

Enzyme use has become more widespread in industry. This study evaluated the molecular basis and effectiveness of ribosome engineering in markedly enhancing enzyme production (>1,000-fold). This method, due to its simplicity, wide applicability, and scalability for large-scale production, should facilitate not only industrial enzyme production but also the identification of novel enzymes, because microorganisms contain many silent or weakly expressed genes which encode novel antibiotics or enzymes. Furthermore, this study provides a new mechanism for strain improvement, with a consistent rather than transient high expression of the key gene(s) involved in enzyme production.
KEYWORDS: enzyme production, silent genes, ribosome engineering, drug resistance, metabolome analysis
ABSTRACT
This study shows that sequential introduction of drug resistance mutations substantially increased enzyme production in Paenibacillus agaridevorans. The triple mutant YT478 (rsmG Gln225→stop codon, rpsL K56R, and rpoB R485H), generated by screening for resistance to streptomycin and rifampin, expressed a 1,100-fold-larger amount of the extracellular enzyme cycloisomaltooligosaccharide glucanotransferase (CITase) than the wild-type strain. These mutants were characterized by higher intracellular S-adenosylmethionine concentrations during exponential phase and enhanced protein synthesis activity during stationary phase. Surprisingly, the maximal expression of CITase mRNA was similar in the wild-type and triple mutant strains, but the mutant showed greater CITase mRNA expression throughout the growth curve, resulting in enzyme overproduction. A metabolome analysis showed that the triple mutant YT478 had higher levels of nucleic acids and glycolysis metabolites than the wild type, indicating that YT478 mutant cells were activated. The production of CITase by the triple mutant was further enhanced by introducing a mutation conferring resistance to the rare earth element, scandium. This combined drug resistance mutation method also effectively enhanced the production of amylases, proteases, and agarases by P. agaridevorans and Streptomyces coelicolor. This method also activated the silent or weak expression of the P. agaridevorans CITase gene, as shown by comparisons of the CITase gene loci of P. agaridevorans T-3040 and another cycloisomaltooligosaccharide-producing bacterium, Paenibacillus sp. strain 598K. The simplicity and wide applicability of this method should facilitate not only industrial enzyme production but also the identification of dormant enzymes by activating the expression of silent or weakly expressed genes.
IMPORTANCE Enzyme use has become more widespread in industry. This study evaluated the molecular basis and effectiveness of ribosome engineering in markedly enhancing enzyme production (>1,000-fold). This method, due to its simplicity, wide applicability, and scalability for large-scale production, should facilitate not only industrial enzyme production but also the identification of novel enzymes, because microorganisms contain many silent or weakly expressed genes which encode novel antibiotics or enzymes. Furthermore, this study provides a new mechanism for strain improvement, with a consistent rather than transient high expression of the key gene(s) involved in enzyme production.
INTRODUCTION
At present, industrially produced enzymes are used in a broad variety of production processes, including the production of pulp and paper (e.g., cellulase, xylanase, and laccase), leather (e.g., protease and lipase), textiles (e.g., catalase and arylesterase), detergents (e.g., protease, cellulase, and lipase), food and beverages (e.g., amylase, protease, lipase, and pectinase), animal feed (e.g., xylanase, protease, and phytase), pharmaceuticals (e.g., lipolase and d-amino acid oxidase), fine chemicals (e.g., lipase), cosmetics (e.g., lipase), and biodiesel (e.g., lipase) (1). This has increased interest in methods that enhance enzyme production and activity (2–4). Classically, enzyme production by microorganisms has been enhanced by random mutagenesis, including mutants induced by chemicals such as N-methyl-N′-nitro-N-nitrosoguanidine (NTG) and ethyl methanesulfonate (EMS) and by UV radiation (5, 6). Strain improvement processes have been described for producing various industrial enzymes, including lipases, chitinases, cellulases, glucoamylases, and proteases (7–12). Enzymes for industrial use are produced by growing bacteria and fungi under submerged or solid-state conditions. In practice, the great majority of microbial enzymes come from a very limited number of genera, especially Aspergillus, Trichoderma, Bacillus, and Kluyveromyces. Most of the strains used by the food industry have either been employed for many years or were derived from such strains by mutation and selection (13). However, these approaches require much time, cost, and labor.
Genome shuffling is an alternative that enables the directed evolution of entire organisms via recursive recombination at the genome level, thus shortening the time required for strain improvement (14, 15). Unlike mutagenesis techniques, genetic engineering methods, such as gene cloning and the transformation of host organisms, are often effective in improving enzyme production, although genetic information is required beforehand (16, 17). The industrial production of recombinant enzymes is preceded by an extensive research and development phase that culminates in the construction of an optimal strain for enzyme production (16). The desired industrial goal, however, is often difficult to achieve using the native form of the enzyme. Recent developments in protein engineering have revolutionized the transformation of commercially available enzymes to better industrial catalysts. Protein engineering aims to modify the sequence of a protein, and hence its structure, to create enzymes with improved functional properties, such as stability, specific activity, inhibition by reaction products, and selectivity toward nonnatural substrates (17).
We previously demonstrated that modulating the cellular translational and transcriptional apparatus, by introducing spontaneous drug resistance mutations (ribosome engineering), is effective in activating silent or weakly expressed secondary metabolite genes, enabling the discovery of novel antibiotics (18–22). The concept of ribosome engineering was derived from findings using a Streptomyces lividans strain with an altered ribosomal S12 protein that confers resistance to streptomycin (23). This strain produced abundant quantities of the blue-pigmented antibiotic actinorhodin, which is not normally produced by S. lividans due to the dormancy of the antibiotic biosynthesis genes (23). Furthermore, the bacterial alarmone ppGpp, produced on the ribosome in response to nutrient starvation, was found to bind to RNA polymerase (24–26), eventually initiating the production of antibiotics (19, 27, 28). These results suggested that modified RNA polymerase, produced by introducing a rifampin resistance mutation, may mimic the ppGpp-bound form, activating the expression of biosynthetic gene clusters (29, 30).
We hypothesized that bacterial gene expression may be increased dramatically by altering transcription and translation pathways. That is, a ribosome engineering method targeting the ribosomal protein S12, other ribosomal proteins, and translation factors or RNA polymerases may activate or enhance the production of secondary metabolites. Using this method, strains with mutations in the genes rsmG (encoding the 16S rRNA methyltransferase), rpsL (encoding the ribosomal protein S12), and rpoB (encoding the RNA polymerase β-subunit) were isolated by selection with streptomycin and rifampin. Cumulative drug resistance mutations (e.g., octuple mutations) were found effective in markedly enhancing antibiotic production in Streptomyces species (31, 32). These methods do not require genetic information. This study reports that ribosome engineering can enhance enzyme production, using as an example cycloisomaltooligosaccharide glucanotransferase (CITase; EC 2.4.1.248) of Paenibacillus agaridevorans, an extracellular enzyme that catalyzes the formation of cycloisomaltooligosaccharides (CIs; cyclodextrans) from dextran and is utilized by the food industry as a masking agent for bitter substances.
RESULTS
Isolation and characterization of drug-resistant mutants.
P. agaridevorans wild-type strain T-3040 (formerly Bacillus circulans T-3040) produces little CITase (≤1 unit), suggesting that the gene encoding this enzyme is silent or weakly expressed (Fig. 1). The introduction of a spontaneous drug resistance mutation (rsmG, rpsL, or rpoB) by selecting for resistance to streptomycin or rifampin, thereby activating silent genes involved in the synthesis of secondary metabolites in actinomycetes, enhanced the production of CITase up to 215-fold (Table 1). Notably, the rsmG mutants, many of which resulted in frameshifts or the introduction of a stop codon, displayed similar resistance levels to streptomycin and the constitutive ability to produce 90- to 113-fold more CITase than T-3040, whereas the rpsL and rpoB mutants showed various levels of CITase production. Mutations in position Gln469 of rpoB resulted in the production of 174- to 215-fold more CITase than from T-3040. CITase has been produced industrially in Japan using the P. agaridevorans strain G22-10, which was derived from strain T-3040 by treatment with NTG and selecting for low-level resistance to streptomycin (33). As expected, strain G22-10 harbored a mutation (673C→T [Gln225→stop codon]) within the rsmG gene (Table 2), with high CITase productivity, equivalent to that of spontaneous rsmG mutants (Table 1).
FIG 1.
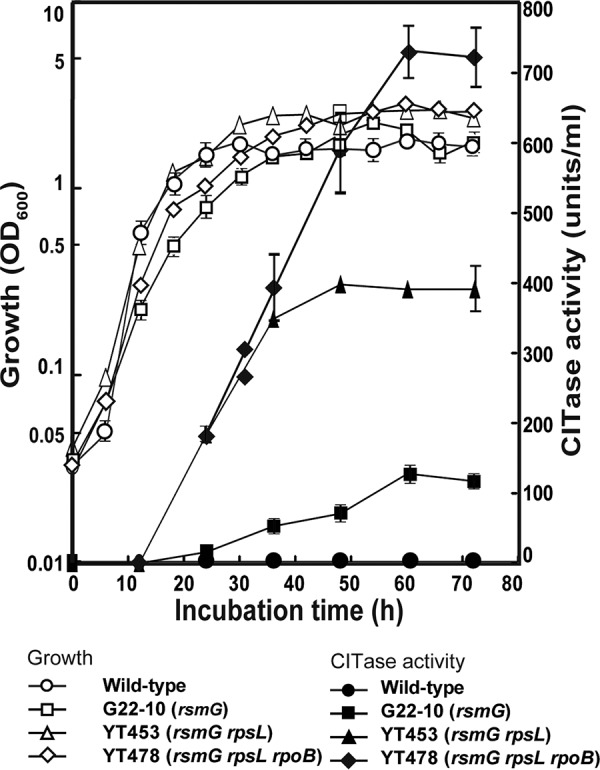
Growth and CITase production by wild-type strain T-3040, mutant strain G22-10, double mutant strain YT453, and triple mutant strain YT478. Strains were grown in liquid LB-Dex medium. Culture samples were withdrawn at the indicated times, and cell densities (growth) and CITase activities were measured. One unit of CITase activity was defined as the amount of enzyme that produced 1 nmol of the sum of CI-7, CI-8, and CI-9 per min from dextran at pH 5.5 at 40°C. Each strain was cultured in six culture flasks, and the results are expressed as means ± standard deviations.
