

Antibiotic treatment of P. aeruginosa residing in the lung of cystic fibrosis patients is ineffective. Treatment failure is attributed in part to antibiotic-tolerant phenotypic variants known as persister cells. Understanding how these cells emerge will likely inform future therapeutic strategies. In the current study, we identified carB, which codes for the large subunit of carbamoyl-phosphate synthetase, as a persister gene that contributes to multidrug tolerance in P. aeruginosa. Disruption of carB resulted in a metabolic perturbation that increased cellular ATP and reduced persister formation. Conversely, lowering ATP in the mutant restored antibiotic tolerance. Our data support the hypothesis that a drop in intracellular ATP is a general mechanism of persister formation in bacteria.
KEYWORDS: Tn-seq, antibiotic tolerance, persisters
ABSTRACT
Persisters represent a small subpopulation of cells within a bacterial culture that are tolerant to killing by antibiotics. Persisters have been linked to recalcitrant infections caused by numerous bacterial pathogens, including Pseudomonas aeruginosa. A classic example is the incurable infection of the airways for patients with cystic fibrosis. The genetic mediators of persister formation for P. aeruginosa are poorly understood. We generated a high-density transposon insertion library of P. aeruginosa PAO1 and determined the relative frequency of each insertion following fluoroquinolone treatment using transposon sequencing (Tn-seq). Of the 4,411 disrupted genes included in the screen, 137 had a ≥10-fold impact on survival. The gene disruption that resulted in the lowest survival rate was disruption of carB, which codes for the large subunit of carbamoyl phosphate synthetase (CPSase). CPSase is a metabolic enzyme that is involved in pyrimidine and arginine synthesis. Disruption of carB resulted in survival rates that were reduced by up to 2,500-fold following antibiotic treatment, and this phenotype was abolished by the addition of uracil, highlighting the importance of de novo pyrimidine biosynthesis for persister formation. Disruption of carB resulted in intracellular ATP accumulation, and lowering ATP levels using arsenate restored the antibiotic tolerance profile of the mutant to levels similar to those seen with the wild type. A decrease in ATP would lead to reduced antibiotic target activity and increased survival.
IMPORTANCE Antibiotic treatment of P. aeruginosa residing in the lung of cystic fibrosis patients is ineffective. Treatment failure is attributed in part to antibiotic-tolerant phenotypic variants known as persister cells. Understanding how these cells emerge will likely inform future therapeutic strategies. In the current study, we identified carB, which codes for the large subunit of carbamoyl-phosphate synthetase, as a persister gene that contributes to multidrug tolerance in P. aeruginosa. Disruption of carB resulted in a metabolic perturbation that increased cellular ATP and reduced persister formation. Conversely, lowering ATP in the mutant restored antibiotic tolerance. Our data support the hypothesis that a drop in intracellular ATP is a general mechanism of persister formation in bacteria.
INTRODUCTION
Antibiotic resistance poses an immediate threat to human health. Persistent bacterial infections, however, are commonly caused by difficult-to-eradicate pathogens that have not evolved classical resistance (1). In this context, a small subset of antibiotic-tolerant cells known as persisters have been implicated in treatment failure and recurrent bacterial infections (2–4). A prime example is Pseudomonas aeruginosa, which causes an incurable airway infection and is the major cause of mortality associated with cystic fibrosis (CF) (5). Over the course of infection, P. aeruginosa has been shown to evolve toward a high-persister state, producing more antibiotic-tolerant cells (4), and yet we know surprisingly little about the processes that contribute to their existence.
Our knowledge of the molecular aspects of persister physiology comes primarily from the model organism Escherichia coli. In vitro evolution experiments produced stable high-persister mutants and allowed identification of the first bona fide persister gene, hipA (6). HipA is a serine-protein kinase that phosphorylates glutamyl-tRNA synthase and, in concert with HipB, forms a toxin-antitoxin (TA) module (7, 8). HipA inhibits protein synthesis, which induces cell dormancy and subsequent antibiotic tolerance. The same gain-in-function mutations that evolved in vitro were identified in patients with urinary tract infections (UTI), linking hipA to the clinical manifestation of recalcitrant infection (3). A second toxin-mediated mechanism of persister formation involves the antimicrobial peptide toxin TisB. Ciprofloxacin causes DNA damage that induces the expression of tisB. TisB forms channels in the cell membrane that disrupt the proton motive force (PMF), resulting in a drop in ATP levels (9). A decrease in cellular energy leads to drug tolerance, as the activity of antibiotic targets is typically ATP dependent (10, 11).
Until recently, it was widely accepted that stress-induced expression of mRNA interferases from 10 type II TA loci was a major contributor to persister formation in E. coli. In this model, the alarmone ppGpp inhibits the PPX phosphatase, resulting in increased polyphosphate levels and subsequent activation of Lon protease. The Lon protease was proposed to degrade antitoxins, resulting in enhanced toxin activity and antibiotic tolerance. Careful reexamination of mutants lacking 10 TAs (Δ10TA), Lon protease (Δlon ΔsulA), and the polyphosphate operon (Δppx Δppk) did not reveal persister phenotypes, disproving the model (11–13). As a corollary to these findings, at the time of writing, no published report had directly implicated TAs in persister formation for P. aeruginosa and its genome had not been shown to harbor predicted hipA or tisB orthologs. This suggests that additional, undefined mechanisms are responsible for persister formation in P. aeruginosa.
We have illustrated the usefulness of transposon sequencing (Tn-seq) for identifying persister genes in bacteria (14). In the current study, we applied this technique to P. aeruginosa and identified carB, encoding the small subunit of carbamoylphosphate synthetase (CPSase), as a persister gene.
RESULTS
Tn-seq identifies P. aeruginosa persister genes.
