

Summary
Hepatocellular carcinoma (HCC) is the most common primary liver cancer and the second most frequent cause of cancer-related mortality worldwide. The multikinase inhibitor sorafenib is the only treatment option for advanced HCC. Due to tumor heterogeneity, its efficacy greatly varies between patients and is limited due to adverse effects and drug resistance. Current in vitro models fail to recapitulate key features of HCCs. We report the generation of long-term organoid cultures from tumor needle biopsies of HCC patients with various etiologies and tumor stages. HCC organoids retain the morphology as well as the expression pattern of HCC tumor markers and preserve the genetic heterogeneity of the originating tumors. In a proof-of-principle study, we show that liver cancer organoids can be used to test sensitivity to sorafenib. In conclusion, organoid models can be derived from needle biopsies of liver cancers and provide a tool for developing tailored therapies.
Keywords: hepatocellular carcinoma, cholangiocellular carcinoma, liver, patient-derived organoids, patient-derived xenografts, needle biopsy, Biobank, genetic heterogeneity, drug response
Graphical Abstract
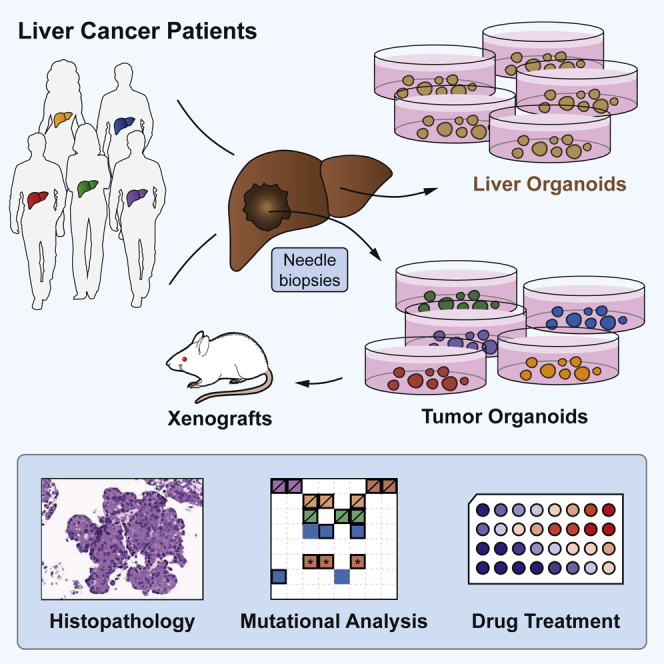
Highlights
-
•
Organoids can be derived from tumor needle biopsies of liver cancers
-
•
Organoids retain the morphology and tumor marker expression of the original tumors
-
•
Tumor organoids preserve the genetic heterogeneity of the originating tumors
-
•
Tumor organoids provide a tool for developing tailored therapies
Nuciforo et al. report the generation of patient-derived liver cancer organoids. Tissue was collected from all disease stages and major etiologies using an ultrasound-guided coaxial needle biopsy technique. Tumor organoids preserve the morphology and genetic heterogeneity of their originating tumors and provide opportunities to develop targeted therapies for hepatocellular carcinoma (HCC).
Introduction
Hepatocellular carcinoma (HCC) is the most common primary liver cancer, accounting for 90% of all liver cancers, and is the second most frequent cause of cancer-related mortality worldwide (Marquardt et al., 2015). Main risk factors include infection with hepatitis B virus, hepatitis C virus, alcoholic liver disease, nonalcoholic fatty liver disease, and nonalcoholic steatohepatitis. Intrahepatic cholangiocellular carcinoma (CCC) represents the second most common primary liver cancer with main risk factors including primary sclerosing cholangitis, cysts of the biliary duct, and parasitic infection with liver flukes (Marquardt et al., 2015).
Currently available treatment options for HCC are unsatisfactory. In the past, conventional chemotherapies have been extensively tested, but none of them have improved survival (Chen et al., 2015). Major progress came with the introduction of the multikinase inhibitor sorafenib in 2008. In a landmark trial, sorafenib was found to significantly prolong median survival from 7.9 in the control group to 10.7 months in the sorafenib treatment group (Llovet et al., 2008). In the following years >10 additional targeted drugs were tested, but all failed to meet clinical endpoints in phase III trials (Llovet and Hernandez-Gea, 2014). More recently, the sorafenib derivative regorafenib (Bruix et al., 2017) and the immune-checkpoint inhibitor nivolumab (El-Khoueiry et al., 2017) showed efficacy in second-line treatments for advanced HCC. However, given the limited efficacy of current HCC treatment options, there is an urgent need for new therapies for HCC.
A major obstacle in preclinical drug development is the lack of appropriate cell culture model systems. Current in vitro cell culture models of HCC are based on conventional hepatoma and hepatocarcinoma cell lines that fail to recapitulate key features of tumor tissues such as three-dimensional tumor architecture, cellular heterogeneity, and cell-cell interactions. The recently developed organoid technology could overcome these limitations because it allows differentiation of tissue stem cells into functional organ-like structures (Clevers, 2016). Indeed, the generation of three-dimensional organoid cultures from patient-derived tumors has been a major breakthrough in cancer biology. Over the past 3 years, tumor-derived organoids have been described for prostate (Gao et al., 2014), pancreatic (Boj et al., 2015), colorectal (van de Wetering et al., 2015), breast (Sachs et al., 2018), and bladder cancers (Lee et al., 2018).
In this study, we report the successful generation of tumor organoid cultures from needle biopsies obtained from patients with HCC. We demonstrate that HCC organoids recapitulate the histological features of the originating tumor in vitro. Moreover, injection of HCC organoids into immunodeficient mice results in the formation of tumors that also recapitulate the histological features of the original biopsy. Additionally, we show that HCC organoids maintain the genomic features of their originating tumors during long-term culturing for up to 32 weeks. Finally, we demonstrate that HCC organoids respond to sorafenib treatment with variable sensitivity.
Results
Establishment of HCC Organoid Cultures
We obtained tumor and non-tumor liver samples from patients undergoing diagnostic needle biopsies for suspected HCCs (Figures 1A and 1B). Biopsies were performed under ultrasound guidance using a coaxial needle biopsy technique that allows for obtaining up to five samples from the same location in a tumor (described in the Experimental Procedures). This multiple sampling procedure allowed a comprehensive characterization of all samples by histopathology for clinical diagnosis and Edmondson staging (Edmondson and Steiner, 1954), by immunohistochemical staining to identify tumor markers, and by whole-exome sequencing and RNA sequencing (RNA-seq) for molecular analysis. In total, we established 10 HCC-derived organoid lines from eight patients (Table 1). For patient 5, we generated organoid lines from two different tumor nodules (5-A and 5-B). For patient 12, we established two tumor organoid lines from two different locations of the same large tumor nodule (12-I and 12-II; Table 1). We also established organoid cultures from non-tumor liver biopsies in all patients (Figures 1C and S1A; Table S1). HCC organoids present morphologically as compact spheroids without a lumen but occasionally forming pseudoglands (Figure 1C), whereas non-tumor liver-derived organoids, originating from cholangiocytes, grow as single-cell layered cysts resembling the ductal epithelium (Huch et al., 2015) (Figure S1A). The underlying disease spectrum of our patient cohort encompasses the major risk factors for HCC, viral hepatitis, nonalcoholic fatty liver disease (NAFLD), and alcoholic liver disease (ALD) (Table 1). Furthermore, the cohort represents all different clinical stages of HCC according to the Barcelona Clinic Liver Cancer (BCLC) staging system (Llovet et al., 1999) (Table 1).
Figure 1.

Establishment of Organoid Cultures from Needle Biopsies of Hepatocellular Carcinoma and Paired Non-tumor Liver Tissues
(A) Schematic workflow of organoid generation from needle biopsies.
(B) Representative biopsy pieces of tumor and liver tissue used for organoid generation.
(C) Representative bright-field images of tumor and paired non-tumor liver tissue organoids from three different patients. Tumor organoids form compact spheroids, whereas liver organoids from non-tumor liver tissue grow as cystic structures. Organoids were imaged at the indicated passage numbers. Scale bar: 500 μm.
Table 1.