TABLE 1.
Characterization of streptomycin- and rifampin-resistant mutants of P. agaridevorans T-3040
| Strain | Antibiotic (μg/ml) used for selection | Mutationa |
Amino acid substitution | Resistance (μg/ml)b |
CITase activity (U/ml)c | |||
|---|---|---|---|---|---|---|---|---|
| rsmG | rpsL | rpoB | Streptomycin | Rifampin | ||||
| T-3040 (wild type) | 20 | 0.5 | ≥1 | |||||
| YT362 | Streptomycin (30) | 118–121→CGAA→Δ | Frameshift (position 40→stop codon) | 150 | 0.5 | 90 ± 41 | ||
| YT359 | Streptomycin (30) | 119G→1-bp deletion | Frameshift | 150 | 0.5 | 109 ± 13 | ||
| YT361 | Streptomycin (30) | 148G→AA | Frameshift | 150 | 0.5 | 96 ± 10 | ||
| YT360 | Streptomycin (30) | 544G→T | Glu182→stop codon | 150 | 0.5 | 107 ± 11 | ||
| YT373 | Streptomycin (30) | 685C→T | Arg229→Cys | 150 | 0.5 | 113 ± 16 | ||
| YT382 | Streptomycin (100) | 167A→G | Lys56→Arg | 3000 | 0.5 | 140 ± 38 | ||
| YT374 | Streptomycin (100) | 192G→T | Lys64→Asn | 50 | 0.5 | 114 ± 8 | ||
| YT420 | Streptomycin (100) | 301A→G | Lys101→Glu | 1000 | 0.5 | 35 ± 16 | ||
| YT421 | Streptomycin (100) | 302A→G | Lys101→Arg | 500 | 0.5 | 99 ± 11 | ||
| YT385 | Streptomycin (100) | 302A→T | Lys101→Met | 500 | 0.5 | 109 ± 16 | ||
| YT386 | Streptomycin (100) | 314G→A | Gly105→Asp | 200 | 0.5 | 72 ± 12 | ||
| YT504 | Rifampin (2) | 1393T→C | Ser465→Pro | 20 | 5 | 32 ± 18 | ||
| YT510 | Rifampin (2) | 1405C→A | Gln469→Lys | 20 | 300 | 197 ± 62 | ||
| YT399 | Rifampin (2) | 1406A→T | Gln469→Leu | 20 | 30 | 215 ± 67 | ||
| YT393 | Rifampin (2) | 1406A→G | Gln469→Arg | 20 | 300 | 174 ± 3 | ||
| YT506 | Rifampin (2) | 1415A→T | Asp472→Leu | 20 | 2 | 3 ± 20 | ||
| YT395 | Rifampin (2) | 1433C→A | Ala478→Asp | 20 | 30 | 59 ± 12 | ||
| YT398 | Rifampin (2) | 1460C→A | Ser487→Thr | 20 | 30 | 110 ± 25 | ||
| YT394 | Rifampin (2) | 1460C→T | Ser487→Phe | 20 | 30 | 80 ± 8 | ||
Numbered from the start codon of the open reading frame of B. subtilis.
Determined after incubating for 3 days on NG plates.
Strains were grown in LB-Dex medium at 30°C for 3 days. One unit of CITase was defined as the amount of enzyme capable of producing 1 nmol of the sum of CI-7, CI-8, and CI-9 per min from dextran at pH 5.5 at 40°C. Experiments were performed in triplicate, and the data are expressed as means ± standard deviations.
TABLE 2.
Sequential introduction of streptomycin and rifampin resistance mutations into the P. agaridevorans rsmG mutant G22-10
| Straina | Antibiotic (μg/ml) used for selection | Mutationb |
Amino acid substitution | Resistance (μg/ml)c |
CITase activity (U/ml)d | |||
|---|---|---|---|---|---|---|---|---|
| rsmG | rpsL | rpoB | Streptomycin | Rifampin | ||||
| G22-10d | 673C→T | Gln225→stop codon | 300 | 0.5 | 117 ± 8 | |||
| YT453 | Streptomycin (2,000) | 673C→T | 167A→G | Lys56→Arg | 3,000 | 0.5 | 518 ± 77 | |
| YT483 | Rifampin (2) | 673C→T | 167A→G | 1406A→G | Gln469→Arg | 3,000 | 300 | 168 ± 142 |
| YT474 | Rifampin (2) | 673C→T | 167A→G | 1433C→A | Ala478→Asp | 3,000 | 30 | 432 ± 72 |
| YT477 | Rifampin (2) | 673C→T | 167A→G | 1444C→G | His482→Asp | 3,000 | 100 | 644 ± 45 |
| YT502 | Rifampin (2) | 673C→T | 167A→G | 1444C→T | His482→Tyr | 3,000 | 100 | 195 ± 42 |
| YT503 | Rifampin (2) | 673C→T | 167A→G | 1445C→G | His482→Arg | 3,000 | 100 | 112 ± 99 |
| YT478 | Rifampin (2) | 673C→T | 167A→G | 1454G→A | Arg485→His | 3,000 | 30 | 1,104 ± 143 |
| YT475 | Rifampin (2) | 673C→T | 167A→G | 1460C→T | Ser487→Phe | 3,000 | 30 | 605 ± 66 |
G22-10 is the parent strain, from which YT453 was derived. YT483 to YT475 were derived from YT453.
Numbered from the start codon of the open reading frame of B. subtilis.
Determined after incubation for 3 days on NG plates.
Strains were grown in LB-Dex medium at 30°C for 3 days. One unit of CITase was defined as the amount of enzyme capable of producing 1 nmol of the sum of CI-7, CI-8, and CI-9 per min from dextran at pH 5.5 at 40°C. Experiments were performed in triplicate, and the data are expressed as means ± standard deviations.
Construction of combined drug-resistant mutants.
Starting from the industrial strain G22-10 (rsmG), we attempted to further increase CITase production by introducing cumulative drug resistance mutations. The introduction of an rpsL mutation (Lys56→Arg) by selecting for high-level resistance to streptomycin increased CITase production 4.4-fold. CITase production was further increased by introducing a third mutation, rpoB (Arg485→His), which confers resistance to rifampin, with the triple mutant YT478 producing 1,104 units/ml CITase, more than 1,000-fold higher than that of the wild-type strain T-3040 (Fig. 2A and Table 2). This amount corresponds to approximately 100 mg CITase per liter (or 0.01%), as determined by protein assay of culture supernatants. The triple mutant strain grew as well as the wild-type strain and produced CITase even after entering into stationary-growth phase (Fig. 1). Unexpectedly, several rpoB mutations (Gln469→Arg and Ala478→Asp), which enhanced CITase production when introduced into the wild-type strain, did not enhance CITase production when introduced into the double mutant (Table 2). These findings suggested that only a limited number of rpoB mutations can have positive effects in improved strains, such as the double mutant YT453, because improved strains with multiple mutations would become more sensitive metabolically. This mechanism may also explain the reduced CITase productivity of YT502 and YT503. The CITase production profiles of the wild-type and mutant strains were confirmed by SDS-PAGE and zymography (Fig. 2B). The molecular size of CITase was predicted to be 103 kDa, similar to that of the protein band on Coomassie brilliant blue R-250 (CBB)-stained gels and the halos observed by zymography. A smaller-sized band detected at 96 kDa with reduced enzyme activity may be the C-terminal-truncated form of CITase, which was partially digested by proteases secreted into the medium (33). Notably, dextran, but not other carbohydrates, induced CITase production by wild-type (T-3040) and mutant (YT453 and YT478) strains (see Fig. S1 in the supplemental material).
FIG 2.

Detection and analysis of CITase. (A) CITase production by wild-type and single, double, and triple mutant strains. One unit of CITase was defined as the amount of enzyme that produced 1 nmol of the sum of CI-7, CI-8, and CI-9 per min from dextran at pH 5.5 at 40°C. Experiments were performed in triplicate, and the results are expressed as means ± standard deviations. (B) SDS-PAGE (left) and zymography (right) of CITase. The arrows indicate native CITase and the 96 kDa protein.
Transcriptional analysis of the mutants.
Transcriptional analysis was performed to assess the mechanism underlying CITase overproduction. The expression of CITase mRNA was transient in the wild-type strain T-3040, being expressed only during transition phase (Fig. 3A). Strikingly, its periods of expression were greater in single (G22-10), double (YT453), and triple (YT478) mutant strains, in that order, although their maximum expression levels did not differ markedly, accounting for the constitutive production of CITase by YT453 and YT478 (Fig. 1). This contrasts with secondary metabolites, in that the enhanced antibiotic production by drug resistance mutants was almost always accompanied by much higher maximal expression levels of antibiotic biosynthesis genes (20, 21, 31, 32). A primer extension analysis of cit (see Fig. S2) confirmed these results, by demonstrating the absence of additional mutations as well as by detecting the transcriptional start points, at −10 and −35 hexamers, and the ribosome binding site (Shine-Dalgarno [SD] sequence). However, codons 2 (GTG) and 5 (ATG) may be potential start codons, as GTG is often used as a translation start codon (Fig. 4A and B).