We challenged a saturated transposon mutant pool of P. aeruginosa PAO1 with a fluoroquinolone antibiotic and compared the frequencies of each insertion mutation before and after treatment using next-generation sequencing. The input pool contained approximately 100,000 unique transposon insertion sites, which equates to an insertion every 60 bp (Fig. 1A). Ciprofloxacin challenge resulted in typical biphasic killing (Fig. 1B), and surviving persisters were washed and plated following 3 h of treatment (the output). A survival index (SI; normalized measure of the frequency of a gene mutation after treatment divided by the frequency before treatment [14]) was generated for nonessential genes that contained more than three insertion sites across each biological replicate (4,411 genes in total) (Fig. 1C; see also Table S1 in the supplemental material).
FIG 1.

Transposon sequencing reveals persister genes of P. aeruginosa. (A) Distribution of Tn insertions for the mutagenized P. aeruginosa PAO1 input pool. (B) Antibiotic killing kinetics of the P. aeruginosa PAO1 Tn pool. Cultures were grown in LB medium to the exponential phase in biological triplicate and treated with 1 μg/ml ciprofloxacin. Data are represented as means and standard deviations. (C) A survival index (Tn insertion frequency after treatment/before treatment) was generated for 4,411 nonessential genes. Black symbols represent the mean survival index for a given gene; gray bars represent standard deviations (n = 3). (D) PseudoCAP functional analysis of 137 genes with a 10-fold change in the SI. FA, fatty acid; LPS, lipopolysaccharide; ncRNA, noncoding RNA.
In total, 137 genes had ≥10-fold changes in SI (118 with increased SI and 19 with decreased SI) (Table S1). Grouped on the basis of PseudoCAP function (15), genes involved in metabolism were overrepresented (Fig. 1D). Similarly to what was observed previously for E. coli (14), 12 genes encoding amino acid biosynthesis enzymes had decreased SI, including genes of the tryptophan synthesis operon (trpABC) (Table S1). Genes coding for important regulators of energy and carbon metabolism were also associated with decreased survival following antibiotic treatment, including glpR, which codes for the repressor of the glp regulon, apaH, encoding diadenosine tetraphosphatase, and the crc gene encoding a catabolite repression control protein, each of which had been previously associated with persister formation in bacteria (16–18). Additional transcriptional regulators had increased SI, most notably, repressors of efflux systems, including those encoded by mexR, mexZ, nalC, and nfxB (19–21). In support of these findings, each of the genes encoding the MexXY multidrug efflux pump, which extrudes fluoroquinolones, had a decreased SI (Fig. 1C; see also Table S1).
The carB gene is important for multidrug tolerance.
The gene with the lowest SI was carB, encoding the large subunit of carbamoyl phosphate synthetase (CPSase). CPSase catalyzes the ATP-dependent synthesis of carbamoyl phosphate, which is the shared precursor for pyrimidine and arginine biosynthesis in bacteria. To confirm the Tn-seq findings and to determine if carB contributes to multidrug tolerance, we performed time-dependent killing assays of wild-type PAO1 and a carB transposon mutant (PAO1carB::Tn). The carB mutant produced fewer persister cells when challenged with ofloxacin, tobramycin, rifampin, and meropenem in both the exponential and stationary phases of growth (see Fig. S1 in the supplemental material).
The effect of carB disruption was greatest in the stationary phase. The carB mutant produced 2,436-fold, 36-fold, and 18-fold-fewer persisters than PAO1 when challenged with tobramycin, ofloxacin, and meropenem, respectively (Fig. 2). To confirm that the decrease in the levels of persisters for PAO1carB::Tn was due to the absence of a functional copy of carB and not due to other polar effects, we generated an inducible carB expression vector and introduced it into the PAO1 and PAO1carB::Tn strains. Survival following antibiotic treatment for the PAO1carB::Tn(pcarB) complementation strain was increased upon induction of carB (Fig. 2; see also Fig. S2). The survival of the PAO1(pcarB) overexpression strain was higher than that of the wild-type strain for each antibiotic tested; however, the differences were not found to be statistically significant (P = 0.3; two-way analysis of variance [ANOVA], Tukey's multiple-comparison test).
FIG 2.
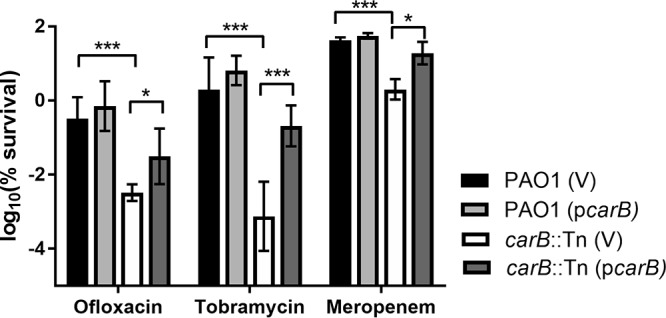
The effects of carB deletion on persister formation in P. aeruginosa. A PAO1 strain containing a vector control [PAO1 (V)], a PAO1 strain with an inducible copy of carB [PAO1 (pcarB)], a carB::Tn strain containing a vector control [carB::Tn (V)], and a carB::Tn strain with an inducible copy of carB [carB::Tn (pcarB)] were grown to the stationary phase (16 to 18 h) in LB medium at 37°C and were treated with ofloxacin (20 μg/ml) or tobramycin (10 μg/ml) or meropenem (5 μg/ml). IPTG (10 mM) was added to induce Ptac and express carB. Survival was determined by spot plating and colony counting. Asterisks denote statistical significance as determined by two-way ANOVA followed by Tukey's multiple-comparison test. *, P < 0.05; ***, P < 0.001.
To discount the possibility that carB is a mediator of intrinsic antimicrobial resistance, we tested the antimicrobial MIC of the carB mutant and compared it to that of PAO1. The MICs for ofloxacin and tobramycin were the same as those measured for PAO1 (2 μg/ml and 0.5 μg/ml, respectively), and the MIC for meropenem was 2-fold lower (0.5 μg/ml). The intrinsic error associated with MIC determination is 2-fold (22); therefore, the difference in the meropenem MIC was deemed not statistically significant. These data suggest that carB is a persister gene that contributes to antibiotic tolerance rather than to antibiotic resistance.