HCC and CCC Patient Data Table
| Patient | Biopsy ID | Sex | Age (Years) | Tumor | Liver Disease(s) | Cirrhosis | BCLC | Edmondson | Growth Pattern | AFP (IU/mL) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | C655 | male | 55 | HCC | HCV; ALD | no | C | III | trabecular-pseudoglandular | 269 |
| 2 | C798 | male | 73 | HCC | NAFLD | no | C | III | solid-trabecular | 20’377 |
| 5-A | C948 | male | 57 | HCC | HCV; ALD | yes | C | III | trabecular | 12′054 |
| 5-B | C949 | III | trabecular | |||||||
| 9 | C975 | male | 59 | HCC | HCV; ALD | yes | B | III | solid | 250 |
| 12-I | D045 | male | 69 | HCC | HCV | no | A | III | solid-trabecular | 7’852 |
| 12-II | D046 | III | solid-trabecular | |||||||
| 13 | D091 | female | 61 | CCC | none | no | – | – | – | 4.4 |
| 16 | D141 | male | 59 | LELCC | HBV | yes | – | – | – | 2.1 |
| 20 | D178 | female | 63 | CCC | none | no | – | – | – | 3.1 |
| 25 | D324 | male | 58 | HCC | HCV | yes | D | III | solid-trabecular | 104’710 |
| 27 | D359 | male | 86 | HCC | NAFLD | no | A | III | trabecular | 5’917 |
| 29-A | D386 | male | 80 | HCC | ALD; NAFLD | no | A | IV | solid | 49.8 |
Clinical staging was done according to the Barcelona Clinic Liver Cancer (BCLC) staging system (Llovet et al., 1999). Serum alpha-fetoprotein (AFP) concentrations were obtained from the clinical charts of the patients. Edmondson grade (Edmondson and Steiner, 1954) and the growth pattern were determined in each biopsy on H&E-stained sections by two experienced hepato-pathologists (M.S.M. and L.M.T.). All CCC tumors were poorly differentiated. AFP, alpha-fetoprotein; ALD, alcoholic liver disease; BCLC, Barcelona Clinic Liver Cancer; CCC, cholangiocellular carcinoma; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; LELCC, lymphoepithelioma-like cholangiocarcinoma; NAFLD, nonalcoholic fatty liver disease.
The success rate for the generation of HCC organoids was 26% based on the number of cultured tumor biopsies (10 out of a total of 38 HCC biopsies). HCC organoids were obtained in 8 of the 24 HCC patients included in the study (33%) (Table S1). We did not find a correlation between a number of clinically relevant patient characteristics and the success or failure to generate HCC organoids from their tumors (Figure 2A). On the other side, there was a strong correlation with the histopathological grading of the HCCs: all HCC organoids are derived from poorly differentiated tumors (Edmondson grades III and IV) (Figure 2A; Table 1). Furthermore, KI-67 staining of tumor biopsies showed significantly higher cancer cell proliferation rates in samples that could be propagated as tumor organoids compared with samples that failed (Figures S2A and S2B).
Figure 2.

Clinical, Histopathological, and Molecular Features of HCC Biopsies Used for Organoid Generation
(A) Color-coded table of patient characteristics of all biopsies (n = 38) used for organoid generation. Edmondson grade (Edmondson and Steiner, 1954) and the growth pattern were determined in each biopsy on H&E-stained sections by two experienced hepato-pathologists (M.S.M. and L.M.T.). Clinical data were extracted from the electronic patient information system of the hospital. Of note, only the Edmondson grade III was a significant determinant of successful organoid generation (p = 0.01, Fisher’s exact test, two-sided). For the Edmondson grade, calculations were performed per biopsy, whereas all other parameters were calculated per patient. ALD, alcoholic liver disease; AFP, alpha-fetoprotein; BCLC, Barcelona Clinic Liver Cancer; HBV, hepatitis B virus; HCV, hepatitis C virus; MVI, macrovascular invasion; NAFLD, nonalcoholic fatty liver disease.
(B and C) Unsupervised hierarchical clustering analysis.
(B) Biopsy (organoid) cohort (this study) combined with all HCCs from TCGA cohort.
(C) Biopsy (organoid) cohort (this study) combined with high-grade (Edmondson grades III and IV) HCCs from TCGA cohort.
The transcriptome data of the tumor biopsies were used to analyze the distribution of our samples within a reference set from The Cancer Genome Atlas (TCGA) (Cancer Genome Atlas Research Network, 2017) using an unsupervised hierarchical clustering analysis. Overall, our samples distributed evenly in the entire reference dataset, but the eight samples from which we could derive HCC organoids preferentially located in a subclass located at the left end of the clustering tree (Figure 2B). Because all of our HCC organoids originated from poorly differentiated HCCs (Edmondson grades III and IV), we also performed the clustering analysis using the subset of poorly differentiated HCCs from TCGA HCC database as a reference. In this analysis, our samples distributed over the entire spectrum of the tree (Figure 2C). We conclude that the organoid model system strongly selects for Edmondson grade III and IV HCCs, but within this group of poorly differentiated HCCs, there seems to be no further selection of a specific molecular subtype.
HCC Organoids Recapitulate the Histopathological Characteristics of the Originating Tumor
To investigate whether the histological characteristics of the originating tumors were preserved in the HCC organoids, two expert pathologists with expertise in hepatopathology performed histological analysis and diagnostic evaluation on the original biopsies and their tumor organoids on paraffin-embedded sections. Notably, HCC organoids maintained the growth pattern and differentiation grade of the originating primary tumors (Figure 3A). For example, HCC organoids derived from patient 2 displayed a solid growth pattern with an Edmondson differentiation grade III as in the originating tumor (Figure 3A). Likewise, tumor organoids from patient 12 formed pseudoglands (Figure 3A), a feature that was also present in the original HCC of this patient. Importantly, long-term culturing up to 1 year did not alter the histological properties of the HCC organoids (Figure S1B). As expected, immune cell infiltrates and tumor stromal cells were not propagated in the organoids.
Figure 3.

Histopathological Characteristics of HCC and CCC Organoids and Their Primary Tumors
(A) Histological sections of HCC and CCC organoids and their original tumors stained with H&E. The originating tumors display primarily a solid-trabecular architectural pattern with poor differentiation (Edmondson grades III and IV), features that are maintained in the corresponding HCC organoids. Arrowheads indicate pseudoglandular structures in HCC organoids and intracytoplasmic lumen in CCC organoids.
(B) AFP expression detected by immunohistochemistry on organoids and original biopsies.
(C) Expression of biliary markers KRT7 and KRT19 detected by immunohistochemistry on organoids and original biopsies. Organoids were imaged at the indicated passage numbers.
Scale bars: 100 μm.
We next assessed whether the expression of alpha-fetoprotein (AFP), a tumor marker for HCC (Table 1), was maintained in the corresponding organoids. Immunohistochemical analysis revealed consistent distribution and expression intensity of AFP between organoids and their original tumor biopsy tissue (Figure 3B). The same was true for three additional biomarkers commonly used for histological HCC diagnosis, Glypican 3, glutamine synthetase, and heat shock protein 70 (GPC3, GS, and HSP70, respectively) (Di Tommaso et al., 2009) (Figure S1C). Some of the HCCs also stained positive for the biliary cell markers Keratin 7 (KRT7) and Keratin 19 (KRT19). Again, the expression of these markers was maintained in the organoids (Figure 3C). Taken together, these results demonstrate that HCC organoids retain the phenotypic characteristics of their originating tumors.
HCC Organoids Give Rise to Tumors upon Injection into Immunodeficient Mice
To assess whether HCC organoids retained the ability to form bona fide tumors in mice, we injected HCC organoids subcutaneously into immunodeficient mice. So far, 6 of the 10 HCC organoids could be stably propagated in mice (Figures 4A and 4D). Two organoids failed to grow despite repeated transplantations. Two organoids were injected recently and the outcome could not yet be determined (Figure 4D). Of note, all successfully transplanted organoids gave rise to xenograft tumors that recapitulated the histopathological features and the tumor marker expression (Figure 4B) of the originating organoids and the original tumors. In contrast, and as expected, none of the paired non-tumor liver organoids gave rise to neoplasms.
Figure 4.