FIG 3.

Functional analysis of mutant strains. (A) Transcriptional analysis of the gene encoding CITase. Strains were grown in LB-Dex medium at 30°C to exponential phase (OD600 of 0.2), transition phase (OD600 of 0.7), early stationary phase (12 h after transition phase), and late stationary phase (24 h after transition phase). (B) In vitro protein synthesis activities of mutant strains as determined by chloramphenicol acetyltransferase (CAT) activity. Strains were grown to late exponential phase (OD600 of 0.4), and cell-free synthesis of CAT by washed ribosomes was performed as described in Materials and Methods. Experiments were performed in triplicate, and the results are expressed as means ± standard deviations.
FIG 4.

Molecular analysis of the CITase gene. (A) Schematic representation of gene loci containing cit in the CI-producing bacteria. The gene loci (contig_0325 [PAT3040_05184 to PAT3040_05199]) of P. agaridevorans T-3040 (top) and Paenibacillus sp. 598K (GenBank accession no. LC160266.1 [bottom]) are indicated. Genes encoding CITase (cit), 6-α-glucosyltransferase (6gt31a), and other proteins are shown as black, white, and gray arrows, respectively. Pcit, promoter region of the cit gene. (B) Promoter region of the cit gene. The transcriptional start points for the cit gene are indicated by asterisks. The −10 and −35 hexamers are underlined, and the ribosome binding site (SD sequence) is indicated by a dotted underline.
Translational analysis of the mutants.
We next studied the protein synthesis activity and intracellular S-adenosylmethionine (SAM) levels of mutant and wild-type strains. To avoid the possible effects of rpoB and/or rpsL mutations, the single mutant YT362 (rsmG) and the double mutant YT453 (rsmG rpsL), rather than the triple mutant YT478, were compared with the wild type. The rsmG rpsL double mutant YT453 was found to have a 2.5-fold higher in vitro protein synthesis activity than the wild type, an increase apparently due to the rpsL mutation, as determined by chloramphenicol acetyltransferase activity (Fig. 3B; see also Fig. S3). In addition, the expression of the metK gene, which encodes the enzyme SAM synthetase, was significantly enhanced in the rsmG mutant YT362 (and in other rsmG mutants G22-10, YT453, and YT478) (Fig. 5), resulting in higher intracellular SAM levels (see Fig. S4). These results are consistent with findings showing that, in Streptomyces and Bacillus, protein synthesis activity during transition and stationary phases and intracellular SAM levels are both crucial for the activation or enhancement of secondary metabolism, including antibiotic production, as demonstrated by rpsL, rsmG, and mthA mutants (18, 34–37).
FIG 5.
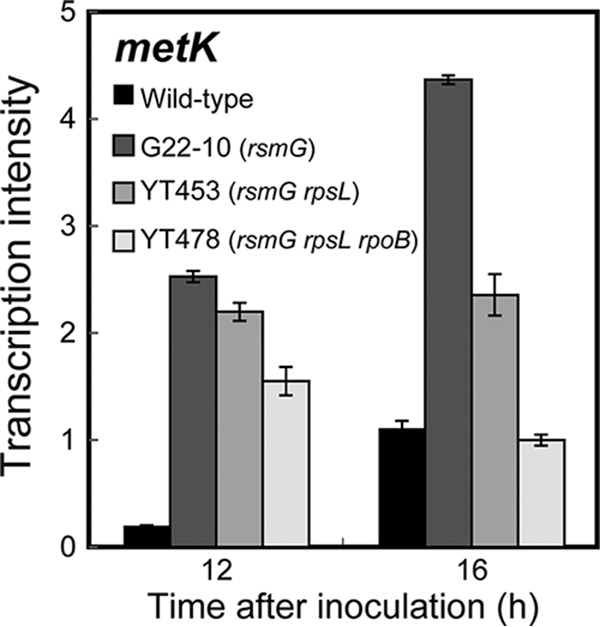
Transcriptional analysis of the metK gene encoding S-adenosylmethionine synthetase in the wild-type and mutant (G22-10, YT453, and YT478) strains harboring the rsmG mutation. Transcription of metK was determined as described in Materials and Methods. Experiments were performed in triplicate, and the results are expressed as means ± standard deviations.
Metabolome analysis of the mutants.
Metabolome analysis was performed to assess the metabolic changes resulting from the triple mutations in rsmG, rpsL, and rpoB. A comparison of the triple mutant YT478 with wild-type strain T-3040 showed many differences in metabolism (see Data Set S1). YT478 had higher levels of nucleic acids and glycolysis metabolites during mid-exponential phase. This was especially pronounced for the nucleoside triphosphates (ATP, GTP, UTP, and CTP), the concentrations of which were 1.5- to 3-fold higher in YT478 than in the wild type. The differences in key metabolites between wild-type and triple mutant strains are summarized in Table 3. Notably, the level of intracellular SAM during mid-exponential phase (12 h) was 2-fold higher in the triple mutant than in the wild type (Table 3), consistent with the increased expression of metK in YT478 (Fig. 5) and suggesting the significance of the SAM level in activating enzyme production. These metabolic changes may be a reflection of the “activated” YT478 mutant cells and may play at least a partial role in the overproduction of CITase. For example, the elevated concentrations of nucleoside triphosphates, especially ATP and GTP, may contribute to the cellular energy status required for CITase overproduction. Moreover, many metabolites, including pyruvic acid, 2-oxoglutaric acid, citric acid, gluconic acid, ribulose 5-phosphate, sedoheptulose 7-phosphate, GMP, guanosine, inosine, and hypoxanthine, were significantly enriched at 48 h in the wild type but not in YT478. Although the association of these events with CITase productivity remains unclear, marked metabolic differences were observed between the wild-type and triple mutant strains. The lower levels of almost all amino acids observed at 48 h in the triple mutant YT478 may be explained by the active CITase production at this growth phase, which would consume intracellular amino acids.
TABLE 3.
Intracellular concentrations of key metabolites during exponential (12 h) growth of the wild-type (T-3040) and triple mutant (YT478) strainsa
| Metabolite | Concn (pmol/OD unit)b |
|
|---|---|---|
| Wild-type (T-3040) | Mutant (YT478) | |
| SAMc | 134 ± 11 | 274 ± 19 |
| ATP | 970 ± 45 | 1,431 ± 330 |
| GTP | 159 ± 45 | 304 ± 158 |
| UTP | 247 ± 9.7 | 457 ± 115 |
| CTP | 234 | 756 ± 71 |
| Glucose 6-phosphate | 207 ± 167 | 527 ± 229 |
| Fructose 1,6-diphosphate | 185 ± 147 | 1,000 ± 572 |
| 3-Phosphoglyceric acid | 465 ± 226 | 1,565 ± 406 |
| 2-Phosphoglyceric acid | 68 ± 42 | 157 ± 24 |
| Phosphoenolpyruvic acid | 133 ± 59 | 446 ± 77 |
Strains were grown to mid-exponential (12 h) phase in LB-Dex medium. Intracellular metabolites were extracted and subjected to metabolome analysis.
All experiments were performed in triplicate, and the data are expressed as means ± standard deviations.
SAM, S-adenosylmethionine.
Further enhancement of CITase production by scandium resistance mutations.
We previously reported that certain mutations conferring resistance to rare earth elements such as scandium effectively increase antibiotic production by Streptomyces spp. (19) and amylase production by Bacillus subtilis (38). As expected, the introduction of certain scandium resistance mutations further increased CITase production. More than half of the 18 scandium-resistant mutants derived from the triple mutant YT478 showed higher production of CITase than the parental strain YT478. The representative strain SC9, which was resistant up to 500 μg/ml of ScCl3·6H2O, displayed a 1.4-fold-greater production of CITase than YT478, although the mutated gene has not yet been identified. These results show that scandium resistance mutations not only enhance secondary metabolism (as represented by antibiotic production) but enhance the production of certain extracellular enzymes.
Gene locus for CI biosynthesis.
To compare the CITase gene cit in P. agaridevorans with that of another CI-producing bacterium, Paenibacillus sp. strain 598K, we sequenced the genome of the wild-type strain T-3040. The draft genome of T-3040 contained 467 contigs with a total length of 8,694,911 bp and a coverage depth of 108×. The largest contig was 597,596 bp, the N50 contig length was 117,306 bp, and the genome contained 6,887 open reading frames (ORFs). A genome analysis revealed that this strain possesses only one gene encoding CITase (locus tag PAT3040_05188). According to the CAZy database (39), the CITase in T-3040 belongs to the glycoside hydrolase family 66 (GH66), which consists of CITases and dextranases (EC 3.2.1.11). As reported previously (40), a gene in the AraC family encoding a regulatory protein (PAT3040_05187) was found upstream of cit, and genes encoding putative sugar ABC transporters were located on the opposite strand of cit both upstream (PAT3040_05184 to PAT3040_05186) and downstream (PAT3040_05190 to PAT3040_05192) (Fig. 4A). A gene (putative 6gt31a [PAT3040_05195 and PAT3040_05196]) encoding a putative glycoside hydrolase with approximately 60% amino acid sequence identity to Paenibacillus sp. 598K 6-α-glucosyltransferase, which is essential for the production of the substrate of CITase from starch (40), was found 7,625 bp downstream from cit on the opposite strand. Moreover, a distinct AraC regulator gene (PAT3040_05197) was found upstream of the putative 6gt31a. Importantly, the draft genome contained no potential dextranase genes other than cit (see Table S2).