Pyrimidine supplementation restores antibiotic tolerance.
CPSase catalyzes the initial step in the de novo synthesis of pyrimidines. As such, the carB mutant is auxotrophic for pyrimidines in minimal medium and shows a growth defect in LB medium for both P. aeruginosa (Fig. 3A) and E. coli (Fig. S3). The growth of the carB mutant is arrested prematurely, most likely due to exhaustion of the nucleotide pools required for DNA replication and RNA synthesis. Supplementing the medium with uracil, which is metabolized to UMP via the pyrimidine salvage pathway, restored the growth profile of the carB mutant to wild-type status. Furthermore, addition of uracil to PAO1carB::Tn cultures during growth arrest (t = 465 min for P. aeruginosa; t = 360 min for E. coli) facilitated rapid resumption of growth and allowed the mutant to reach a final density similar to that seen with each respective wild-type strain. Pyrimidine starvation was also responsible for the reduced persister profile of strain PAO1carB::Tn. When P. aeruginosa was challenged with 20 μg/ml ofloxacin, uracil supplementation increased the survival of PAO1carB::Tn by 100-fold, to levels similar to those seen with the wild-type strain (Fig. 3B).
FIG 3.
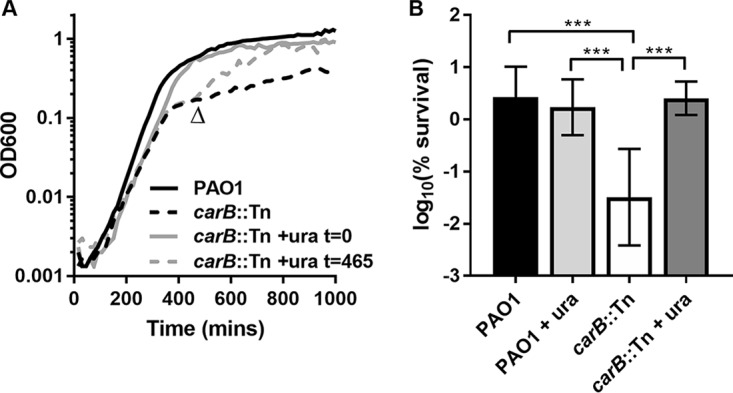
Pyrimidine supplementation restores growth and antibiotic tolerance of PAO1carB::Tn to levels similar to those seen with the wild type. (A) PAO1 and carB::Tn strains were grown in LB medium at 37°C. To determine the growth effect of pyrimidine supplementation, 2.5 mM uracil (ura) was added at t = 0 or during the stationary phase (uracil was added at the item indicated by the arrowhead). Data represent means from triplicate measures. (B) Stationary-phase cultures of P. aeruginosa were treated with 20 μg/ml ofloxacin, and survival was determined after 20 h of incubation. Bacteria were grown in LB medium with and without 2.5 mM uracil supplementation. Experiments were performed twice in biological triplicate (n = 6). Error bars represent standard deviations. Asterisks denote statistical significance as determined by one-way ANOVA followed by Tukey's multiple-comparison test. ***, P < 0.001.
Antibiotic tolerance of P. aeruginosa is associated with a drop in ATP.
We had reasoned previously that a drop in cellular energy in the form of a drop in ATP levels may represent a general mechanism of persister formation in bacteria (10, 11). The bactericidal antibiotics used in this study kill by corrupting energy-dependent processes. A drop in ATP levels would result in antibiotic target inactivity and subsequent multidrug tolerance. To test whether this is true for P. aeruginosa, we first correlated intracellular ATP levels with antibiotic tolerance at different stages of growth. As expected, the intracellular ATP concentration was close to 7 times lower in stationary phase than in exponential phase (Fig. 4A) and this was associated with the presence of more persisters tolerant to three antibiotic classes (Fig. 4B). When arsenate was added to exponentially growing cultures, intracellular ATP concentrations dropped to levels similar to those seen in the stationary phase (Fig. 4A), and this correlated with increased antibiotic tolerance (Fig. 4B), suggesting that the cellular ATP concentration is also a persister determinant for P. aeruginosa. These data support results from a recent study that used carbonyl cyanide m-chlorophenylhydrazone (CCCP) to induce P. aeruginosa persisters tolerant to antimicrobial peptides by disrupting the proton motive force, which would subsequently reduce the rate of ATP synthesis (23).
FIG 4.

Accumulation of intracellular ATP is responsible for the low-persister phenotype of the carB mutant. (A) Intracellular ATP levels were determined for exponential (exp.) and stationary (stat.) cultures of PAO1. Exponential cultures were treated with 5 mM arsenate (ars) for 30 min to reduce ATP to levels similar to those seen with stationary cultures. (B) PAO1 (V) was grown to the exponential or stationary phase and treated independently with three classes of antibiotic at 10× MIC. Survival was determined following 20 h of incubation. Where indicated, ars was added to exponential cultures for 30 min to reduce ATP levels. (C) Intracellular ATP was quantified for stationary cultures of the PAO1 and carB::Tn strains and of the complemented strain, the carB::Tn(pcarB) strain. For carB::Tn (V) + ura, growth medium was supplemented with 2.5 mM uracil. For carB::Tn (V) + ars, cultures were pretreated with 5 mM arsenate for 30 min. (D) PAO1 and carB::Tn strains were grown to the stationary phase and treated with 10× MIC for three classes of antibiotic, and survival was determined following 20 h of incubation. For carB::Tn + ars, cultures of carB::Tn were pretreated with 5 mM arsenate for 30 min. All experiments were performed at least twice in biological triplicate (n ≥ 6). Data represent means ± standard deviations. Significance was determined using one-way ANOVA (A and C) or two-way ANOVA (B and D) and Tukey's multiple-comparison tests. **, P < 0.01; ***, P < 0.001.