Histological Analysis of Xenografts Derived from HCC and CCC Organoids
(A) Growth curves of the xenograft tumors.
(B) Histological sections of xenograft tumors derived from HCC and CCC organoids stained with H&E. The HCC marker AFP and the biliary marker KRT7 were detected by immunohistochemistry. Scale bar: 100 μm.
(C) Trichrome and Alcian blue-PAS staining on biopsy, derivative organoids, and xenograft of patient 20. Collagen-rich areas representing the desmoplastic stroma reaction are colored in blue in Trichrome-stained sections. Mucin production appears light blue in sections stained with Alcian blue-PAS (arrowheads). Organoids were imaged at the indicated passage numbers. Scale bars: 100 μm.
(D) Statistics of the xenograft experiments.
HCC Organoids Retain the Somatic Genetic Alterations of the Originating Tumor
To assess whether the HCC organoids recapitulate the genetic alterations of the originating tumor, we subjected DNA from seven HCC organoid lines, their originating tumor biopsies, as well as the paired non-tumor biopsies to whole-exome sequencing (WES). WES was performed to median depths of 85×, 95×, and 50× in the organoids, biopsies, and non-tumor counterparts, respectively (Table S2). The number of somatic mutations in organoids (median 165, range 117–180) did not significantly differ from that of the corresponding tumor biopsies (median 146, range 127–207; p = 0.78, Mann-Whitney U test; Table S3).
Of the total somatic and the subgroup of non-synonymous somatic mutations found in the HCC biopsies, a median of 88% and 90%, respectively, was observed in the corresponding HCC organoids at early passage of culturing (P3–P4) (Figures 5A, S3, and S4; Tables S3 and S4). Similar proportions (all somatic: 86%; non-synonymous somatic: 88%) were observed in three representative cases where late-passage HCC organoids (≥P8) were profiled (Figures S3 and S7A; Tables S3 and S4). Nearly all non-synonymous somatic mutations in bona fide cancer genes, including all of those in TP53 (p.Arg209fs in patient 2, hotspot p.Arg342∗ in patient 9, and p.Val157Phe in patient 12-II), ARID1A (c.5125-2A > T in patients 5-A and 5-B), CTNNB1 (hotspot p.Ser45Ala and p.Arg528Cys in patients 5-A and 5-B), TSC1 (p.Gln767∗ in patient 2), and LRP1B (p.Cys2903Arg in patient 9), were found in the organoids at both early and late passages (Figures 5B, S3, and S7B). Of all the non-synonymous mutations in bona fide cancer genes, only two were lost in the corresponding HCC organoid (BRD7 p.Phe340Ile in patient 9; ARHGAP35 p.Glu1273Ala in patient 12-I). However, these mutations were subclonal in the originating HCC biopsies (Figures S3D and S4A), have not been previously reported in HCC, and are predicted to be passenger mutations (Table S4). Overall, we identified a median of 19 (range 8–29) and 14 (range 5–24) novel somatic and non-synonymous somatic mutations in the HCC organoids that were not present in the originating biopsies, representing a median of 15% and 12% of the mutations present in the HCC organoids, respectively. Most of these novel mutations did not occur in bona fide cancer genes. Indeed, in four of the seven tumor organoid lines, no additional non-synonymous mutations in cancer genes were identified. In patient 5-A, an ASXL1 mutation was found in both early- and late-passage HCC organoids, a PDGFRA mutation was found only in the early passage, and an AXIN2 mutation only in the late passage, but not in the originating HCC biopsies (Figures 5B and S7B). However, 23%–46% of the organoid-specific mutations were present in both early- and late-passage organoids originating from patient 2, patient 5-A, and patient 5-B (Table S3), strongly suggesting that a substantial proportion of the HCC organoid-specific mutations were likely present in the originating tumors at low frequencies, rather than being genuinely novel.
Figure 5.

Repertoire of Genetic Alterations Found in the HCC and CCC Organoids and Their Originating Tumors
(A) Venn diagrams illustrate the number of somatic non-synonymous mutations present in each HCC biopsy and their derivative HCC organoids. The dashed line denotes CCC-derived tumors and corresponding organoids.
(B) Repertoire of somatic non-synonymous mutations affecting cancer genes (Fujimoto et al., 2016, Kandoth et al., 2013, Lawrence et al., 2014, Cancer Genome Atlas Research Network, 2017). The effects of the mutations are color-coded according to the legend, with hotspots (Chang et al., 2016, Gao et al., 2017) colored in red. Multiple non-synonymous mutations in the same gene are indicated by an asterisk. Loss of heterozygosity of the wild-type allele of a mutated gene is represented by a diagonal bar, and mutations found to be clonal by ABSOLUTE (Carter et al., 2012) are indicated by a black box.
(C) Contour plots illustrate the distribution of the cancer cell fractions (CCFs) of somatic mutations in the tumors and their corresponding organoids, with the increasing shades of red indicating higher number of somatic mutations at a given CCF.
A detailed analysis of the cancer cell fraction (CCF) of the somatic mutations (i.e., the proportion of cancer cells harboring a given genetic alteration) between the organoid cultures and their matched originating tumor indicated that both harbored subclonal mutations (Figures 5C and S3–S6). For example, we observed similar extents of intratumor heterogeneity between the biopsies and organoids of patient 5 (Figures 5C and S3–S6). This has been previously observed in colorectal cancer organoids (van de Wetering et al., 2015) and is likely to be a genuine advantage of organoid cultures compared with cancer cell lines.
Copy number analysis showed that most amplifications were preserved in HCC organoids, and the overall patterns of copy number alterations were similar between the biopsies and the derivative HCC organoids at early and late passage (Figures S7C and S7D). For instance, the amplifications of chr1q21.3 (encompassing MCL1, SETDB1, ARNT, and MLLT11) in patient 5 (A and B), 8q24.13-24.23 (MYC and NDRG1) in patient 2, and 11q13.2-13.4 (CCND1, FGF19, FGF4, and FGF3) in patient 12-II were all found in their corresponding organoids. However, the heterogeneity observed at mutational level was also present at the copy number alteration (CNA) level. In fact, an amplification on 18q12.2 restricted to the HCC biopsy of patient 9, but not seen in the respective HCC organoids and two amplifications on 19q12 (CCNE1) and 19q13.2 (MAP3K10 and AKT2), was found only in the derivative HCC organoids.
We next investigated whether the biological and chemical processes that shape the mutational landscape were maintained in the organoid cultures. The analysis of the mutational signatures demonstrated that the mutational landscape of the HCC biopsies and the organoids was largely driven by mutational processes associated with signatures 1 (associated with aging), 3 (homologous recombination DNA repair deficiency), 6 (mismatch repair deficiency), and 16 (previously found in liver cancer with unknown etiology) (Alexandrov et al., 2013) (Figure S8). The mutational signatures were remarkably consistent between the organoids and the originating HCC biopsies. These results suggest that the mutational processes that drive tumor development were maintained in the organoids. Of note, the same patterns of mutational signatures were also maintained in late-passage organoids (Figure S8).
Taken together, these results demonstrate that the HCC organoids derived from tumor biopsies largely maintain the genetic alterations and mutational signatures observed in their originating HCCs. Importantly, mutations and amplifications affecting bona fide cancer genes found in the biopsies were preserved in the organoids. Furthermore, in line with previously published reports (Lee et al., 2018, van de Wetering et al., 2015), each HCC organoid line retained a remarkable intratumoral mutational heterogeneity.
Generation of Organoids from Intrahepatic CCCs
In our consecutive series of patients with suspected primary liver cancer who had a diagnostic needle biopsy (Table S1) there were four cases of intrahepatic CCCs and one case of a rare variant of CCC, a lymphoepithelioma-like cholangiocarcinoma. All five cases were poorly differentiated CCCs. In three cases we successfully established CCC-derived tumor organoids. Morphologically, CCC organoids resembled HCC organoids and formed compact spheroids (Figures 1C and 3A), whereas the corresponding non-tumor liver organoids formed single-cell layered epithelial cysts as expected (Figures 1C and S1A). CCC organoids displayed similar histological properties such as trabecular and/or solid growth with cytoplasmic eosinophilia and highly atypical cells like the poorly differentiated adenocarcinomas from which they were derived (Figure 3A). Furthermore, some cells within CCC organoids and their originating biopsies formed intracytoplasmic lumens and produced Mucin, two characteristic features of adenocarcinomas (Figures 3A and 4C). As expected, AFP expression was not detected in CCC tumor biopsies and organoids (Figure 3B). All CCC organoid lines expressed typical biliary markers such as KRT7 and KRT19 consistent with the expression pattern in the originating tumor biopsies (Figure 3C).