Industrial applicability of the triple mutant YT478.
The productivity of microbial metabolites is often markedly decreased under conditions of large-scale production. This may be due, for example, to mechanical reasons in fermentation and/or the genetic instability of the microorganism. We therefore assessed the productivity of CITase by P. agaridevorans by using a 90-liter jar fermentor and a 300-liter tank. The levels of CITase produced by the triple mutant YT478 were as high under large-scale conditions as under laboratory conditions (97% in the 90-liter jar fermentor and 78% in the 300-liter tank compared with that in shake flask fermentation). Moreover, the CITase productivity of YT478 was 6- to 10-fold higher than that of the industrial strain G22-10 under large-scale conditions (for example, 802 units/ml [YT478] versus 110 units/ml [G22-10] in 90-liter jar fermentors). These results indicate that the YT478 strain may be used for industrial applications.
Applicability for production of other enzymes.
The introduction into B. subtilis of certain rpsL and rpoB mutations has been reported to enhance the production of amylases and proteases 1.3- to 3.5-fold (41, 42). Similarly, the introduction of the rpsL K88R mutation into a marine-derived strain of Streptomyces viridochromogenes was found to increase xylanase production 14% (43). The combined drug resistance mutation method was found to be widely applicable to enhance the production of certain extracellular enzymes. For example, P. agaridevorans production of amylase was enhanced 4.3-fold by introducing the rsmG, rpsL, and rpoB mutations (Fig. 6A). The introduction of certain rsmG, rpsL, and rpoB mutations into Streptomyces coelicolor enhanced agarase production 6.3-fold, but had a less pronounced effect on protease production, as shown by a 2.2-fold increase (Fig. 6B; see also Fig. S5). To normalize productivity, the production of these enzymes per numbers of cells, as assessed by optical density at 600 nm (OD600) or mg dry cell weight, was also determined (see Fig. S6). Unlike dormant or weakly expressed enzymes (such as CITase), a >3-fold increase in the productivity of any highly expressed enzyme (including amylase, protease, and agarase) is not trivial, as the improvement of high-production strains has been reported to be difficult.
FIG 6.

Enzyme production by the wild-type and mutant strains of P. agaridevorans and S. coelicolor. (A) Production of amylase by P. agaridevorans wild-type and single, double, and triple mutant strains. Strains were grown at 30°C for the indicated periods in LB medium supplemented with 1.5% glucose and 1% soluble starch. (B) Production of protease and agarase by Streptomyces coelicolor A3(2) and its mutant strains. Strains were grown at 30°C for 2 days in liquid skim milk medium (0.5% skim milk in deionized water) to measure protease production (left) or for 3 days in AYM medium, consisting of GYM medium (75) with 0.4% glucose replaced by 1% sodium aspartate, supplemented with 0.1% agarose to measure agarase production (right). Experiments were performed in triplicate, and the results are expressed as means ± standard deviations.
DISCUSSION
Microbial enzymes are of great importance in the development of industrial bioprocesses. The global market for these enzymes is continuing to grow and was estimated to be $5 billion in 2015 (13). Because enzymes for industrial use must be produced at low cost, many techniques have been developed for strain improvement (5, 6, 14–16). Currently, CIs are commercially produced from dextran using CITase obtained by culturing P. agaridevorans G22-10. We have shown that certain mutations that alter the ribosome (i.e., rsmG and rpsL mutations) and RNA polymerase β-subunit (i.e., rpoB mutations) cumulatively enhance enzyme production. Apart from such technical aspects, this study provides a new mechanism for strain improvement, resulting in constitutive, rather than transient, high expression of the key gene(s) involved in enzyme production. Similar findings have been observed in assessing antibiotic production by B. subtilis, in that strains carrying certain drug resistance (mthA) mutants associated with bacilysin overproduction expressed bacilysin biosynthesis genes throughout all growth phases (37, 44).
Strikingly, we found that the rpsL K56R mutation in the ribosomal protein S12 of P. agaridevorans, similar to the rpsL K56R mutation of B. subtilis and the rpsL K87E mutation of Escherichia coli (41, 45), resulted in enhanced protein synthesis activity during late exponential phase (Fig. 3B). Certain mutations within S12 activate or enhance antibiotic production in a variety of bacteria, and the rpsL K88E mutation (corresponding to position 87 in E. coli) enhanced the protein synthesis activity of S. coelicolor during transition and stationary phases, resulting in the overproduction of the blue-pigmented polyketide antibiotic actinorhodin (36, 46). In Pseudomonas putida, the rpsL K88R mutation conferred enhanced tolerance to toxic organic chemicals (47). The mechanisms underlying the generation of these mutants have been studied extensively in Streptomyces and Bacillus. The streptomycin-resistant (Smr) rpsL mutant ribosomes, carrying an amino acid substitution in ribosomal protein S12 that confers a high level of resistance to streptomycin, are more stable than wild-type ribosomes under stress conditions, such as amino acid starvation and low magnesium concentration, indicating that increased stability may enhance protein synthesis in stationary phase (35, 41). This characteristic may result in the enhancement during stationary phase of protein production from newly transcribed genes, such as those involved in antibiotic production. These findings suggest that both the greater stability of 70S ribosomes and the elevated levels of ribosome recycling factor resulting from the K88E mutation are responsible for enhanced protein synthesis in stationary phase, with the latter being responsible for antibiotic overproduction by the K88E mutant (35). The K88E mutation in S12, as well as rsmG mutations, result in the preferential transcription during stationary phase of S. coelicolor of specific genes, such as pathway-specific regulatory genes (actII-ORF4 and redD) governing antibiotic production (31, 36, 48). Although the mechanism by which the rpsL K88E mutation mediates such preferential gene transcription remains unclear, higher-order regulatory proteins may govern the expression of these pathway-specific regulatory genes.
Unlike rpsL mutations, mutations in rsmG (encoding 16S rRNA methyltransferase) have been shown to cause low-level streptomycin resistance in E. coli, Mycobacterium tuberculosis, S. coelicolor, and B. subtilis (32, 36, 49, 50). The loss of the m7G modification in 16S rRNA results in resistance to streptomycin, providing a molecular basis for rsmG mutation-induced streptomycin resistance. Our finding that SAM may play an essential role in triggering the onset of enzyme production is not unprecedented. For example, rsmG mutants in S. coelicolor exhibit enhanced expression of the metK gene encoding SAM synthetase, accompanied by increased protein synthesis during stationary phase and eventually leading to the overproduction of antibiotics (36). Thus, increases in SAM synthetase (i.e., increased SAM levels) resulting from rsmG mutations likely activated secondary metabolism in S. coelicolor (32, 36). Furthermore, the addition of SAM resulted in the overproduction of antibiotics by streptomycetes (51, 52), indicating that SAM may be a common intracellular signal molecule for the onset of secondary metabolism in Streptomyces and Bacillus (34, 36, 37). The rpsL K88E mutant did not show any enhancement of SAM synthetase expression (T. Hosaka and K. Ochi, unpublished data). The physiological significance of SAM has been discussed in B. subtilis and Streptomyces spp. (53–55). SAM is synthesized from methionine and ATP by a SAM synthetase encoded by the metK gene and plays a central role in many cellular functions (56–58). In contrast to Streptomyces cells (34), extracellular SAM cannot be incorporated by E. coli or B. subtilis cells (59, 60). Intracellular SAM levels are likely tightly regulated in native B. subtilis cells, as a 2-fold increase was found to markedly alter cellular physiology and morphology (37). SAM is the methyl donor for the methylation of cytosine and adenosine bases in DNA, rRNA, and tRNA, various proteins, and small molecules important for both lower and higher organisms (56, 61, 62). In E. coli, SAM was shown to be a corepressor of the methionine regulation system (57). Although there is no evidence to date for the existence of a methyltransferase involved in the regulation of antibiotic or enzyme production, SAM-dependent protein methylation may play a role in controlling the activity of regulatory proteins that control the expression of antibiotic biosynthesis or enzyme-encoding genes. Alternatively, DNA or RNA methylation may be involved in regulating the expression of these genes (63, 64).
In contrast to rpsL and rsmG mutations, certain rifampin-resistant (Rifr) rpoB mutations, which yield RNA polymerases with mutant β-subunits, activate antibiotic production by increasing the affinities of mutant RNA polymerases for promoters of certain genes in S. coelicolor, eventually enhancing the transcription of antibiotic biosynthesis genes (18). A subset of rifampin resistance (rpoB) mutations was found to result in the marked overproduction of antibiotics in various actinomycetes, with H437Y and H437R rpoB mutations being most frequently effective. Moreover, the rpoB mutations markedly activated (up to 70-fold at the transcriptional level) the cryptic/silent secondary metabolite biosynthetic gene clusters of these actinomycetes (20). Our present study with P. agaridevorans provides a molecular basis for the observed increase in the transcripts of cit in the rpoB R485H mutant. Taken together, these findings indicate that certain rpoB mutations increase the productivity of antibiotics or enzymes mainly, if not solely, by enhancing the affinity of mutant RNA polymerases to the relevant promoters (reviewed by Alifano et al. [65] and Ochi [22]).
Because little CITase is produced by the wild-type strain T-3040, cit should be weakly expressed in this strain. P. agaridevorans T-3040 produces CIs from starch very slowly, and the amount in culture of 6-α-glucosyl transferase, which produces dextran-like α-(1→6)-glucosyl chains from starch, was small but negligible (40). A comparison of the cit gene loci of P. agaridevorans T-3040 and Paenibacillus sp. 598K showed that the 6gt31a gene, which encodes 6-α-glucosyl transferase, is located immediately downstream from cit in the Paenibacillus sp. 598K genome and that these two genes are coexpressed by the same promoter (40). In contrast, the putative 6gt31a gene in P. agaridevorans T-3040 is located distant from cit and on the opposite strand. Moreover, a member of the AraC family seems to regulate the transcription of the putative 6gt31a gene, a regulator different from that for cit, suggesting that the CI production system of T3040 does not function well.
