
- 59.Phipps JC, Aronoff DM, Curtis JL, Goel D, O’Brien E, Mancuso P. Cigarette smoke exposure impairs pulmonary bacterial clearance and alveolar macrophage complement-mediated phagocytosis of Streptococcus pneumoniae. Infect Immun 78: 1214–1220, 2010. doi: 10.1128/IAI.00963-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pugin J, Verghese G, Widmer MC, Matthay MA. The alveolar space is the site of intense inflammatory and profibrotic reactions in the early phase of acute respiratory distress syndrome. Crit Care Med 27: 304–312, 1999. doi: 10.1097/00003246-199902000-00036. [DOI] [PubMed] [Google Scholar]
- 61.Ralston AC, Webb RK, Runciman WB. Potential errors in pulse oximetry. I. Pulse oximeter evaluation. Anaesthesia 46: 202–206, 1991. doi: 10.1111/j.1365-2044.1991.tb09410.x. [DOI] [PubMed] [Google Scholar]
- 62.Romanovsky AA, Shido O, Sakurada S, Sugimoto N, Nagasaka T. Endotoxin shock: thermoregulatory mechanisms. Am J Physiol 270: R693–R703, 1996. [DOI] [PubMed] [Google Scholar]
- 63.Roviezzo F, Tsigkos S, Kotanidou A, Bucci M, Brancaleone V, Cirino G, Papapetropoulos A. Angiopoietin-2 causes inflammation in vivo by promoting vascular leakage. J Pharmacol Exp Ther 314: 738–744, 2005. doi: 10.1124/jpet.105.086553. [DOI] [PubMed] [Google Scholar]
- 64.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med 353: 1685–1693, 2005. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 65.Saetta M, Di Stefano A, Turato G, Facchini FM, Corbino L, Mapp CE, Maestrelli P, Ciaccia A, Fabbri LM. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 157: 822–826, 1998. doi: 10.1164/ajrccm.157.3.9709027. [DOI] [PubMed] [Google Scholar]
- 66.Sakhatskyy P, Gabino Miranda GA, Newton J, Lee CG, Choudhary G, Vang A, Rounds S, Lu Q. Cigarette smoke-induced lung endothelial apoptosis and emphysema are associated with impairment of FAK and eIF2α. Microvasc Res 94: 80–89, 2014. doi: 10.1016/j.mvr.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sakhatskyy P, Wang Z, Borgas D, Lomas-Neira J, Chen Y, Ayala A, Rounds S, Lu Q. Double-hit mouse model of cigarette smoke priming for acute lung injury. Am J Physiol Lung Cell Mol Physiol 312: L56–L67, 2017. doi: 10.1152/ajplung.00436.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sauve C, Azoulay-Dupuis E, Moine P, Darras-Joly C, Rieux V, Carbon C, Bédos JP. Efficacies of cefotaxime and ceftriaxone in a mouse model of pneumonia induced by two penicillin- and cephalosporin-resistant strains of Streptococcus pneumoniae. Antimicrob Agents Chemother 40: 2829–2834, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shapiro SD. Proteinases in chronic obstructive pulmonary disease. Biochem Soc Trans 30: 98–102, 2002. doi: 10.1042/bst0300098. [DOI] [PubMed] [Google Scholar]
- 70.Shen P, Morissette MC, Vanderstocken G, Gao Y, Hassan M, Roos A, Thayaparan D, Merlano M, Dorrington MG, Nikota JK, Bauer CMT, Kwiecien JM, Labiris R, Bowdish DME, Stevenson CS, Stämpfli MR. Cigarette smoke attenuates the nasal host response to Streptococcus pneumoniae and predisposes to invasive pneumococcal disease in mice. Infect Immun 84: 1536–1547, 2016. doi: 10.1128/IAI.01504-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sterling TD, Dimich H, Kobayashi D. Indoor byproduct levels of tobacco smoke: a critical review of the literature. J Air Pollut Control Assoc 32: 250–259, 1982. doi: 10.1080/00022470.1982.10465397. [DOI] [Google Scholar]
- 72.Su X, Lee JW, Matthay ZA, Mednick G, Uchida T, Fang X, Gupta N, Matthay MA. Activation of the alpha7 nAChR reduces acid-induced acute lung injury in mice and rats. Am J Respir Cell Mol Biol 37: 186–192, 2007. doi: 10.1165/rcmb.2006-0240OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thomas WR, Holt PG, Keast D. Cigarette smoke and phagocyte function: effect of chronic exposure in vivo and acute exposure in vitro. Infect Immun 20: 468–475, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Torres A, Blasi F, Dartois N, Akova M. Which individuals are at increased risk of pneumococcal disease and why? Impact of COPD, asthma, smoking, diabetes, and/or chronic heart disease on community-acquired pneumonia and invasive pneumococcal disease. Thorax 70: 984–989, 2015. doi: 10.1136/thoraxjnl-2015-206780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Toy P, Gajic O, Bacchetti P, Looney MR, Gropper MA, Hubmayr R, Lowell CA, Norris PJ, Murphy EL, Weiskopf RB, Wilson G, Koenigsberg M, Lee D, Schuller R, Wu P, Grimes B, Gandhi MJ, Winters JL, Mair D, Hirschler N, Sanchez Rosen R, Matthay MA; TRALI Study Group . Transfusion-related acute lung injury: incidence and risk factors. Blood 119: 1757–1767, 2012. doi: 10.1182/blood-2011-08-370932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tuomanen E, Hengstler B, Rich R, Bray MA, Zak O, Tomasz A. Nonsteroidal anti-inflammatory agents in the therapy for experimental pneumococcal meningitis. J Infect Dis 155: 985–990, 1987. doi: 10.1093/infdis/155.5.985. [DOI] [PubMed] [Google Scholar]