The genetic analysis revealed hyper-mutator phenotypes in patient 16 (with the lymphoepithelioma-like cholangiocarcinoma) and in patient 20 (Figure 5). Both tumors had >500 somatic mutations. In patient 16, most of the mutations were maintained in the organoids, whereas in patient 20, most were lost during the derivation of organoids (Figure 5A). Interestingly, the vast majority of somatic mutations in patient 16 were present in >80% of the tumor cells, whereas in patient 20, most of them were found only in subclonal cell populations (Figures S5 and S6; Table S4). It is likely that most of the subclones in the originating tumor were lost during the early steps of organoid culture. Patient 13 had 85 non-synonymous somatic mutations in the tumor. 36 were not preserved in the organoids. Most of them were subclonal and were not in bona fide cancer genes. The presumed cancer driver mutations were preserved (Figure 5).
We could successfully transplant the CCC organoids from patients 16 and 20 into immunodeficient mice. No xenografts could be established from patient 13 despite repeated transplantation attempts (Figure 4D). Surprisingly, histologic analysis of the xenograft tissue revealed tumor areas with a desmoplastic stroma reaction, a typical CCC feature that was also present in the originating tumors, but not in the organoids due to the lack of stromal cells (Figure 4C). This demonstrates that the capability to induce a desmoplastic reaction and thus to reproduce the tumor microenvironment in vivo is intrinsically programmed in CCC cells. Finally, Mucin-producing cells were also detected in the xenograft tumors from CCC organoids (Figure 4C).
HCC and CCC Organoids Display Variable Sensitivity to Sorafenib
In order to assess whether HCC-derived organoids would be a suitable system for preclinical drug development, we treated HCC organoid cultures with different concentrations of sorafenib and monitored cell viability with CellTiter-Glo. The concentration range was based on pharmacokinetic data from patients treated with sorafenib (Nexavar) (Abou-Alfa et al., 2006). In our in vitro assay, sorafenib reduced HCC organoid growth in a dose-dependent manner (Figure 6A) with half-maximal inhibitory concentration (IC50) values that varied by 2.5-fold from 2.0 to 5.0 μM (Figures 6B and 6C). Direct comparison of in vitro sorafenib activity with the clinical response was not feasible because none of the patients from whom we generated organoid cultures were treated with sorafenib. The validation of the organoid models as test systems for sorafenib response in vivo will require the recruitment of many more patients and the generation of a sizable number of additional HCC organoids derived from patients treated with sorafenib.
Figure 6.

Differential Drug Responses in Patient-Derived HCC and CCC Organoids under Sorafenib Treatment
HCC and CCC organoids were exposed to sorafenib at the indicated concentration for 6 days. DMSO-treated tumor organoids were used as control.
(A) Representative bright-field images of sorafenib-treated HCC organoids (patient 5-B). Scale bar: 200 μm.
(B) Sorafenib reduces cell viability of HCC and CCC organoids in a dose-dependent manner. The dashed line represents the IC50. Data are presented as the percentage of control DMSO-treated tumor organoids and are the mean of at least two independent experiments performed in duplicate.
(C) Differential IC50 (in μM) of HCC and CCC organoids shown as mean ± SEM. Patient numbers correspond to Table 1.
We also tested the efficacy of sorafenib on the three CCC organoid lines. Notably, a CCC organoid derived from a rare subtype of CCC (lymphoepithelioma-like CCC, patient 16) responded to sorafenib treatment in vitro with IC50 values comparable with sorafenib-sensitive HCC organoids (Figures 6B and 6C). Sorafenib is not an established treatment for CCCs, but it has recently been explored in a multicenter prospective study that showed a modest effect on disease control rate (Luo et al., 2017).
Taken together, our results demonstrate that organoids derived from biopsies of primary liver cancers can be used to test tumor-specific sensitivities to growth-inhibitory substances.
Discussion
In vitro studies of HCC biology have so far been restricted to a limited number of hepatoma and HCC cell lines. Most of them have been established decades ago, and it is unclear how well they represent the tumor biology of HCCs. HCC organoids overcome many of the limitations of these cell lines. They can be directly linked to a patient and to spatiotemporal information such as a specific tumor nodule or a metastasis, or a specific time during the evolution of the cancer in a patient. Importantly, we found that the organoids are polyclonal and thereby preserve an important feature of the originating tumor that is linked to cancer evolution, immune evasion, and resistance to oncostatic and targeted therapies. Patient-derived tumor organoids have recently been described for prostate (Gao et al., 2014), pancreatic (Boj et al., 2015), colorectal (van de Wetering et al., 2015), breast (Sachs et al., 2018), and bladder cancer (Lee et al., 2018). Broutier et al. (2017) reported the establishment of organoid cultures derived from resections of primary liver cancers. In our study, we report the establishment of organoids from HCC needle biopsies, overcoming a major limitation of approaches that use surgically resected HCC specimens. Indeed, surgical resection is a treatment option for the minority of HCC patients with early tumor stages and/or a non-cirrhotic liver. These patients usually do not receive systemic therapies. In contrast, biopsies can also be obtained from patients with intermediate and advanced tumor stages who frequently receive systemic therapies. The use of biopsies therefore allows for establishment of an organoid biobank that reflects the entire spectrum of HCCs. Furthermore, such a biobank can be enriched to include clinical data such as response to treatments, resistance development, and survival.
The use of tumor biopsies also allowed a comparison of tumor and organoids. Of note, several biopsies from the same location in a tumor could be obtained using an ultrasound-guided coaxial biopsy technique. The resulting set of biopsies is mutually representative and can be used for multidimensional analysis. We found a striking similarity between originating tumors and organoids in routine histopathology and immunostaining analysis. Moreover, the individual tumor features were also maintained after transplantations into immunodeficient mice. We conclude that these morphological features are inherently programmed in the tumor cells and are not subject to the tumor environment (Figure 3). Furthermore, the expression of well-established HCC biomarkers, as well as the mutational landscape, is preserved in HCC organoids (Figure 5). When compared with the previous study by Broutier et al. (2017) that used resected specimens, our tissue collection procedure allowed for collection of non-tumor liver biopsies at a site distant from the tumor nodule(s) to generate non-tumor liver organoids for all patients and to perform patient-specific normalization of our genomic and transcriptomic data.
In our series of tumor biopsies we derived HCC organoids with a success rate of ∼26% (per number of biopsies) and 33% (per number of patients). This is lower compared with the reported success rates for pancreatic cancer (75%–83%) (Boj et al., 2015) and colorectal cancers (90%) (van de Wetering et al., 2015), possibly because the cell of origin of HCCs, the hepatocytes, lack features of epithelial stem cells that favor their propagation in the organoid culture system.
However, our success rate is in line with a recent study by Pauli et al. (2017) reporting an average success rate of ∼38% across different tumor types. In their report, the authors used small-needle biopsies as tumor tissue source in some of their cases and conclude that the major limitation to establish tumor organoids was the insufficient amount of fresh tissue. Indeed, given the limited amount of starting material (needle biopsies), we could not set up a systematic screening of different culture systems to improve the derivation efficiency. Broutier et al. (2017) tried to optimize the culture conditions for the generation of liver cancer organoids by removing some of the growth factors contained in the original media recipe in order to reduce the outgrowth of normal liver organoids. However, the changes they performed did not result in organoid generation from all of their tumor specimen. Based on informed guesses, we tried to optimize culture conditions in a small number of cases where we had more than one biopsy as a starting material. For example, we removed compounds with potential negative effects on HCC proliferation such as Forskolin, N-acetyl-L-cysteine, nicotinamide, and HGF from the medium, and added FGF19, a factor with potential growth-promoting effects for HCCs. However, these limited efforts did not result in the establishment of additional HCC organoid lines. Nevertheless, we anticipate that additional efforts in refining the media recipes (Fujii et al., 2016), and possibly also the use of tailored 3D matrices (Gjorevski et al., 2016), to specifically accommodate the growth of HCC- and CCC-derived organoids, respectively, will improve the success rate. This would be a prerequisite for using HCC organoids as patient-specific in vitro models of drug sensitivity with the aim to inform treatment decisions on an individualized basis. Of note, the time required to expand the organoid cultures for drug testing is presently 4–12 weeks. Urgent treatment decisions will therefore not rely on in vitro drug testing of individual patient-derived organoids, even if the success rate should be much higher in the future. More realistically, systematic drug testing in a large enough number of HCC organoids will allow to predict patient responses to different treatments based on matching molecular characteristics of tumor biopsies and organoids (Drost and Clevers, 2018).