Cyclodextrin and cycloalternan are other types of bacterial cyclic oligosaccharides consisting of α-d-glucoses. Klebsiella pneumoniae has been reported to produce cyclodextrins extracellularly and intracellularly (66); these cyclodextrins are digested by a cyclodextrinase and metabolized by the maltose degradation pathway (66, 67). Cycloalternan-producing Bacillus sp. strain NRRL B-21195 has been reported to express an intracellular cycloalternan-degrading enzyme (68). CI-degrading enzymes must be dextranases, cleaving endo-type α-(1→6)-glucosidic bonds. In the CAZy database (39), dextranases were found in the GH66 and GH49 families. Because the draft genome of P. agaridevorans T-3040 contained no potential dextranase genes, except for cit, the lack of systems responsible for the production and digestion of CIs may have made CIs useless for strain T-3040, resulting in the degeneration of CITase-producing systems in this organism. This hypothesis may account for the silent or weak expression of the CITase gene in T-3040.
Sequencing of the genomes of Streptomyces, fungi, and myxobacteria has shown that each strain contains genes encoding the enzymes necessary to synthesize various potential secondary metabolites, but that these strains express only a fraction of these genes during fermentation (69, 70). The activation of silent or sleeping genes for the biosynthesis of bacterial secondary metabolites may therefore result in new methods applicable to industrial microbiology (18, 71, 72). Because the rsmG mutation activated the expression of the gene involved in CITase production, altering intracellular SAM levels by introducing an rsmG mutation may be feasible for activating dormant genes. Likewise, the introduction of rpsL and rpoB mutations could be used for a similar purpose. The discovery of a novel enzyme from a microbial source takes from months to years. Our method, involving activating the expression of silent or weakly expressed genes encoding dormant enzymes, does not require induced mutagenesis or gene-engineering techniques. Thus, the simplicity, wide applicability, and scalability of this method should facilitate not only industrial enzyme production but also the identification of novel enzymes from microbial sources.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Paenibacillus agaridevorans T-3040 (assigned on the basis of 16S rRNA sequencing data [DDBJ accession no. LC042199] [73]), with 99.9% identity to that of the type strain, and formerly called Bacillus circulans T-3040 (FERMB-4132 [NBRC]), was obtained from Tetsuya Oguma (Kikkoman Co). Strain G22-10, a high producer of CITase, was derived from T-3040 by treating the spores with NTG (33). Spontaneous streptomycin-resistant and rifampin-resistant mutants were isolated by spreading the spores onto modified nutrient broth glucose (NG) agar plates (10 g nutrient broth, 10 g glucose, 5 mg CuSO4·5H2O, 7.5 mg FeSO4·7H2O, 3.6 mg MnSO4·5H2O, 50 mg MgSO4·7H2O, 15 mg CaCl2·2H2O, and 9 mg ZnSO4·7H2O per liter) (74) containing various concentrations of the respective drugs. Similarly, spontaneous scandium-resistant mutants were isolated by plating onto modified NG agar plates containing 300 to 400 μg/ml of ScCl3·6H2O (purity, 99.9%). The growth of the parental strain was entirely inhibited by 300 μg/ml of ScCl3·6H2O. All mutant strains were stored in 20% (vol/vol) glycerol at −80°C and routinely cultured on NG agar medium at 30°C for 3 to 5 days. LB-Dex medium (Luria-Bertani [LB] medium containing 20 g dextran 40 [GE Healthcare] per liter) (33) was used for the production of CITase.
Determination of MICs.
To determine MICs, full growth cultures were dotted on NG plates containing various concentrations of a drug and incubated at 30°C for 72 h. The minimum drug concentration able to fully inhibit growth was defined as the MIC.
Mutation analysis of the rsmG, rpsL, and rpoB genes.
The rsmG, rpsL, and rpoB genes were each amplified by PCR, using the genomic DNA of each mutant strain as a template, synthetic oligonucleotide primers designed on the basis of sequences in several Bacillus strains (see Table S1 in the supplemental material), and Ex Taq (TaKaRa). Purified PCR products were directly sequenced with BigDye Terminator cycle sequencing kits (Perkin-Elmer Applied Biosystems, Foster City, CA).
Isolation of drug-resistant mutant strains from Streptomyces coelicolor A3(2).
The rsmG mutant (KO-179) and rsmG rpsL double mutant (KO-945) were obtained as spontaneous drug-resistant mutant strains, as described previously (32). The triple (rsmG rpsL rpoB) mutant KO-1252 was derived from the double mutant KO-945 as a spontaneous rifampin-resistant colony, which developed by growing for 10 days on a glucose-yeast extract-malt extract (GYM) plate (75) containing 100 μg/ml of rifampin. Strain KO-1252 contained a mutation (1279G→A [Asp427→Asn]) in the rpoB gene, as determined by DNA sequencing.
Assay of CITase activity.
CITase activity in the medium was determined as described previously (33). One unit of CITase activity was defined as the amount of enzyme that produced 1 nmol of the sum of CI-7, CI-8, and CI-9 per minute. The amount of CI (CI-7 to CI-12) in the culture supernatants was measured by high-pressure liquid chromatography (HPLC), as described previously (33). All CITase assays were repeated at least three times. The concentrations of protein (CITase) secreted into the medium were estimated using Bio-Rad Bradford protein assay kits (Bio-Rad, Hercules, CA), with bovine serum albumin (Sigma-Aldrich, St. Louis, MO) as the standard.
Assay of amylase, protease, and agarase activities.
The activities of amylase, protease, and agarase in the medium were determined as described previously (76, 77).
(i) Amylase.
Culture supernatants (50 μl) obtained after centrifugation were mixed with 0.1 ml of soluble starch solution (50 mM Tris-HCl [pH 6.8], 0.5% soluble starch) and incubated at 30°C. A 4-μl aliquot of each reaction mixture was added to 0.1 ml of Lugol solution, and its absorbance at 700 nm (A700) was measured. Units of amylase activity were expressed as (Ablank − Asample) × Ablank−1 × t−1 × 100, where t indicates the time (min) of the reaction and Ablank and Asample indicate the absorbances at 700 nm of the blank (water) and the sample, respectively.
(ii) Protease.
Culture supernatants (20 μl) obtained after centrifugation were mixed with reaction buffer (1.25% azocasein, 2 mM CaC12, 50 mM Tris-HCl [pH 7.5]) and incubated at 30°C for 20 min. The reactions were stopped by adding 0.2 ml of 10% trichloroacetic acid. The mixtures were centrifuged, and the absorbance of each supernatant was measured at 365 nm (A365). Units of protease activity were expressed as (Asample − Ablank) × t−1 × 100, where t indicates the time of the reaction and Ablank and Asample indicate the absorbances at 365 nm of the blank (water) and the sample, respectively.
(iii) Agarase.
Culture supernatants (40 μl) obtained after centrifugation were mixed with 360 μl of 50 mM sodium phosphate buffer (pH 7.0) containing 0.2% agarose (Agarose S; Nippon Gene Co., Ltd.). After incubating at 37°C for 30 to 60 min, the sample (100 μl) was mixed with 100 μl of dinitrosalicylic acid solution (650 mg dinitrosalicylic acid, 32.5 ml 2 M NaOH, 4.5 ml glycerol in 100 ml ultrapure water) and heated in boiling water for 10 min until the color developed. The reaction was stopped by cooling the sample on ice. Absorbance was measured at 540 nm (A540). One unit of enzyme activity was defined as the amount of activity producing an A540 of 0.001 per minute and per ml enzyme solution.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and zymography.
SDS-PAGE and enzyme zymography were performed as described previously (33). For SDS-PAGE, proteins were separated on 8% polyacrylamide gels and stained with Coomassie brilliant blue R-250 (CBB). For zymography, proteins were separated on 8% SDS-PAGE gels containing 1% blue dextran 2000 (GE Healthcare). The gels were subsequently washed twice with water to renature the proteins and soaked in 50 mM sodium acetate buffer (pH 5.5) at 37°C until active CITase was detected by the formation of a clear band on a blue background.
Total RNA preparation.
Cells were treated with 1 ml chilled killing buffer (20 mM Tris-HCl [pH 7.5], 5 mM MgCl2, 20 mM NaN3), collected by centrifugation, and kept at −80°C. Total cellular RNA was prepared using Isogen reagent (Nippon Gene). Each frozen cell pellet was resuspended in 1 ml of Isogen reagent and incubated at 50°C for 10 min. After cooling, 0.2 ml of chloroform was added, and the sample was vortex and centrifuged at 16,000 × g for 10 min. A 0.6-ml aliquot of each aqueous phase (top layer) was transferred to a clean tube, 0.4 ml of chloroform was added, and the sample was mixed by repeated inversion and centrifuged at 16,000 × g for 10 min. A 0.6-ml aliquot of the aqueous phase (top layer) was transferred to a clean tube. The RNA in each tube was precipitated with an equal volume of isopropanol, rinsed with 70% ethanol, and suspended in an appropriate volume of diethyl pyrocarbonate (DEPC)-treated water.
Real-time quantitative PCR.