Given the altered growth profile of the carB mutant and its central role in metabolism, we tested whether CPSase disruption had an impact on intracellular ATP levels. In stationary-phase cultures, the PAO1carB::Tn strain accumulated four times more ATP per CFU than the wild-type strain (Fig. 4C). Ectopic expression of carB and uracil supplementation each abolished ATP accumulation for the carB mutant (Fig. 4C). To determine whether increased the ATP levels were the cause of the low-persister profile of the carB mutant, we depleted the intracellular ATP of the carB mutant to levels similar to wild-type levels using arsenate (Fig. 4C), and we subsequently restored antibiotic tolerance, increasing the survival of the PAO1carB::Tn strain by up to 2,500-fold (Fig. 4D).
carB disruption results in reduced antibiotic tolerance in other bacterial species.
We next reasoned that disrupting CPSase may cause a metabolic perturbation that results in ATP accumulation and reduced persister formation in other bacterial species. To test this, we assessed the intracellular ATP concentration and antibiotic tolerance of carB mutants of the model Gram-negative organism E. coli and in the Gram-positive pathogen Staphylococcus aureus. Intracellular ATP levels were 2.4-fold higher in the E. coli mutant and 4.9-fold higher in the S. aureus mutant than in the respective wild-type strains (Fig. 5A). This increase in ATP was associated with decreased ciprofloxacin and gentamicin tolerance in E. coli (Fig. 5B) and decreased oxacillin and gentamicin tolerance in S. aureus (Fig. 5C).
FIG 5.

Disruption of carB results in ATP accumulation and reduced persister formation in diverse bacteria. (A) Intracellular ATP concentrations were determined for wild-type and carB mutants of E. coli MG1655 and S. aureus HG003. (B) E. coli was grown to the stationary phase in LB medium and then treated with antibiotics at 10× the MIC. Survival was determined following 20 h of incubation. (C) S. aureus was grown to the late exponential phase in TSB medium and then treated with 1 μg/ml ofloxacin for 24 h or grown to the late exponential phase in Mueller-Hinton broth (MHB) medium and then treated with 10 μg/ml gentamicin for 48 h. All experiments were performed twice in biological triplicate (n = 6); data represent means ± standard deviations. Significance was determined using a t test (A) or two-way ANOVA with Tukey's multiple-comparison test (B and C). *, P < 0.05; ***, P < 0.001.
Despite growth arrest, the carB mutant continued to transcribe and translate protein.
It is well established that persister cells are dormant (24, 25) and that the dormancy is of paramount importance to antibiotic tolerance due to drug target inactivity (26). For example, the effectiveness of β-lactams is intimately linked to growth rates (27). β-Lactams do not kill stationary cells, because the cell wall synthesis machinery that they target is inactive. Similarly, reduced translation rates have been shown to increase tolerance of aminoglycosides, which target the ribosome (14). It is not obvious why the PAO1carB::Tn strain has reduced antibiotic tolerance, given that it entered an apparent dormant state prematurely (Fig. 3A). Our growth experiments gave us a first indication that the PAO1carB::Tn strain has an atypical stationary phase. During growth arrest, pyrimidine supplementation allowed the rapid resumption of growth, which suggests that the mutant was not classically dormant. To investigate this further, we used a plasmid-borne inducible PlacZ::gfp construct in the model organism E. coli to report transcription and translation for the MG1655∆carB(pgfp) strain. As expected, the MG1655∆carB(pgfp) strain entered the stationary phase earlier than the wild type and did not reach the same final cell density (Fig. 6A). When PlacZ::gfp was induced at time zero, green fluorescent protein (GFP) accumulated in the wild type throughout the exponential phase and then plateaued into the stationary phase. In contrast, the MG1655∆carB(pgfp) strain continued to translate GFP well into the stationary phase (Fig. 6B). Inducing PlacZ::gfp in the stationary phase (t = 360 min) did not increase the GFP signal for the wild-type strain; however, the GFP signal increased sharply for the MG1655∆carB(pgfp) strain, suggesting that the strain was both transcriptionally and translationally active (Fig. 6C).
FIG 6.
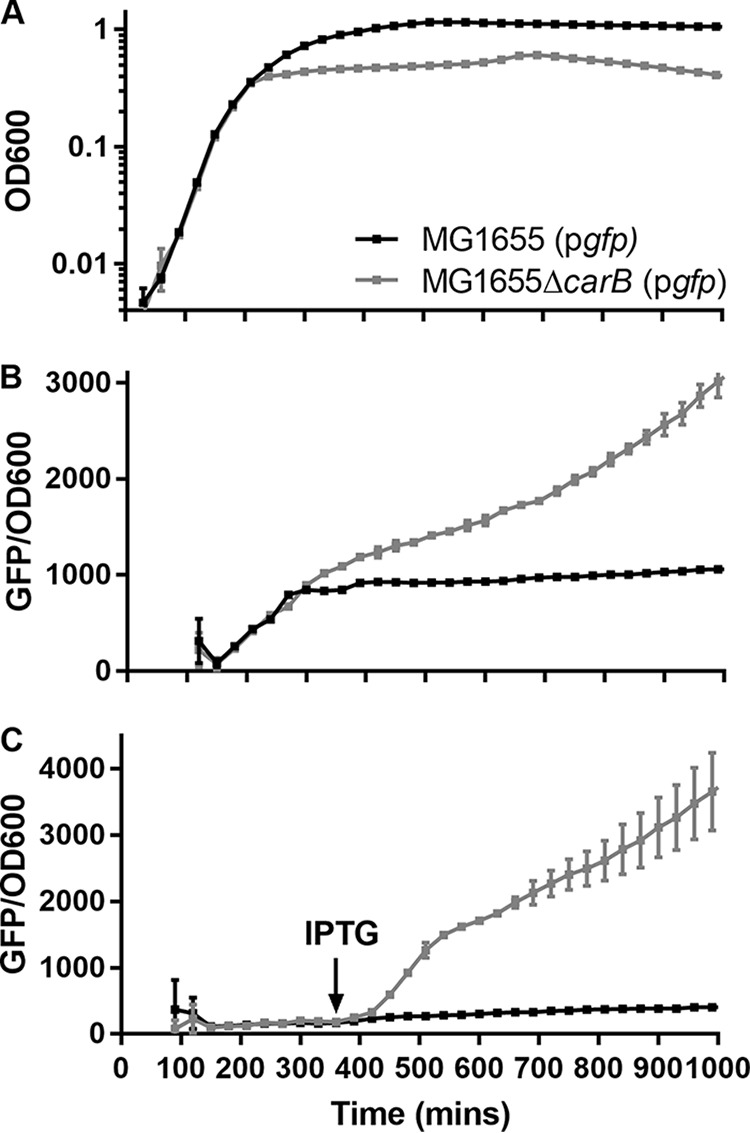
The carB mutant strain is actively transcribing and translating during growth arrest in E. coli. (A) Growth profiles of E. coli MG1655 and MG1655ΔcarB, each containing a plasmid-borne PlacZ::gfp fusion [called MG1655(pgfp) and MG1655ΔcarB(pgfp), respectively]. Strains were grown in LB medium at 37°C with aeration. (B and C) GFP fluorescence (excitation wavelength, 485 nm; emission wavelength, 528 nm) was determined throughout growth. IPTG (1 mM) was added at time zero (B) or after 360 min (C). Data represent means and standard deviations of triplicate measures.