One other potential reason for the limited success rate could be that HCC organoids can be generated only from a restricted subset of HCCs. We therefore compared clinical, histopathological, and molecular features of HCCs that could be propagated as organoids with HCCs that could not. No significant correlations were found with a comprehensive set of clinical data (Figure 2A). Indeed, HCC organoids could be derived from patients with all major underlying liver diseases and different clinical stages of HCC, demonstrating the potential of the organoid technique for building up larger biobanks representing the entire clinical spectrum of liver cancer. On the other side, there was a significant correlation with one of the histopathological features, the Edmondson grade of the tumors. HCC organoids could only be generated from poorly differentiated tumors. It is conceivable that the generation of HCC organoids requires a cell proliferation rate threshold that is not reached in highly differentiated, slow-growing Edmondson grade I and II tumors. This is supported by our finding that the proliferation marker KI-67 and cell-cycle pathway genes were upregulated in tumor biopsies with successful organoid derivations compared with those where organoids could not be generated (Figures S2A and S2B; Table S5). These data are in line with those from Broutier et al. (2017) showing that only moderately to poorly differentiated HCCs with a KI-67 index >5% were able to generate organoids. Of note, other histopathological features in our HCC cohort such as the percentage of viable tumor cells in the biopsy, the amount of stroma or immune cell infiltration, the growth pattern of the tumor, or the degree of tumor necrosis were not correlated with success or failure of organoid derivation (Table S1).
Finally, to investigate whether HCC organoids can only be derived from a specific molecular subtype of HCC, we compared transcriptome data of all of our tumor biopsies with a reference set of poorly differentiated HCCs from the TCGA database (Cancer Genome Atlas Research Network, 2017). We found no enrichment of our samples in distinct subclasses of HCCs (Figure 2C). We conclude that the organoid technique described in this manuscript allows the generation of a heterogeneous tumor organoid biobank that is a representative sample of the entire clinical, histopathological, and molecular spectrum of poorly differentiated HCCs.
The analysis of the relative frequency of non-synonymous somatic mutations in the tumor biopsies and the HCC organoids revealed the expected preservation of highly prevalent putative driver mutations. However, it also revealed that mutations present in only a subset of tumor cells, i.e., subclone-specific mutations, were also preserved, often with surprisingly similar relative frequencies between tumor biopsies and HCC organoids (Figures 5C and S3–S6; Table S4). This is somewhat unexpected, because it does not support a model where tumor organoids are derived from a single cancer stem cell. There is compelling evidence that intestinal organoids can be derived from single LGR5-positive stem cells (Clevers, 2016). For liver-derived organoids, this is less clear (Huch et al., 2015). It remains to be investigated whether the stem cell model is applicable for HCC tumor organoids. In any case, the coexistence of different cancer cell subclones with different sensitivities to targeted therapies is an important factor linked to therapy failure (Fisher et al., 2013). Therefore, we believe that organoid models will play a major role in the development of novel drug candidates able to target different genetic subclones within tumors to impede the selection of resistant cells present at low frequencies at therapy onset. Furthermore, the knowledge of the patient-specific genetic background will allow the unique opportunity to correlate response to specific drugs with putative driver mutations, a prerequisite for future efforts of personalized management of targeted therapies.
Experimental Procedures
Human Tissues and Biopsy Procedure
Human tissues were obtained from patients undergoing diagnostic liver biopsy at the University Hospital Basel. Written informed consent was obtained from all patients. The study was approved by the ethics committee of the northwestern part of Switzerland (Protocol Number EKNZ 2014-099). Ultrasound (US)-guided needle biopsies were obtained from tumor lesion(s) with a coaxial liver biopsy technique that allows taking several biopsy samples through a single biopsy needle tract. After local anesthesia, the introducer needle was advanced 2–3 cm into the liver parenchyma. In case of a focal lesion, the needle was positioned precisely at the tumor border. The trocar of the introducer needle was removed, and up to five cylindrical biopsies of ∼1 mm diameter and 10–30 mm in length were obtained with an automatic spring-loaded biopsy needle (BioPince). The introducer needle was kept in place during the entire procedure to ensure that all specimens came from the same area of the tumor. One cylinder was fixed in formalin and paraffin-embedded for diagnosis and staging. Additional cylinders were immediately snap-frozen in liquid nitrogen for later use in DNA and RNA extraction or embedded in O.C.T. (Tissue-Tek) and frozen using standard procedures. For organoid generation, biopsy pieces were placed in advanced DMEM/F-12 (GIBCO). For control tissue, all patients who underwent US-guided HCC biopsy also got a biopsy of the liver parenchyma at a site distant from the tumor. The needle tract was filled with absorbable gelatin sponge before removal of the introducer needle.
Organoid Culture
Tumor biopsy fragments designated for organoid generation typically measured ∼1 mm × 5–10 mm corresponding to a volume of ∼3.9–7.9 mm3. They were transported to the laboratory on ice and further processed for organoid generation within 20 min after collection. For tumor organoid generation, biopsies underwent a limited digestion to small-cell clusters. We avoided complete digestion into single cells because it has been reported that preservation of cell-cell contacts enhances derivation efficiency (Kondo et al., 2011). Tumor tissue was minced and shortly (maximum [max.] 2–4 min) digested with 2.5 mg/mL collagenase IV (Sigma), 0.1 mg/mL DNase (Sigma) at 37°C. The yield of the procedure varied because of differences in the size of the tumor biopsy available for the generation of organoids and the variable content of viable tumor cells in the biopsies. Cell clusters were then seeded into reduced growth factor BME2 (Basement Membrane Extract, Type 2; Amsbio). After polymerization of BME2, expansion medium (Huch et al., 2015) was added to the cells. The composition is advanced DMEM/F-12 (GIBCO) supplemented with 1:50 B-27 (GIBCO), 1:100 N-2 (GIBCO), 10 mM nicotinamide (Sigma), 1.25 mM N-acetyl-L-cysteine (Sigma), 10 nM [Leu15]-gastrin (Sigma), 10 μM forskolin (Tocris), 5 μM A83-01 (Tocris), 50 ng/mL EGF (PeproTech), 100 ng/mL FGF10 (PeproTech), 25 ng/ml HGF (PeproTech), 10% RSpo1-conditioned medium, (homemade), and 30% Wnt3a-conditioned medium (homemade). In the few cases for which enough biopsy material was available, we tried an adapted version of the culture medium in comparison with the normal one. The adapted medium lacked some of the original components reported to have a negative effect on HCC cell proliferation (forskolin, N-acetyl-L-cysteine, nicotinamide, HGF) and contained FGF19 because of the frequent amplification of the FGF19 gene detected in HCCs and its positive effect on proliferation of HCC cells. However, these attempts did not result in the establishment of additional HCC organoid lines. Organoid cultures from non-tumor liver biopsies were generated as previously described (Huch et al., 2015). Tumor organoids were passaged after dissociation with 0.25% Trypsin-EDTA (GIBCO). Non-tumor liver organoids were passaged by mechanical dissociation through a fire-polished Pasteur-pipette or incubation in 0.25% Trypsin-EDTA (GIBCO) for 2 min. Cryovials were prepared at regular intervals by dissociating organoids and resuspending in Recovery Cell Culture Freezing Medium (GIBCO) prior to freezing.
We could prepare frozen stocks of early (≤P4) passages from all the samples that yielded tumor organoids. All organoid lines could be kept in long-term cultures with regular splitting for at least 1 year. All organoid cultures were regularly tested for Mycoplasma contamination with the MycoAlert Mycoplasma Detection Kit (Lonza) according to the manufacturer’s instructions.