Total RNA was prepared as described. Contaminating DNA was removed by incubating each total RNA sample (1 mg) with 1 U of DNase I (Invitrogen) at 25°C for 15 min. DNase I was inactivated by adding EDTA and heating to 65°C for 10 min. The RNA was reverse-transcribed using high-capacity RNA-to-cDNA kit reverse transcriptase (Applied Biosystems) and random hexamers according to the manufacturer's instructions. After terminating the reaction, the samples were diluted with an appropriate volume of water and analyzed using the 7300 real-time PCR system and Power SYBR green PCR master mix (Applied Biosystems). The synthetic oligonucleotide primers used to amplify the CITase S-adenosylmethionine (SAM) synthetase gene (metK) and 16S rRNA (internal control) genes are listed in Table S1.
Primer extension.
Total RNAs were prepared and the contaminating DNA was removed as described above. After ethanol precipitation, 50 μg RNA was incubated at 80°C for 15 min with 1 pmol of an infrared dye-labeled primer for the CITase gene (cit), IRD800-CITase (5′-IRD800-GGAGGGGATACGACGGATGCGACG-3′) (Aloka). After annealing, 5 μl of 0.1 M dithiothreitol, 10 μl of 5-fold first-strand buffer (250 mM Tris-HCl [pH 8.3], 375 mM KCl, and 15 mM MgCl2), and 5 μl of deoxynucleoside triphosphate (dNTP) mix (10 mM [each] dATP, dGTP, dCTP, and dTTP) were added. To 50 μl of this reaction mixture, 200 U of SuperScript II (Invitrogen) was added and the mixture was incubated at 42°C for 1 h. Subsequently, 1 μl of RNase mixture (Ambion) was added, and the reaction mixture was further incubated at 37°C for 30 min. After ethanol precipitation, primer extension and sequencing reactions with the corresponding primers were run on polyacrylamide gels, followed by analysis using the DNA sequencing system LIC-4200L(S)-2 (LI-COR).
In vitro protein synthesis.
Cells grown to late exponential phase (A600 of 0.4) in LB-Dex medium were collected by centrifugation and washed with standard buffer (10 mM Tris-HCl [pH 7.7] containing 2 mM phenylmethylsulfonyl fluoride, 10 mM magnesium acetate, 30 mM ammonium acetate, and 6 mM 2-mercaptoethanol). Washed ribosomes were prepared (41), followed by the cell-free synthesis of chloramphenicol acetyltransferase (CAT), as described previously (78).
Determination of SAM level.
The levels of intracellular SAM in P. agaridevorans wild-type and mutant strains were determined by reverse-phase HPLC, as described previously (34, 79). Cells grown to various growth phases were harvested, transferred to petri dishes containing 10 ml of 1 M formic acid, and allowed to stand at 4°C for 1 h. The formic acid was collected, filtered through a membrane filter (pore size, 0.45 μm), and lyophilized. Each lyophilized sample was dissolved in a small volume of water and analyzed on a TSKgel ODS-100V column (4.6 mm by 250 mm; Tosoh, Tokyo, Japan). To normalize the number of picomoles of SAM per milliliter of culture to the number of picomoles per number of cells, intracellular SAM levels were expressed as pmol/optical density (OD) unit, where 1 OD unit was defined as the number of cells that would produce an absorbance of 1 at 600 nm (A600 of 1.0) if suspended in 1 ml.
Metabolome analysis.
Cells grown at 30°C for 12 h or 48 h in LB-Dex medium were harvested by filtration using membrane filters (pore size, 0.45 μm; Millipore), washed twice with deionized water, immediately transferred to a petri dish containing 10 ml of 1 M formic acid, and allowed to stand at 4°C for 1 h. The formic acid was collected, filtered through a membrane filter, and lyophilized. The lyophilized samples were dissolved in a small amount of deionized water for metabolome analysis. Metabolome analysis was performed by a facility service at Human Metabolome Technology Inc. (Tsuruoka, Japan). Capillary electrophoresis-time of flight mass spectrometry (CE-TOFMS) was performed using an Agilent CE capillary electrophoresis system (Agilent Technologies, Waldbronn, Germany), equipped with an Agilent 6210 time of flight mass spectrometer, Agilent 1100 isocratic HPLC pump, Agilent G1603A CE-MS adapter kit, and Agilent G1607A CE-electrospray ionization (ESI)-MS sprayer kit. The systems were controlled by Agilent G2201AA ChemStation software version B.03.01 for CE. The metabolites were analyzed using a fused silica capillary column (50-μm inner diameter by 80-cm total length), with commercial electrophoresis buffers (solution identifiers H3301-1001 for cation analysis and H3302-1021 for anion analysis; Human Metabolome Technologies) as the electrolyte. Samples were injected at a pressure of 5 kPa for 10 s (approximately 10 nl) for cation analysis and for 25 s (approximately 25 nl) for anion analysis. The spectrometer was scanned from m/z 50 to 1,000. Other conditions were as described previously (80, 81). Peaks were extracted using MasterHands automatic integration software (Keio University, Tsuruoka, Japan) to obtain peak information, including m/z, migration time for CE-TOFMS measurement (MT), and peak area (82). Signal peaks corresponding to isotopomers, adduct ions, and other product ions of known metabolites were excluded, and the remaining peaks were annotated with putative metabolites from the HMT metabolite database, based on their MTs and m/z values determined by TOFMS. The tolerance range for the peak annotation was configured at ±0.5 min for MT and ±10 ppm for m/z. Peak areas were normalized against those of the internal standards, and the resultant relative areas were further normalized by sample amount. Hierarchical cluster analysis (HCA) and principal-component analysis (PCA) were performed using PeakStat and SampleStat proprietary software, respectively. To normalize the number of picomoles of metabolites per milliliter of culture to the number of picomoles per number of cells, intracellular metabolite levels were expressed as pmol/optical density (OD) unit, where 1 OD unit was defined as the number of cells that would produce an A600 of 1 if suspended in 1 ml.
Draft genome analysis.
A DNA library with a median insert size of 500 bp was constructed in a multiplexed paired-end read format, according to the Illumina protocols. Whole-genome sequencing, generating 100-bp paired-end reads, was performed using an Illumina Genome Analyzer IIx according to the manufacturer's protocol. De novo assembly was performed using Velvet (83) with parameters optimized by the VelvetOptimiser (https://github.com/tseemann/VelvetOptimiser). Coding sequences were predicted by MiGAP (http://www.migap.org/). Gene functions were annotated by a blastp search against an in-house protein database consisting of all predicted proteins in RefSeq representative genomes. Carbohydrate-related enzymes were also annotated using the CAZy database (http://www.cazy.org/) (39) and the dbCAN annotation server (http://csbl.bmb.uga.edu/dbCAN/) (84).
Large-scale production of CITase.
Fermentation was scaled up by C-I-Bio Ltd. (Okinawa), using a 90-liter jar fermentor (medium, 60 liters; MSJ-U2W type; B. E. Marubishi Co., Kasugai, Japan) and a 300-liter tank (medium 200 L; Tsukayama Stainless Co., Okinawa, Japan).
Reproducibility.
All experiments were performed at least three times to confirm reproducibility, except for the experiments in Data Set S1, which were performed only once.
Accession number(s).
The draft genome sequence of Paenibacillus agaridevorans T-3040 has been deposited in the DDBJ/EMBL/GenBank databases under accession numbers BDQX01000001 to BDQX01000467.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants to K.O. from the National Agriculture and Food Research Organization (Programme for Promotion of Basic and Applied Research for Innovations in Bio-oriented Industry) and the MEXT-Supported Program for the Strategic Research Foundation at Private Universities 2014 to 2016 (S1413002), in addition to the Ministry of Education, Culture, Sports, and Technology of the Japanese Government (Effective Promotion of Joint Research of Special Coordination Funds). This work was also supported in part by JSPS KAKENHI grant number 26450133 and Gender Equality Office, University of Yamanashi.
K.O. designed the project, analyzed the data, and wrote the paper; K.F. and Y.T. isolated drug-resistant mutants and analyzed CITase activity. T.H. and T.I. performed biochemical and transcriptional analyses of protein synthesis and cit, respectively. Y.H. assayed intracellular SAM level, and K.M. and K.K. studied the cultivation conditions for CITase production. Y.S., T.M., N.F., and H.Y. conducted genome analysis of P. agaridevorans. S.G. conducted the scale-up experiments.
We thank Yasuko Tanaka, Yu Morimoto, Tomoko Watanabe, Hiroaki Cho, Masumi Izawa, Izumi Yamada, and Yasuyuki Kawabata for their skilled assistance.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00188-18.