- 77.Tuomanen E, Liu H, Hengstler B, Zak O, Tomasz A. The induction of meningeal inflammation by components of the pneumococcal cell wall. J Infect Dis 151: 859–868, 1985. doi: 10.1093/infdis/151.5.859. [DOI] [PubMed] [Google Scholar]
- 78.Urbanowicz RA, Lamb JR, Todd I, Corne JM, Fairclough LC. Enhanced effector function of cytotoxic cells in the induced sputum of COPD patients. Respir Res 11: 76, 2010. doi: 10.1186/1465-9921-11-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vander Top EA, Perry GA, Snitily MU, Gentry-Nielsen MJ. Smoke exposure and ethanol ingestion modulate intrapulmonary polymorphonuclear leukocyte killing, but not recruitment or phagocytosis. Alcohol Clin Exp Res 30: 1599–1607, 2006. doi: 10.1111/j.1530-0277.2006.00192.x. [DOI] [PubMed] [Google Scholar]
- 80.Vila-Corcoles A, Aguirre-Chavarria C, Ochoa-Gondar O, de Diego C, Rodriguez-Blanco T, Gomez F, Raga X, Barnes L, Magarolas R, Esteban L. Influence of chronic illnesses and underlying risk conditions on the incidence of pneumococcal pneumonia in older adults. Infection 43: 699–706, 2015. doi: 10.1007/s15010-015-0801-y. [DOI] [PubMed] [Google Scholar]
- 81.Voss M, Wonnenberg B, Honecker A, Kamyschnikow A, Herr C, Bischoff M, Tschernig T, Bals R, Beisswenger C. Cigarette smoke-promoted acquisition of bacterial pathogens in the upper respiratory tract leads to enhanced inflammation in mice. Respir Res 16: 41, 2015. doi: 10.1186/s12931-015-0204-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Waisman DC, Tyrrell GJ, Kellner JD, Garg S, Marrie TJ. Pneumococcal peritonitis: still with us and likely to increase in importance. Can J Infect Dis Med Microbiol 21: e23–e27, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ware LB, Koyama T, Billheimer DD, Wu W, Bernard GR, Thompson BT, Brower RG, Standiford TJ, Martin TR, Matthay MA; NHLBI ARDS Clinical Trials Network . Prognostic and pathogenetic value of combining clinical and biochemical indices in patients with acute lung injury. Chest 137: 288–296, 2010. doi: 10.1378/chest.09-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ware LB, Lee JW, Wickersham N, Nguyen J, Matthay MA, Calfee CS; California Transplant Donor Network . Donor smoking is associated with pulmonary edema, inflammation and epithelial dysfunction in ex vivo human donor lungs. Am J Transplant 14: 2295–2302, 2014. doi: 10.1111/ajt.12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 342: 1334–1349, 2000. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 86.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 163: 1376–1383, 2001. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]
- 87.Weeks BS, Schnaper HW, Handy M, Holloway E, Kleinman HK. Human T lymphocytes synthesize the 92 kDa type IV collagenase (gelatinase B). J Cell Physiol 157: 644–649, 1993. doi: 10.1002/jcp.1041570326. [DOI] [PubMed] [Google Scholar]
- 88.Welgus HG, Campbell EJ, Cury JD, Eisen AZ, Senior RM, Wilhelm SM, Goldberg GI. Neutral metalloproteinases produced by human mononuclear phagocytes. Enzyme profile, regulation, and expression during cellular development. J Clin Invest 86: 1496–1502, 1990. doi: 10.1172/JCI114867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wiener-Kronish JP, Albertine KH, Matthay MA. Differential responses of the endothelial and epithelial barriers of the lung in sheep to Escherichia coli endotoxin. J Clin Invest 88: 864–875, 1991. doi: 10.1172/JCI115388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yao H, Edirisinghe I, Rajendrasozhan S, Yang S-R, Caito S, Adenuga D, Rahman I. Cigarette smoke-mediated inflammatory and oxidative responses are strain-dependent in mice. Am J Physiol Lung Cell Mol Physiol 294: L1174–L1186, 2008. doi: 10.1152/ajplung.00439.2007. [DOI] [PubMed] [Google Scholar]