Organoid Xenotransplantation
All experiments involving organoid transplantations into mice were performed in strict accordance with Swiss law and were approved by the ethics committee of the northwestern part of Switzerland (Protocol Number EKNZ 2014-099) and the Animal Care Committee of the Canton Basel-Stadt, Switzerland. Tumor organoids, corresponding to 1 × 106 cells, were released from BME2 by incubating in Cell Recovery Solution (Corning), resuspended in ∼100 μL 50:50 (v/v) BME2:expansion medium, and injected subcutaneously into immunodeficient non-obese diabetic (NOD) severe combined immunodeficiency (SCID) gamma (NSG) mice (The Jackson Laboratory) at young age (6–8 weeks). Paired non-tumor liver organoids were used as negative control.
Histology and Immunohistochemistry
Liver biopsies from tumoral and non-tumor tissue, as well as tumor organoid xenografts, were fixed in 4% phosphate-buffered formalin and embedded in paraffin using standard procedures. Additional biopsies were also embedded in O.C.T. (Tissue-Tek) and frozen using standard procedures. Tumor organoids were released from BME2 by incubating in Cell Recovery Solution (Corning) following the manufacturer’s instructions. Organoids were then fixed in freshly prepared 4% formalin solution in PBS for 30 min at room temperature following dehydration and paraffin embedding. Sections were subjected to H&E, Masson’s trichrome, Alcian blue-periodic acid-Schiff (PAS), as well as immunohistochemical staining, using standard procedures. Histopathological evaluation was assessed by two board-certified pathologists (M.S.M. and L.M.T.). Tumors were classified based on architecture and cytological features, and graded according to the Edmondson grading system (Edmondson and Steiner, 1954).
For immunohistochemistry, the following primary antibodies were used for automated staining on a Benchmark XT device (Ventana Medical Systems): AFP (Ventana catalog number [Cat. No.] 760-2603), GS (Ventana Cat. No. 760-4898), GPC3 (Ventana Cat. No. 790-4564), HSP70 (Biocare Medical CM407A), Keratin 7 (Ventana Cat. No. 790-4462), Keratin 19 (Ventana Cat. No. 760-4281), and KI-67 (Ventana Cat. No. 760-4286).
Drug Treatment
Sorafenib tosylate (Cat. No. S-8502) was purchased from LC Laboratories, dissolved in DMSO at 10 mM aliquots, and stored at −20°C. Tumor organoids were plated at a density of 5 × 103 cells in 15 μL BME2 droplets in order to form organoids. At day 6, sorafenib was added to the medium, and cell viability was measured after 6 days using CellTiter-Glo 3D reagent (Promega). Luminescence was measured on a Synergy H1 Multi-Mode Reader (BioTek Instruments). Results were normalized to vehicle (=100% DMSO). Curve fitting was performed using Prism (GraphPad) software and the nonlinear regression equation. All experiments were performed at least two times in duplicate. Results are shown as mean ± SEM.
DNA and RNA Extraction
Genomic DNA from tumor organoids was extracted using the DNeasy Blood & Tissue kit (Qiagen) following the manufacturer’s instructions. Genomic DNA and total RNA from biopsies were extracted using the ZR-Duet DNA and RNA MiniPrep Plus kit (Zymo Research) following the manufacturer’s instructions. Prior to extraction, biopsies were crushed in liquid nitrogen to facilitate lysis. Extracted DNA was quantified using the Qubit Fluorometer (Invitrogen).
Whole-Exome Sequencing
DNA extracted from eight HCC biopsy-derived organoid lines (patients 1, 2, 5-A, 5-B, 9, 12-I, 12-II, and 25), three CCC biopsy-derived organoid lines (patients 13, 16, and 20), the corresponding original biopsies, and the control paired non-tumor biopsies were sequenced using whole-exome sequencing. The eight HCC biopsies were derived from six patients and for three of the biopsies, early- and late-passage organoids were profiled (Table S2). The tumor biopsy sample corresponding to patient 1 had to be excluded from further analyses because of low tumor cell content in the biopsy. Whole-exome capture was performed using the SureSelectXT Clinical Research Exome (Agilent) platform according to the manufacturer’s guidelines. Sequencing was performed on an Illumina HiSeq 2500 at the Genomics Facility Basel according to the manufacturer’s guidelines. Paired-end 101-bp reads were generated.
Whole-Exome Sequencing Analysis
Sequence reads were aligned to the reference human genome GRCh37 using Burrows-Wheeler Aligner (BWA, v0.7.12) (Li and Durbin, 2009). Local realignment, duplicate removal and base quality adjustment were performed using the Genome Analysis Toolkit (GATK, v3.6) (McKenna et al., 2010) and Picard (http://broadinstitute.github.io/picard/). Somatic single nucleotide variants (SNVs) and small insertions and deletions (indels) were detected using MuTect (v1.1.4) (Landau et al., 2013) and Strelka (v1.0.15) (Saunders et al., 2012), respectively. We filtered out SNVs and indels outside of the target regions: those with variant allelic fraction (VAF) of <1% and/or those supported by <3 reads. We excluded variants for which the tumor VAF was <5 times that of the paired non-tumor VAF, as well as those found at >5% global minor allele frequency of dbSNP (build 137). We further excluded variants identified in at least two of a panel of 123 non-tumor samples, including the 4 non-tumor samples included in the current study, captured and sequenced using the same protocols using the artifact detection mode of MuTect2 implemented in GATK. All indels were manually inspected using the Integrative Genomics Viewer (Thorvaldsdóttir et al., 2013). To account for the presence of somatic mutations that may be present below the limit of sensitivity of somatic mutation callers, we used GATK Unified Genotyper to interrogate the positions of all unique mutations in all samples from a given patient to define the presence of additional mutations.
Allele-specific CNAs were identified using FACETS (v0.5.5) (Shen and Seshan, 2016) as previously described (Piscuoglio et al., 2016), which performs a joint segmentation of the total and allelic copy ratio and infers allele-specific copy number states. Somatic mutations associated with the loss of the wild-type allele (i.e., loss of heterozygosity [LOH]) were identified as those where the lesser (minor) copy number state at the locus was 0. All mutations on chromosome X in male patients were considered to be associated with LOH. All gene amplifications and homozygous deletions were visually inspected using plots of raw log2 and allelic copy ratios. Copy number states were collapsed to the gene level based on the median values to coding gene resolution based on all coding genes retrieved from the Ensembl (release GRCh37.p13).
The CCF of each mutation on the autosomes was inferred using the number of reads supporting the reference and the alternate alleles, and the segmented log2 ratio from WES as input for ABSOLUTE (v1.0.6) (Carter et al., 2012). Solutions from ABSOLUTE were manually reviewed as recommended (Carter et al., 2012, Landau et al., 2013). A mutation was classified as clonal if its clonal probability, as defined by ABSOLUTE, was >50%, or if the upper bound of the 95% confidence interval of its CCF crosses 1. Mutations that did not meet the above criteria were considered subclonal.
Cancer genes were annotated according to the cancer gene lists described by Kandoth et al. (2013) (127 significantly mutated genes), Lawrence et al. (2014) (Cancer5000-S gene set), Fujimoto et al. (2016), or the TCGA (Cancer Genome Atlas Research Network, 2017). Mutations affecting hotspot residues (Chang et al., 2016, Gao et al., 2017) were annotated. Pathogenicity of missense mutations was predicted using CHASM (liver cancer predictor, viral or non-viral as appropriate) (Carter et al., 2009) and FATHMM (cancer predictor) (Shihab et al., 2013).
Decomposition of the mutational signature was performed using deconstructSigs (Rosenthal et al., 2016), based on the set of 30 mutational signatures (“signature.cosmic,” based on the signatures at https://cancer.sanger.ac.uk/cosmic/signatures; Alexandrov et al., 2013, Nik-Zainal et al., 2016).
RNA-Seq
RNA extracted from all HCC biopsies (n = 38; Table S1) and the paired non-tumor biopsies were sequenced using RNA-seq. Tumor samples corresponding to patients 1, 7-B (C959), 15-B, and 29-A had to be excluded from further analyses because of low tumor cell content in the biopsy. 200 ng total RNA was used for RNA-seq library prep with the TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Gold (Illumina) according to manufacturer’s specifications. SR126 sequencing was performed on an Illumina HiSeq 2500 using v4 SBS chemistry at the Genomics Facility Basel according to the manufacturer’s guidelines. Primary data analysis was performed with the Illumina RTA version 1.18.66.3.