REFERENCES
- 1.Jegannathan KR, Nielsen PH. 2013. Environmental assessment of enzyme use in industrial production—a literature review. J Clean Prod 42:228–240. doi: 10.1016/j.jclepro.2012.11.005. [DOI] [Google Scholar]
- 2.Gurung N, Ray S, Bose S, Rai V. 2013. A broader view: microbial enzymes and their relevance in industries, medicine, and beyond. Biomed Res Int 2013:329121. doi: 10.1155/2013/329121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adrio JL, Demain AL. 2014. Microbial enzymes: tools for biotechnological processes. Biomolecules 4:117–139. doi: 10.3390/biom4010117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joshi S, Satyanarayana . 2015. In vitro engineering of microbial enzymes with multifarious applications: prospects and perspectives. Bioresour Technol 176:273–283. doi: 10.1016/j.biortech.2014.10.151. [DOI] [PubMed] [Google Scholar]
- 5.Parekh S, Vinci VA, Strobel RJ. 2000. Improvement of microbial strains and fermentation processes. Appl Microbiol Biotechnol 54:287–301. doi: 10.1007/s002530000403. [DOI] [PubMed] [Google Scholar]
- 6.Adrio JL, Demain AL. 2006. Genetic improvement of processes yielding microbial products. FEMS Microbiol Rev 30:187–214. doi: 10.1111/j.1574-6976.2005.00009.x. [DOI] [PubMed] [Google Scholar]
- 7.Koncerova H, Vachova L, Chaloupka J. 1984. Mutants of Bacillus megaterium with altered synthesis of an exocellular neutral proteinase. Folia Microbiol (Praha) 29:99–103. doi: 10.1007/BF02872923. [DOI] [PubMed] [Google Scholar]
- 8.Haki GD, Rakshit SK. 2003. Development in industrially important thermostable enzymes: a review. Bioresour Technol 89:17–34. doi: 10.1016/S0960-8524(03)00033-6. [DOI] [PubMed] [Google Scholar]
- 9.Bapiraju KVVSN, Sujatha P, Ellaiah P, Ramana T. 2004. Mutation induced enhanced biosynthesis of lipase. Afr J Biotechnol 3:618–621. [Google Scholar]
- 10.Karanam SK, Medicherla NR. 2008. Enhanced lipase production by mutation induced Aspergillus japonicus. Afr J Biotechnol 7:2064–2067. doi: 10.5897/AJB2008.000-5054. [DOI] [Google Scholar]
- 11.Zia MA, Khalil-ur-Rahman Sheikh MA, Khan IA. 2010. Chemically treated strain improvement of Aspergillus niger for enhanced production of glucose oxidase. Int J Agric Biol 12:964–966. [Google Scholar]
- 12.Raju EVN. 2013. Bacillus cereus GD55 strain improvement by physical and chemical mutagenesis for enhanced production of fibrinolytic protease. Int J Pharma Sci Res 4:81–93. [Google Scholar]
- 13.Sarrouh B, Santos TM, Miyoshi A, Dias R, Azevedo V. 2012. Up-to-date insight on industrial enzymes applications and global market. J Bioprocess Biotech S4:002. doi: 10.4172/2155-9821.S4-002. [DOI] [Google Scholar]
- 14.Cheng Y, Song X, Qin Y, Qu Y. 2009. Genome shuffling improves production of cellulase by Penicillium decumbens JU-A10. J Appl Microbiol 107:1837–1846. doi: 10.1111/j.1365-2672.2009.04362.x. [DOI] [PubMed] [Google Scholar]
- 15.Biot-Pelletier D, Martin VJ. 2014. Evolutionary engineering by genome shuffling. Appl Microbiol Biotechnol 98:3877–3887. doi: 10.1007/s00253-014-5616-8. [DOI] [PubMed] [Google Scholar]
- 16.Olempska-Beer Z, Merker RI, Ditto MD, DiNovi MJ. 2006. Food-processing enzymes from recombinant microorganisms–a review. Regul Toxicol Pharmacol 45:144–158. doi: 10.1016/j.yrtph.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 17.Singh RK, Tiwari MK, Singh R, Lee J-K. 2013. From protein engineering to immobilization: promising strategies for the upgrade of industrial enzymes. Int J Mol Sci 14:1232–1277. doi: 10.3390/ijms14011232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hosaka T, Ohnishi-Kameyama M, Muramatsu H, Murakami K, Tsurumi Y, Kodani S, Yoshida M, Fujie A, Ochi K. 2009. Antibacterial discovery in actinomycetes strains with mutations in RNA polymerase or ribosomal protein S12. Nat Biotechnol 27:462–464. doi: 10.1038/nbt.1538. [DOI] [PubMed] [Google Scholar]
- 19.Ochi K, Hosaka T. 2013. New strategies for drug discovery: activation of silent or weakly expressed microbial gene clusters. Appl Microbiol Biotechnol 97:87–98. doi: 10.1007/s00253-012-4551-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanaka Y, Kasahara K, Hirose Y, Murakami K, Kugimiya R, Ochi K. 2013. Activation and products of the cryptic secondary metabolite biosynthetic gene clusters by rifampicin resistance (rpoB) mutations in actinomycetes. J Bacteriol 195:2959–2970. doi: 10.1128/JB.00147-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ochi K, Tanaka Y, Tojo S. 2014. Activating the expression of bacterial cryptic genes by rpoB mutations in RNA polymerase or by rare earth elements. J Ind Microbiol Biotechnol 41:403–414. doi: 10.1007/s10295-013-1349-4. [DOI] [PubMed] [Google Scholar]
- 22.Ochi K. 2017. Insights into microbial cryptic gene activation and strain improvement: principle, application and technical aspects. J Antibiot (Tokyo) 70:25–40. doi: 10.1038/ja.2016.82. [DOI] [PubMed] [Google Scholar]
- 23.Shima J, Hesketh A, Okamoto S, Kawamoto S, Ochi K. 1996. Induction of actinorhodin production by rpsL (encoding ribosomal protein S12) mutations that confer streptomycin resistance in Streptomyces lividans and Streptomyces coelicolor A3(2). J Bacteriol 178:7276–7284. doi: 10.1128/jb.178.24.7276-7284.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Artsimovitch I, Patlan V, Sekine S, Vassylyeva MN, Hosaka T, Ochi K, Yokoyama S, Vassylyev DG. 2004. Structural basis for transcription regulation by alarmone ppGpp. Cell 117:299–310. doi: 10.1016/S0092-8674(04)00401-5. [DOI] [PubMed] [Google Scholar]
- 25.Zuo Y, Wang Y, Stritz TA. 2013. The mechanism of E. coli RNA polymerase regulation by ppGpp is suggested by the structure of their complex. Mol Cell 50:430–436. doi: 10.1016/j.molcel.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ross W, Verntas CE, Sanchez-Vazquez P, Gaal T, Gourse RL. 2013. The magic spot: a ppGpp binding site on E. coli RNA polymerase responsible for regulation of transcription initiation. Mol Cell 50:420–429. doi: 10.1016/j.molcel.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bibb MJ. 2005. Regulation of secondary metabolism in streptomycetes. Curr Opin Microbiol 8:208–215. doi: 10.1016/j.mib.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 28.Ochi K. 2007. From microbial differentiation to ribosome engineering. Biosci Biotechnol Biochem 71:1373–1386. doi: 10.1271/bbb.70007. [DOI] [PubMed] [Google Scholar]
- 29.Lai C, Xu J, Tozawa Y, Okamoto-Hosoya Y, Yao X, Ochi K. 2002. Genetic and physiological characterization of rpoB mutations that activate antibiotic production in Streptomyces lividans. Microbiology 148:3365–3373. doi: 10.1099/00221287-148-11-3365. [DOI] [PubMed] [Google Scholar]
- 30.Xu J, Tozawa Y, Lai C, Hayashi H, Ochi K. 2002. A rifampicin resistance mutation in the rpoB gene confers ppGpp-independent antibiotic production in Streptomyces coelicolor A3(2). Mol Genet Genomics 268:179–189. doi: 10.1007/s00438-002-0730-1. [DOI] [PubMed] [Google Scholar]
- 31.Wang G, Hosaka T, Ochi K. 2008. Dramatic activation of antibiotic production in Streptomyces coelicolor by cumulative drug resistance mutations. Appl Environ Microbiol 74:2834–2840. doi: 10.1128/AEM.02800-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka Y, Komatsu M, Okamoto S, Tokuyama S, Kaji A, Ikeda H, Ochi K. 2009. Antibiotic overproduction by rpsL and rsmG mutants of various actinomycetes. Appl Environ Microbiol 75:4919–4922. doi: 10.1128/AEM.00681-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawabata Y, Kimura K, Funane K. 2012. Extracellular production of cycloisomaltooligosaccharide glucanotransferase and cyclodextran by a protease-deficient Bacillus subtilis host-vector system. Appl Microbiol Biotechnol 93:1877–1884. doi: 10.1007/s00253-011-3671-y. [DOI] [PubMed] [Google Scholar]
- 34.Okamoto S, Lezhava A, Hosaka T, Okamoto-Hosoya Y, Ochi K. 2003. Enhanced expression of S-adenosylmethionine synthetase causes overproduction of actinorhodin in Streptomyces coelicolor A3(2). J Bacteriol 185:601–609. doi: 10.1128/JB.185.2.601-609.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hosaka T, Xu J, Ochi K. 2006. Increased expression of ribosome recycling factor is responsible for the enhanced protein synthesis during the late growth phase in an antibiotic-overproducing Streptomyces coelicolor ribosomal rpsL mutant. Mol Microbiol 61:883–897. doi: 10.1111/j.1365-2958.2006.05285.x. [DOI] [PubMed] [Google Scholar]