DISCUSSION
Bacterial infection of the airways contributes to significant mortality in CF patients (5, 28). P. aeruginosa adapts to the lung environment and establishes a chronic infection that is not completely eradicated by antibiotic treatment (29, 30). Therapeutic failure has been attributed to the development of antibiotic resistance, as well as to the presence of antibiotic-tolerant persister cells, the latter of which are poorly understood (4, 31).
Tn-seq is a well-established method for identifying fitness determinants for bacteria under diverse conditions (14, 32–34). In the current study, we utilized this technique to identify genes that contribute to persister cell formation for P. aeruginosa. The gene with the largest impact on survival following antibiotic treatment was carB, which codes for the large subunit of CPSase. CPSase contributes to persister formation due to its indispensable role in pyrimidine biosynthesis (Fig. 3). The absence of de novo pyrimidine synthesis prevents P. aeruginosa from reaching a saturated cell density in the stationary phase. In this state, however, the carB mutant cells are not classically dormant; they have high intracellular ATP levels and are actively transcribing and translating. It is likely that the limited pyrimidines available in the medium are exhausted before other nutrients required for energy generation and maintenance of PMF. Increased cellular activity in the absence of biomass production is analogous to a futile cycle and results in reduced antibiotic tolerance due to increased activity of antibiotic targets (Fig. 7). Reducing ATP using arsenate, or by complementing the ATP phenotype by adding uracil, restored the antibiotic tolerance of the carB mutant to levels similar to wild-type levels, demonstrating causality (Fig. 4).
FIG 7.
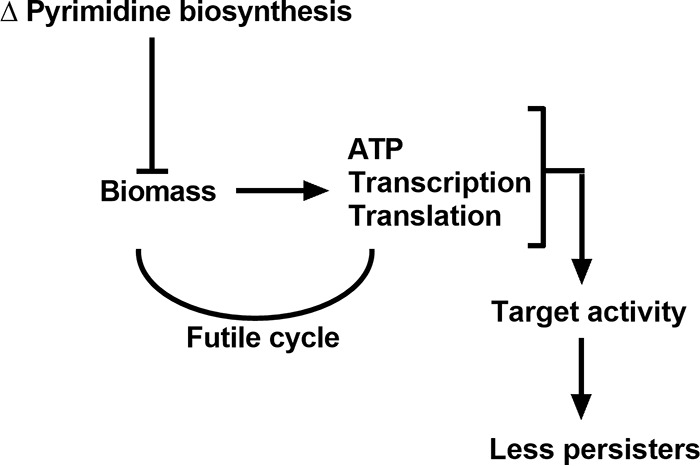
Model explaining the reduced persister phenotype of the carB mutant. Without functional CPSase, carB::Tn cells stop dividing prematurely due to pyrimidine exhaustion. The mutant, however, remains transcriptionally and translationally active during growth arrest, which allows rapid division when cultures are supplemented with pyrimidines. Despite high levels of cellular ATP and active transcription and translation, the carB mutant cannot produce biomass, resulting in a futile cycle. Increased activity of antibiotic targets results in increased killing and fewer persisters.
The molecular pathways that contribute to persister formation are redundant, and screening of gene knockout libraries of E. coli did not identify mutants that lack persisters (1). Variation in intracellular ATP content provides a satisfactory explanation for this redundancy, because an abundance of cellular pathways may contribute to ATP heterogeneity within bacterial pathogens. In this report, we identified a central metabolic pathway that, when mutated, results in accumulation of cellular ATP. It is quite possible that the other metabolic genes identified in this screen (Fig. 1D; see also Table S1 in the supplemental material) also have an impact on persister cell levels in an ATP-dependent manner. Most antibiotics target energy-dependent processes. Quinolones cause DNA damage by targeting DNA gyrase and topoisomerase (35), aminoglycosides cause mistranslation by the ribosome resulting in the generation of toxic peptides (36), rifampin inhibits DNA-dependent RNA polymerase (37), and β-lactams (including carbapenems) cause toxic malfunctioning of cell wall biosynthetic machinery (38). Cells with lower ATP levels survive antibiotic treatment because the targets are inactive. This also explains why bacteria are highly tolerant of the killing effects of antibiotics in the stationary phase of growth, where ATP levels are depleted (Fig. 4A and B).