RNA Sequencing Analysis
Sequence reads were aligned to the human reference genome GRCh37 by STAR (Dobin et al., 2013) using the two-pass approach. Transcript quantification was performed using RSEM (Li and Dewey, 2011). For unsupervised cluster analysis, we retrieved the TCGA Liver dataset RNA-seq data (“V2_MapSpliceRSEM”) from the Genomics Data Commons Data Portal (Cancer Genome Atlas Research Network, 2017). We performed gene-level upper quartile normalization of the combined dataset to the fixed threshold 1,000 as described in the TCGA study (Cancer Genome Atlas Research Network, 2017). Genes whose expression was quantified to zero by RSEM (Li and Dewey, 2011) in >75% of the samples were removed. RSEM values were subsequently log2-transformed, adding 0.5 to RSEM values prior to transformation. To identify genes with variable expression for clustering, genes with standard deviation < 2 were excluded. Batch correction using the edgeR package (Nikolayeva and Robinson, 2014) was performed to correct for systematic biases between the datasets. Cluster analysis was performed using hierarchical clustering using the Ward method and with a 1-Pearson correlation distance (Cancer Genome Atlas Research Network, 2017).
For the TCGA HCC cohort (Cancer Genome Atlas Research Network, 2017), images of diagnostic H&E slides were retrieved from the cbioportal (http://www.cbioportal.org; accessed December 2017) (Gao et al., 2013) and reviewed by two expert hepato-pathologists (M.S.M. and L.M.T.) according to the Edmondson grading system (Edmondson and Steiner, 1954) for comparison purposes. Differential expression analysis between biopsies that did or did not yield organoids was performed using the edgeR package (Nikolayeva and Robinson, 2014). Specifically, genes with <1 count per million in more than five HCC biopsies were removed. Normalization was performed using the “TMM” (weighted trimmed mean) method, and differential expression was assessed using the quasi-likelihood F-test.
Statistical Analysis
p values were calculated with Fisher’s exact test or Mann-Whitney test using Prism (GraphPad) software, as specified in the Results section and in the figure legends.
Data and Software Availability
Sequence data have been deposited at the European Genome-phenome Archive (EGA), which is hosted by the EBI and the CRG, under accession number EGAS00001003115.
Acknowledgments
We thank all the patients who participated in this study, Hans Clevers for providing the Wnt3a cell line and Calvin Kuo for providing the RSpo1-Fc cell line, Petra Hirschmann for performing histopathological staining, Christian Beisel (Genomics Facility Basel) for performing whole-exome and RNA-seq, and Xueya Wang and Sylvia Ketterer for excellent technical assistance. This work was funded by European Research Council Synergy grant 609883 (MERiC) and by SystemsX.ch grant MERiC. L.M.T. and S.P. had support from Oncosuisse (KFS-3995-08-2016) and S.P. from the Swiss National Science Foundation (Ambizione PZ00P3_168165).
Author Contributions
M.H.H., S.N., and I.F. conceived the study and designed experiments; M.H.H. and T. Boldanova recruited patients, performed biopsies, and collected and curated clinical annotation data; S.N., T. Blumer, and D.C. performed the experiments; M.S.M. and L.M.T. performed the histopathological analysis; S.P. and C.K.Y.N. processed, computed, and analyzed the genomics data; F.R. and G.S. provided essential reagents and protocols for establishment of organoid cultures; all authors were involved in data analysis and interpretation of the results; M.H.H. coordinated the study; S.N., I.F., S.W., S.P., C.K.Y.N., and M.H.H. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: July 31, 2018
Footnotes
Supplemental Information includes eight figures and five tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.07.001.
Supplemental Information
Samples were obtained between July 2015 and September 2017 and are ordered chronologically according to the date of the biopsy. If more than one tumor nodule was biopsied in a given patient, the different nodules are designated with letters (e.g., 5-A and 5-B). If more than one biopsy was taken from different locations of the same (large) nodule, the different locations are labeled with roman numbers (e.g., 12-I and 12-II). The five cholangiocellular carcinoma cases are shown in italic font. Sex, age, background liver disease, clinical staging according to the Barcelona Clinic Liver Cancer (BCLC) staging system and alpha-fetoprotein (AFP) serum concentrations were obtained from the clinical charts of the patients. The viable tumor cell content, Edmondson grade and the growth pattern were determined in each biopsy on Hematoxylin and Eosin stained sections by two experienced hepato-pathologists (M.S.M. and L.M.T.). All CCC tumors were poorly differentiated. ALD: Alcoholic Liver Disease; AFP: Alpha-Fetoprotein; BCLC: Barcelona Clinic Liver Cancer; CCC: Cholangiocellular Carcinoma; HBV: Hepatitis B Virus; HCC: Hepatocellular Carcinoma; HCV: Hepatitis C Virus; LELCC: Lymphoepithelioma-like Cholangiocarcinoma; NAFLD: Nonalcoholic Fatty Liver Disease; n.a.: not applicable; n.e.: not evaluable because of insufficient tumor cell content in the biopsy.
References
- Abou-Alfa G.K., Schwartz L., Ricci S., Amadori D., Santoro A., Figer A., De Greve J., Douillard J.Y., Lathia C., Schwartz B. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2006;24:4293–4300. doi: 10.1200/JCO.2005.01.3441. [DOI] [PubMed] [Google Scholar]
- Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Børresen-Dale A.L., Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium. ICGC MMML-Seq Consortium. ICGC PedBrain Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boj S.F., Hwang C.I., Baker L.A., Chio I.I., Engle D.D., Corbo V., Jager M., Ponz-Sarvise M., Tiriac H., Spector M.S. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–338. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broutier L., Mastrogiovanni G., Verstegen M.M., Francies H.E., Gavarró L.M., Bradshaw C.R., Allen G.E., Arnes-Benito R., Sidorova O., Gaspersz M.P. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017;23:1424–1435. doi: 10.1038/nm.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruix J., Qin S., Merle P., Granito A., Huang Y.-H., Bodoky G., Pracht M., Yokosuka O., Rosmorduc O., Breder V., RESORCE Investigators Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389:56–66. doi: 10.1016/S0140-6736(16)32453-9. [DOI] [PubMed] [Google Scholar]
- Carter H., Chen S., Isik L., Tyekucheva S., Velculescu V.E., Kinzler K.W., Vogelstein B., Karchin R. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res. 2009;69:6660–6667. doi: 10.1158/0008-5472.CAN-09-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter S.L., Cibulskis K., Helman E., McKenna A., Shen H., Zack T., Laird P.W., Onofrio R.C., Winckler W., Weir B.A. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M.T., Asthana S., Gao S.P., Lee B.H., Chapman J.S., Kandoth C., Gao J., Socci N.D., Solit D.B., Olshen A.B. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2016;34:155–163. doi: 10.1038/nbt.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.W., Ou T.M., Hsu C.W., Horng C.T., Lee C.C., Tsai Y.Y., Tsai C.C., Liou Y.S., Yang C.C., Hsueh C.W., Kuo W.H. Current systemic treatment of hepatocellular carcinoma: a review of the literature. World J. Hepatol. 2015;7:1412–1420. doi: 10.4254/wjh.v7.i10.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H. Modeling development and disease with organoids. Cell. 2016;165:1586–1597. doi: 10.1016/j.cell.2016.05.082. [DOI] [PubMed] [Google Scholar]
- Di Tommaso L., Destro A., Seok J.Y., Balladore E., Terracciano L., Sangiovanni A., Iavarone M., Colombo M., Jang J.J., Yu E. The application of markers (HSP70 GPC3 and GS) in liver biopsies is useful for detection of hepatocellular carcinoma. J. Hepatol. 2009;50:746–754. doi: 10.1016/j.jhep.2008.11.014. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost J., Clevers H. Organoids in cancer research. Nat. Rev. Cancer. 2018;18:407–418. doi: 10.1038/s41568-018-0007-6. [DOI] [PubMed] [Google Scholar]
- Edmondson H.A., Steiner P.E. Primary carcinoma of the liver: a study of 100 cases among 48,900 necropsies. Cancer. 1954;7:462–503. doi: 10.1002/1097-0142(195405)7:3<462::aid-cncr2820070308>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- El-Khoueiry A.B., Sangro B., Yau T., Crocenzi T.S., Kudo M., Hsu C., Kim T.-Y., Choo S.-P., Trojan J., Welling T.H. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–2502. doi: 10.1016/S0140-6736(17)31046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R., Pusztai L., Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br. J. Cancer. 2013;108:479–485. doi: 10.1038/bjc.2012.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii M., Shimokawa M., Date S., Takano A., Matano M., Nanki K., Ohta Y., Toshimitsu K., Nakazato Y., Kawasaki K. A colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell. 2016;18:827–838. doi: 10.1016/j.stem.2016.04.003. [DOI] [PubMed] [Google Scholar]
- Fujimoto A., Furuta M., Totoki Y., Tsunoda T., Kato M., Shiraishi Y., Tanaka H., Taniguchi H., Kawakami Y., Ueno M. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016;48:500–509. doi: 10.1038/ng.3547. [DOI] [PubMed] [Google Scholar]
- Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D., Vela I., Sboner A., Iaquinta P.J., Karthaus W.R., Gopalan A., Dowling C., Wanjala J.N., Undvall E.A., Arora V.K. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159:176–187. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J., Chang M.T., Johnsen H.C., Gao S.P., Sylvester B.E., Sumer S.O., Zhang H., Solit D.B., Taylor B.S., Schultz N., Sander C. 3D clusters of somatic mutations in cancer reveal numerous rare mutations as functional targets. Genome Med. 2017;9:4. doi: 10.1186/s13073-016-0393-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjorevski N., Sachs N., Manfrin A., Giger S., Bragina M.E., Ordóñez-Morán P., Clevers H., Lutolf M.P. Designer matrices for intestinal stem cell and organoid culture. Nature. 2016;539:560–564. doi: 10.1038/nature20168. [DOI] [PubMed] [Google Scholar]
- Huch M., Gehart H., van Boxtel R., Hamer K., Blokzijl F., Verstegen M.M., Ellis E., van Wenum M., Fuchs S.A., de Ligt J. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell. 2015;160:299–312. doi: 10.1016/j.cell.2014.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C., McLellan M.D., Vandin F., Ye K., Niu B., Lu C., Xie M., Zhang Q., McMichael J.F., Wyczalkowski M.A. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo J., Endo H., Okuyama H., Ishikawa O., Iishi H., Tsujii M., Ohue M., Inoue M. Retaining cell-cell contact enables preparation and culture of spheroids composed of pure primary cancer cells from colorectal cancer. Proc. Natl. Acad. Sci. USA. 2011;108:6235–6240. doi: 10.1073/pnas.1015938108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau D.A., Carter S.L., Stojanov P., McKenna A., Stevenson K., Lawrence M.S., Sougnez C., Stewart C., Sivachenko A., Wang L. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–726. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M.S., Stojanov P., Mermel C.H., Robinson J.T., Garraway L.A., Golub T.R., Meyerson M., Gabriel S.B., Lander E.S., Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.H., Hu W., Matulay J.T., Silva M.V., Owczarek T.B., Kim K., Chua C.W., Barlow L.J., Kandoth C., Williams A.B. Tumor evolution and drug response in patient-derived organoid models of bladder cancer. Cell. 2018;173:515–528.e17. doi: 10.1016/j.cell.2018.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llovet J.M., Hernandez-Gea V. Hepatocellular carcinoma: reasons for phase III failure and novel perspectives on trial design. Clin. Cancer Res. 2014;20:2072–2079. doi: 10.1158/1078-0432.CCR-13-0547. [DOI] [PubMed] [Google Scholar]
- Llovet J.M., Brú C., Bruix J. Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin. Liver Dis. 1999;19:329–338. doi: 10.1055/s-2007-1007122. [DOI] [PubMed] [Google Scholar]
- Llovet J.M., Ricci S., Mazzaferro V., Hilgard P., Gane E., Blanc J.F., de Oliveira A.C., Santoro A., Raoul J.L., Forner A., SHARP Investigators Study Group Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- Luo X., Jia W., Huang Z., Li X., Xing B., Jiang X., Li J., Si A., Yang T., Gao C. Effectiveness and safety of sorafenib in the treatment of unresectable and advanced intrahepatic cholangiocarcinoma: a pilot study. Oncotarget. 2017;8:17246–17257. doi: 10.18632/oncotarget.12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt J.U., Andersen J.B., Thorgeirsson S.S. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat. Rev. Cancer. 2015;15:653–667. doi: 10.1038/nrc4017. [DOI] [PubMed] [Google Scholar]
- McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S., Davies H., Staaf J., Ramakrishna M., Glodzik D., Zou X., Martincorena I., Alexandrov L.B., Martin S., Wedge D.C. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534:47–54. doi: 10.1038/nature17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolayeva O., Robinson M.D. edgeR for differential RNA-seq and ChIP-seq analysis: an application to stem cell biology. Methods Mol. Biol. 2014;1150:45–79. doi: 10.1007/978-1-4939-0512-6_3. [DOI] [PubMed] [Google Scholar]
- Pauli C., Hopkins B.D., Prandi D., Shaw R., Fedrizzi T., Sboner A., Sailer V., Augello M., Puca L., Rosati R. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 2017;7:462–477. doi: 10.1158/2159-8290.CD-16-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piscuoglio S., Ng C.K., Murray M.P., Guerini-Rocco E., Martelotto L.G., Geyer F.C., Bidard F.C., Berman S., Fusco N., Sakr R.A. The genomic landscape of male breast cancers. Clin. Cancer Res. 2016;22:4045–4056. doi: 10.1158/1078-0432.CCR-15-2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R., McGranahan N., Herrero J., Taylor B.S., Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31. doi: 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs N., de Ligt J., Kopper O., Gogola E., Bounova G., Weeber F., Balgobind A.V., Wind K., Gracanin A., Begthel H. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 2018;172:373–386.e10. doi: 10.1016/j.cell.2017.11.010. [DOI] [PubMed] [Google Scholar]
- Saunders C.T., Wong W.S., Swamy S., Becq J., Murray L.J., Cheetham R.K. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- Shen R., Seshan V.E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihab H.A., Gough J., Cooper D.N., Stenson P.D., Barker G.L., Edwards K.J., Day I.N., Gaunt T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013;34:57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327–1341.e23. doi: 10.1016/j.cell.2017.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H., Robinson J.T., Mesirov J.P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering M., Francies H.E., Francis J.M., Bounova G., Iorio F., Pronk A., van Houdt W., van Gorp J., Taylor-Weiner A., Kester L. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161:933–945. doi: 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Samples were obtained between July 2015 and September 2017 and are ordered chronologically according to the date of the biopsy. If more than one tumor nodule was biopsied in a given patient, the different nodules are designated with letters (e.g., 5-A and 5-B). If more than one biopsy was taken from different locations of the same (large) nodule, the different locations are labeled with roman numbers (e.g., 12-I and 12-II). The five cholangiocellular carcinoma cases are shown in italic font. Sex, age, background liver disease, clinical staging according to the Barcelona Clinic Liver Cancer (BCLC) staging system and alpha-fetoprotein (AFP) serum concentrations were obtained from the clinical charts of the patients. The viable tumor cell content, Edmondson grade and the growth pattern were determined in each biopsy on Hematoxylin and Eosin stained sections by two experienced hepato-pathologists (M.S.M. and L.M.T.). All CCC tumors were poorly differentiated. ALD: Alcoholic Liver Disease; AFP: Alpha-Fetoprotein; BCLC: Barcelona Clinic Liver Cancer; CCC: Cholangiocellular Carcinoma; HBV: Hepatitis B Virus; HCC: Hepatocellular Carcinoma; HCV: Hepatitis C Virus; LELCC: Lymphoepithelioma-like Cholangiocarcinoma; NAFLD: Nonalcoholic Fatty Liver Disease; n.a.: not applicable; n.e.: not evaluable because of insufficient tumor cell content in the biopsy.