Identifying persister gene candidates is not trivial. Understanding how persister cells form has enabled us to target them more effectively. We have had some success with acyldepsipeptide (ADEP), which, in combination with conventional antibiotics, killed persisters and eradicated deep-seated S. aureus infection in mice (2). ADEP activates the ClpP protease, itself a persister determinant (39), resulting in self-digestion and cell death. Importantly, when associated with ADEP, the proteolytic activity of ClpP is no longer dependent upon ATP. A second strategy for killing persisters involves reenergizing them via the addition of sugars and tricarboxylic acid (TCA) cycle intermediates such as fumarate (40, 41). Aminoglycoside uptake is dependent on PMF, and this explains why aminoglycosides have limited efficacy against nongrowing, stationary-phase cells (42). Fumarate potentiates the killing of P. aeruginosa by stimulating cellular respiration and enhancing PMF. This same strategy also increases intracellular ATP levels. Whether providing bacteria with nutrients that can be utilized for growth in vivo is a viable means of antibiotic treatment remains to be determined.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Strains and plasmids are listed in Table S2 in the supplemental material. Bacteria were grown in LB medium (P. aeruginosa and E. coli), Mueller-Hinton broth (S. aureus), or tryptic soy broth (TSB) at 37°C with aeration at 225 rpm. When necessary, carbenicillin (150 μg/ml) and tetracycline (10 μg/ml) were used for selection and isopropyl-β-d-1-thiogalactopyranoside (IPTG; 1 or 10 mM) was used for induction of Ptac. Media were supplemented with 2.5 mM uracil where specified.
Model strain PAO1 was used as the wild-type strain for all P. aeruginosa experiments. The P. aeruginosa carB transposon mutant (designated PAO1carB::Tn) was acquired from the Two-Allele Library (PW8995) constructed by Jacobs and colleagues (43). A carB deletion strain was constructed in E. coli MG1655 (MG1655ΔcarB) via P1 transduction from the Keio collection (44). A carB transposon mutant was constructed in S. aureus HG003 (HG003carB::Tn) via 80α transduction from the Nebraska Transposon Mutant Library (45). All mutations were confirmed by PCR. An inducible carB expression vector was generated for P. aeruginosa by amplifying the complete carB coding sequence from PAO1 using primers carB-F (5′-CCCGAGCTCGCAACGAGAGTCCCATGCC) and carB-R (5′-CCCTCTAGAGACGGTCATGGGGAACTTGC) and cloning the gene into the SacI-XbaI site downstream of Ptac in pMMB67EH (46). The plasmid was transformed into E. coli SM10λpir before being introduced into the PAO1 and PAO1carB::Tn strains via biparental conjugation, creating overexpression strain PAO1(pcarB) and complementation strain PAO1carB::Tn(pcarB), respectively. The pMMB67EH plasmid without carB was introduced into strains PAO1 and PAO1carB::Tn, and these strains served as vector controls.
Transposon sequencing (Tn-seq).
Transposon mutant libraries of PAO1 were constructed using the pIT2 plasmid containing a Tn5 derivative, as described previously (43). The pooled Tn mutant library (∼1 × 105 CFU) was grown overnight and then diluted 1:100 in 10 ml LB medium and incubated until the cultures reached the mid-exponential phase (1 × 108 to 2 × 108 CFU/ml). An aliquot (10 μl) was diluted in 5 ml of LB and then plated on 10 150-mm-diameter LB agar plates (the input). The cultures were treated with 1 μg/ml (10× MIC) ciprofloxacin for 3 h. Following treatment, cultures were pelleted, washed with phosphate-buffered saline (PBS), resuspended in 5 ml of LB, and then plated on 10 150-mm-diameter LB agar plates (the output). Plates were incubated overnight at 30°C. Colonies were scraped from each plate, pooled, washed with PBS, and then pelleted for genomic DNA extraction. Given the considerable variation in the levels of the persisters that are observed between experimental replicates (47), we performed Tn-seq in biological triplicate.
DNA was isolated via phenol-chloroform extraction and then fragmented using Fragmentase (NEB) until the majority of the DNA was <1,000 bp in size. Poly(C) tailing reactions were performed for 2 μg of fragmented DNA using terminal deoxynucleotidyl transferase (TdT; Promega). Poly(C)-tailed DNA (5 μl) was used as the template in the first round of nested PCR using Easy A cloning enzyme (Agilent). Products of these reactions (1 μl) were used as the template in the second round of nested PCR. All primer sequences used for Tn-seq are available upon request. Equimolar concentrations of input and output libraries were run on 8 lanes of Illumina HiSeq 2000 at The Broad Institute, Cambridge, MA. Survival indices (SIs) for each gene were determined as described previously (14).
Growth measurements.
Bacteria were grown overnight in LB medium, adjusted to an optical density at 600 nm (OD600) of 0.3, and then diluted 1:100 in 150 μl LB in 96-well microtiter plates. Plates were incubated at 37°C with constant shaking, and OD600 was measured every 15 or 30 min using a Synergy H1 microplate reader (BioTek). A transcription and translation analysis was performed for E. coli strains harboring plasmid-borne IPTG-inducible PlacZ::gfp (48). IPTG (1 mM) was added when specified, and GFP levels were measured (emission wavelength, 528 nm; excitation wavelength, 485 nm) every 30 min.
Antibiotic susceptibility and persister assays.
MICs were determined using broth microdilution. To assess antibiotic tolerance, persister assays were performed. Bacterial strains were grown to the stationary phase (∼16 h). Cultures were serially diluted and plated on agar to determine the initial CFU. P. aeruginosa and E. coli were challenged with antibiotics for 24 h at 10× MIC (for P. aeruginosa, ofloxacin at 20 μg/ml, tobramycin at 5 μg/ml, and meropenem at 5 μg/ml; for E. coli, ciprofloxacin at 1 μg/ml, gentamicin at 50 μg/ml, and ampicillin at 100 μg/ml). Due to the high degree of tolerance of stationary-phase S. aureus, cultures were treated in the late exponential phase with oxacillin (1 μg/ml) for 24 h or gentamicin (10 μg/ml) for 48 h. Following treatment, cultures were serially diluted and spot plated to determine CFU, and percent survival was determined as follows: [(posttreatment CFU/initial CFU) × 100].
ATP quantification.
Intracellular ATP was quantified using a BacTitre Glo kit (Promega) according to the manufacturer's instructions. ATP concentrations were normalized to CFU. Where specified, 5 mM arsenate was added 30 min prior to ATP quantification.
Supplementary Material
ACKNOWLEDGMENTS
We thank Stephen Lory for providing strains and plasmids, Katja Schlegel for advice regarding statistical analyses, and Kakei Sum for excellent technical assistance.
This work was funded in part by a Cystic Fibrosis Foundation Pilot and Feasibility Award.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00303-18.
REFERENCES
- 1.Lewis K. 2010. Persister cells. Annu Rev Microbiol 64:357–372. doi: 10.1146/annurev.micro.112408.134306. [DOI] [PubMed] [Google Scholar]
- 2.Conlon BP, Nakayasu ES, Fleck LE, LaFleur MD, Isabella VM, Coleman K, Leonard SN, Smith RD, Adkins JN, Lewis K. 2013. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 503:365–370. doi: 10.1038/nature12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schumacher MA, Balani P, Min J, Chinnam NB, Hansen S, Vulic M, Lewis K, Brennan RG. 2015. HipBA-promoter structures reveal the basis of heritable multidrug tolerance. Nature 524:59–64. doi: 10.1038/nature14662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mulcahy LR, Burns JL, Lory S, Lewis K. 2010. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J Bacteriol 192:6191–6199. doi: 10.1128/JB.01651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gibson RL, Burns JL, Ramsey BW. 2003. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 168:918–951. doi: 10.1164/rccm.200304-505SO. [DOI] [PubMed] [Google Scholar]
- 6.Moyed HS, Bertrand KP. 1983. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J Bacteriol 155:768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korch SB, Hill TM. 2006. Ectopic overexpression of wild-type and mutant hipA genes in Escherichia coli: effects on macromolecular synthesis and persister formation. J Bacteriol 188:3826–3836. doi: 10.1128/JB.01740-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Germain E, Castro-Roa D, Zenkin N, Gerdes K. 2013. Molecular mechanism of bacterial persistence by HipA. Mol Cell 52:248–254. doi: 10.1016/j.molcel.2013.08.045. [DOI] [PubMed] [Google Scholar]
- 9.Dörr T, Vulic M, Lewis K. 2010. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol 8:e1000317. doi: 10.1371/journal.pbio.1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conlon BP, Rowe SE, Gandt AB, Nuxoll AS, Donegan NP, Zalis EA, Clair G, Adkins JN, Cheung AL, Lewis K. 2016. Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat Microbiol 1:16051. doi: 10.1038/nmicrobiol.2016.51. [DOI] [PubMed] [Google Scholar]
- 11.Shan Y, Brown Gandt A, Rowe SE, Deisinger JP, Conlon BP, Lewis K. 2017. ATP-dependent persister formation in Escherichia coli. mBio 8:e02267-16. doi: 10.1128/mBio.02267-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harms A, Fino C, Sorensen MA, Semsey S, Gerdes K. 2017. Prophages and growth dynamics confound experimental results with antibiotic-tolerant persister cells. mBio 8:e01964-17. doi: 10.1128/mBio.01964-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chowdhury N, Kwan BW, Wood TK. 2016. Persistence increases in the absence of the alarmone guanosine tetraphosphate by reducing cell growth. Sci Rep 6:20519. doi: 10.1038/srep20519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shan Y, Lazinski D, Rowe S, Camilli A, Lewis K. 2015. Genetic basis of persister tolerance to aminoglycosides in Escherichia coli. mBio 6:e00078-15. doi: 10.1128/mBio.00078-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winsor GL, Griffiths EJ, Lo R, Dhillon BK, Shay JA, Brinkman FS. 2016. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res 44:D646–D653. doi: 10.1093/nar/gkv1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spoering AL, Vulic M, Lewis K. 2006. GlpD and PlsB participate in persister cell formation in Escherichia coli. J Bacteriol 188:5136–5144. doi: 10.1128/JB.00369-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hansen S, Lewis K, Vulic M. 2008. Role of global regulators and nucleotide metabolism in antibiotic tolerance in Escherichia coli. Antimicrob Agents Chemother 52:2718–2726. doi: 10.1128/AAC.00144-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L, Chiang WC, Gao Q, Givskov M, Tolker-Nielsen T, Yang L, Zhang G. 2012. The catabolite repression control protein Crc plays a role in the development of antimicrobial-tolerant subpopulations in Pseudomonas aeruginosa biofilms. Microbiology 158:3014–3019. doi: 10.1099/mic.0.061192-0. [DOI] [PubMed] [Google Scholar]
- 19.Masuda N, Sakagawa E, Ohya S, Gotoh N, Tsujimoto H, Nishino T. 2000. Contribution of the MexX-MexY-oprM efflux system to intrinsic resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother 44:2242–2246. doi: 10.1128/AAC.44.9.2242-2246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsuo Y, Eda S, Gotoh N, Yoshihara E, Nakae T. 2004. MexZ-mediated regulation of mexXY multidrug efflux pump expression in Pseudomonas aeruginosa by binding on the mexZ-mexX intergenic DNA. FEMS Microbiol Lett 238:23–28. doi: 10.1111/j.1574-6968.2004.tb09732.x. [DOI] [PubMed] [Google Scholar]
- 21.Poole K, Tetro K, Zhao Q, Neshat S, Heinrichs DE, Bianco N. 1996. Expression of the multidrug resistance operon mexA-mexB-oprM in Pseudomonas aeruginosa: mexR encodes a regulator of operon expression. Antimicrob Agents Chemother 40:2021–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turnidge J, Paterson DL. 2007. Setting and revising antibacterial susceptibility breakpoints. Clin Microbiol Rev 20:391–408. doi: 10.1128/CMR.00047-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grassi L, Di Luca M, Maisetta G, Rinaldi AC, Esin S, Trampuz A, Batoni G. 2017. Generation of persister cells of Pseudomonas aeruginosa and Staphylococcus aureus by chemical treatment and evaluation of their susceptibility to membrane-targeting agents. Front Microbiol 8:1917. doi: 10.3389/fmicb.2017.01917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bigger JW. 1944. Treatment of staphylococcal infections with pencillin by intermittent sterilisation. Lancet 244:497–500. doi: 10.1016/S0140-6736(00)74210-3. [DOI] [Google Scholar]
- 25.Shah D, Zhang Z, Khodursky A, Kaldalu N, Kurg K, Lewis K. 2006. Persisters: a distinct physiological state of E coli. BMC Microbiol 6:53. doi: 10.1186/1471-2180-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewis K. 2007. Persister cells, dormancy and infectious disease. Nat Rev Microbiol 5:48–56. doi: 10.1038/nrmicro1557. [DOI] [PubMed] [Google Scholar]
- 27.Tuomanen E, Cozens R, Tosch W, Zak O, Tomasz A. 1986. The rate of killing of Escherichia coli by beta-lactam antibiotics is strictly proportional to the rate of bacterial growth. J Gen Microbiol 132:1297–1304. [DOI] [PubMed] [Google Scholar]
- 28.Folkesson A, Jelsbak L, Yang L, Johansen HK, Ciofu O, Hoiby N, Molin S. 2012. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat Rev Microbiol 10:841–851. doi: 10.1038/nrmicro2907. [DOI] [PubMed] [Google Scholar]
- 29.Ramsey BW, Pepe MS, Quan JM, Otto KL, Montgomery AB, Williams-Warren J, Vasiljev KM, Borowitz D, Bowman CM, Marshall BC, Marshall S, Smith AL. 1999. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N Engl J Med 340:23–30. [DOI] [PubMed] [Google Scholar]
- 30.Mena A, Smith EE, Burns JL, Speert DP, Moskowitz SM, Perez JL, Oliver A. 2008. Genetic adaptation of Pseudomonas aeruginosa to the airways of cystic fibrosis patients is catalyzed by hypermutation. J Bacteriol 190:7910–7917. doi: 10.1128/JB.01147-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hancock RE, Speert DP. 2000. Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and impact on treatment. Drug Resist Updat 3:247–255. doi: 10.1054/drup.2000.0152. [DOI] [PubMed] [Google Scholar]
- 32.Gallagher LA, Shendure J, Manoil C. 2011. Genome-scale identification of resistance functions in Pseudomonas aeruginosa using Tn-seq. mBio 2:e00315-10. doi: 10.1128/mBio.00315-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Opijnen T, Bodi KL, Camilli A. 2009. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods 6:767–772. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turner KH, Wessel AK, Palmer GC, Murray JL, Whiteley M. 2015. Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc Natl Acad Sci U S A 112:4110–4115. doi: 10.1073/pnas.1419677112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hooper DC. 2001. Mechanisms of action of antimicrobials: focus on fluoroquinolones. Clin Infect Dis 32:S9–S15. doi: 10.1086/319370. [DOI] [PubMed] [Google Scholar]
- 36.Davis BD. 1987. Mechanism of bactericidal action of aminoglycosides. Microbiol Rev 51:341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hartmann G, Honikel KO, Knusel F, Nuesch J. 1967. The specific inhibition of the DNA-directed RNA synthesis by rifamycin. Biochim Biophys Acta 145:843–844. doi: 10.1016/0005-2787(67)90147-5. [DOI] [PubMed] [Google Scholar]
- 38.Cho H, Uehara T, Bernhardt TG. 2014. Beta-lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell 159:1300–1311. doi: 10.1016/j.cell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Springer MT, Singh VK, Cheung AL, Donegan NP, Chamberlain NR. 2016. Effect of clpP and clpC deletion on persister cell number in Staphylococcus aureus. J Med Microbiol 65:848–857. doi: 10.1099/jmm.0.000304. [DOI] [PubMed] [Google Scholar]
- 40.Meylan S, Porter CBM, Yang JH, Belenky P, Gutierrez A, Lobritz MA, Park J, Kim SH, Moskowitz SM, Collins JJ. 2017. Carbon sources tune antibiotic susceptibility in Pseudomonas aeruginosa via tricarboxylic acid cycle control. Cell Chem Biol 24:195–206. doi: 10.1016/j.chembiol.2016.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koeva M, Gutu AD, Hebert W, Wager JD, Yonker LM, O'Toole GA, Ausubel FM, Moskowitz SM, Joseph-McCarthy D. 22 November 2017. An antipersister strategy for treatment of chronic Pseudomonas aeruginosa infections. Antimicrob Agents Chemother doi: 10.1128/AAC.00987-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taber HW, Mueller JP, Miller PF, Arrow AS. 1987. Bacterial uptake of aminoglycoside antibiotics. Microbiol Rev 51:439–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, Will O, Kaul R, Raymond C, Levy R, Chun-Rong L, Guenthner D, Bovee D, Olson MV, Manoil C. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 100:14339–14344. doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537-12. doi: 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fürste JP, Pansegrau W, Frank R, Blöcker H, Scholz P, Bagdasarian M, Lanka E. 1986. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene 48:119–131. doi: 10.1016/0378-1119(86)90358-6. [DOI] [PubMed] [Google Scholar]
- 47.Wiuff C, Zappala RM, Regoes RR, Garner KN, Baquero F, Levin BR. 2005. Phenotypic tolerance: antibiotic enrichment of noninherited resistance in bacterial populations. Antimicrob Agents Chemother 49:1483–1494. doi: 10.1128/AAC.49.4.1483-1494.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zaslaver A, Bren A, Ronen M, Itzkovitz S, Kikoin I, Shavit S, Liebermeister W, Surette MG, Alon U. 2006. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat Methods 3:623–628. doi: 10.1038/nmeth895. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.