- 36.Nishimura K, Hosaka T, Tokuyama S, Okamoto S, Ochi K. 2007. Mutations in rsmG, encoding a 16S rRNA methyltransferase, result in low-level streptomycin resistance and antibiotic overproduction in Streptomyces coelicolor A3(2). J Bacteriol 189:3876–3883. doi: 10.1128/JB.01776-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tojo S, Kim JY, Tanaka Y, Inaoka T, Hiraga Y, Ochi K. 2014. The mthA mutation conferring low-level resistance to streptomycin enhances antibiotic production in Bacillus subtilis by increasing the S-adenosylmethionine pool size. J Bacteriol 196:1514–1524. doi: 10.1128/JB.01441-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inaoka T, Ochi K. 2012. Undecaprenyl pyrophosphate involvement in susceptibility of Bacillus subtilis to rare earth elements. J Bacteriol 194:5632–5637. doi: 10.1128/JB.01147-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Funane K, Ichinose H, Araki M, Suzuki R, Kimura K, Fujimoto Z, Kobayashi M, Kimura A. 2014. Evidence for cycloisomaltooligosaccharide production from starch by Bacillus circulans T-3040. Appl Microbiol Biotechnol 98:3947–3954. doi: 10.1007/s00253-014-5515-z. [DOI] [PubMed] [Google Scholar]
- 41.Kurosawa K, Hosaka T, Tamehiro N, Inaoka T, Ochi K. 2006. Improvement of α-amylase production by modulation of ribosomal component protein S12 in Bacillus subtilis 168. Appl Environ Microbiol 72:71–77. doi: 10.1128/AEM.72.1.71-77.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kubo Y, Inaoka T, Hachiya T, Miyake M, Hase S, Nakagawa R, Hasegawa H, Funane K, Sakakibara Y, Kimura K. 2013. Development of a rifampicin-resistant Bacillus subtilis strain for natto-fermentation showing enhanced exoenzyme production. J Biosci Bioeng 115:654–657. doi: 10.1016/j.jbiosc.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 43.Liu Z, Zhao X, Bai F. 2013. Production of xylanase by an alkaline-tolerant marine-derived Streptomyces viridochromogenes strain and improvement by ribosome engineering. Appl Microbiol Biotechnol 97:4361–4368. doi: 10.1007/s00253-012-4290-y. [DOI] [PubMed] [Google Scholar]
- 44.Tojo S, Tanaka Y, Ochi K. 2015. Activation of antibiotic production in Bacillus spp. by cumulative drug resistance mutations. Antimicrob Agents Chemother 59:7799–7804. doi: 10.1128/AAC.01932-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hosaka T, Tamehiro N, Chumpolkulwong N, Hori-Takemoto C, Shirouzu M, Yokoyama S, Ochi K. 2004. The novel mutation K87E in ribosomal protein S12 enhances protein synthesis activity during the late growth phase in Escherichia coli. Mol Genet Genomics 271:317–324. doi: 10.1007/s00438-004-0982-z. [DOI] [PubMed] [Google Scholar]
- 46.Okamoto-Hosoya Y, Hosaka T, Ochi K. 2003. An aberrant protein synthesis activity is linked with antibiotic overproduction in rpsL mutants of Streptomyces coelicolor A3(2). Microbiology 149:3299–3309. doi: 10.1099/mic.0.26490-0. [DOI] [PubMed] [Google Scholar]
- 47.Hosokawa K, Park NH, Inaoka T, Itoh Y, Ochi K. 2002. Streptomycin-resistant (rpsL) or rifampicin-resistant (rpoB) mutation in Pseudomonas putida KH146-2 confers enhanced tolerance to organic chemicals. Environ Microbiol 4:703–712. doi: 10.1046/j.1462-2920.2002.00348.x. [DOI] [PubMed] [Google Scholar]
- 48.Wang G, Inaoka T, Okamoto S, Ochi K. 2009. A novel insertion mutation in Streptomyces coelicolor ribosomal S12 protein results in paromomycin resistance and antibiotic overproduction. Antimicrob Agents Chemother 53:1019–1026. doi: 10.1128/AAC.00388-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okamoto S, Tamaru A, Nakajima C, Nishimura K, Tanaka Y, Tokuyama S, Suzuki Y, Ochi K. 2007. Loss of a conserved 7-methylguanosine modification in 16S rRNA confers low-level streptomycin resistance in bacteria. Mol Microbiol 63:1096–1106. doi: 10.1111/j.1365-2958.2006.05585.x. [DOI] [PubMed] [Google Scholar]
- 50.Nishimura K, Johansen SK, Inaoka T, Hosaka T, Tokuyama S, Tahara Y, Okamoto S, Kawamura F, Douthwaite S, Ochi K. 2007. Identification of the RsmG methyltransferase target as 16S rRNA nucleotide G527 and characterization of Bacillus subtilis rsmG mutants. J Bacteriol 189:6068–6073. doi: 10.1128/JB.00558-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huh JH, Kim DJ, Zhao XQ, Li M, Jo YY, Yoon TM, Shin SK, Yong JH, Ryu YW, Yang YY, Suh JW. 2004. Widespread activation of antibiotic biosynthesis by S-adenosylmethionine in streptomycetes. FEMS Microbiol Lett 238:439–447. doi: 10.1111/j.1574-6968.2004.tb09787.x. [DOI] [PubMed] [Google Scholar]
- 52.Saito N, Kurosawa K, Xu J, Okamoto S, Ochi K. 2003. Effect of S-adenosylmethionine on antibiotic production in Streptomyces griseus and Streptomyces griseoflavus. Actinomycetologica 17:47–49. doi: 10.3209/saj.17_47. [DOI] [Google Scholar]
- 53.Mäder U, Zig L, Kretschmer J, Homuth G, Putzer H. 2008. mRNA processing by RNases J1 and J2 aggects Bacillus subtilis gene expression on a global scale. Mol Microbiol 70:183–196. doi: 10.1111/j.1365-2958.2008.06400.x. [DOI] [PubMed] [Google Scholar]
- 54.Sello JK, Buttner MJ. 2008. The gene encoding RNase III in Streptomyces coelicolor is transcribed during exponential phase and is required for antibiotic production and for proper sporulation. J Bacteriol 190:4079–4083. doi: 10.1128/JB.01889-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee J-H, Gatewood ML, Jones GH. 2013. RNase III is required for actinomycin production in Streptomyces antibioticus. Appl Environ Microbiol 79:6447–6451. doi: 10.1128/AEM.02272-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parveen N, Cornell KA. 2011. Methylthioadenosine/S-adenosylhomocysteine nucleosidase, a critical enzyme for bacterial metabolism. Mol Microbiol 79:7–20. doi: 10.1111/j.1365-2958.2010.07455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Greene RC. 1996. Biosynthesis of methionine, p 542–560. In Neidhardt FC, Curtiss R III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE (ed), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed ASM Press, Washington, DC. [Google Scholar]
- 58.Iwig DF, Booker SJ. 2004. Insight into the polar reactivity of the onium chalcogen analogues of S-adenosyl-l-methionine. Biochemistry 43:13496–13509. doi: 10.1021/bi048693+. [DOI] [PubMed] [Google Scholar]
- 59.Wang S, Arends SJ, Weiss DS, Newman EB. 2005. A deficiency in S-adenosylmethionine synthetase interrupts assembly of the septal ring in Escherichia coli K-12. Mol Microbiol 58:791–799. doi: 10.1111/j.1365-2958.2005.04864.x. [DOI] [PubMed] [Google Scholar]
- 60.Grove TL, Benner JS, Radle MI, Ahlum JH, Landgraf BJ, Krebs C, Booker SJ. 2011. A radically different mechanism for S-adenosylmethionine-dependent methyltransferases. Science 332:604–607. doi: 10.1126/science.1200877. [DOI] [PubMed] [Google Scholar]
- 61.Chiang PK, Gordon RK, Tal J, Zeng GC, Doctor BP, Pardhasaradhi K, McCann PP. 1996. S-Adenosylmethionine and methylation. FASEB J 10:471–480. doi: 10.1096/fasebj.10.4.8647346. [DOI] [PubMed] [Google Scholar]
- 62.Reisenauer A, Kahng LS, McCollum S, Shapiro L. 1999. Bacterial DNA methylation: a cell cycle regulator? J Bacteriol 181:5135–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van der Woude M, Braaten B, Low D. 1996. Epigenetic phase variation of the pap operon in Escherichia coli. Trends Microbiol 4:5–9. doi: 10.1016/0966-842X(96)81498-3. [DOI] [PubMed] [Google Scholar]
- 64.McDaniel BA, Grundy FJ, Artsimovitch I, Henkin TM. 2003. Transcription termination control of the S box system: direct measurement of S-adenosylmethionine by the leader RNA. Proc Natl Acad Sci U S A 100:3083–3088. doi: 10.1073/pnas.0630422100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alifano P, Palumbo C, Pasanisi D, Talà A. 2015. Rifampicin-resistance, rpoB polymorphism and RNA polymerase genetic engineering. J Biotechnol 202:60–77. doi: 10.1016/j.jbiotec.2014.11.024. [DOI] [PubMed] [Google Scholar]
- 66.Feederle R, Pajatsch M, Kremmer E, Böck A. 1996. Metabolism of cyclodextrins by Klebsiella oxytoca M5a1: purification and characterisation of a cytoplasmically located cyclodextrinase. Arch Microbiol 165:206–212. doi: 10.1007/BF01692863. [DOI] [PubMed] [Google Scholar]
- 67.Fiedler G, Pajatsch M, Böck A. 1996. Genetics of a novel starch utilisation pathway in Klebsiella oxytoca. J Mol Biol 256:279–291. doi: 10.1006/jmbi.1996.0085. [DOI] [PubMed] [Google Scholar]
- 68.Kim YK, Kitaoka M, Hayashi K, Kim CH, Côté GL. 2004. Purification and characterization of an intracellular cycloalternan-degrading enzyme from Bacillus sp. NRRL B-21195. Carbohydr Res 339:1179–1184. doi: 10.1016/j.carres.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 69.Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang CH, Kieser T, Larke L, Murphy L, Oliver K, O'Neil S, Rabbinowitsch E, Rajandream MA, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA. 2002. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 70.Goldman BS, Nierman WC, Kaiser D, Slater SC, Durkin AS, Eisen JA, Ronning CM, Barbazuk WB, Blanchard M, Field C, Halling C, Hinkle G, Iartchuk O, Kim HS, Machenzie C, Madupu R, Miller N, Shvartsbeyn A, Sullivan SA, Vaudin M, Wiegand R, Kaplan HB. 2006. Evolution of sensory complexity recorded in a myxobacterial genome. Proc Natl Acad Sci U S A 103:15200–15205. doi: 10.1073/pnas.0607335103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hopwood DA. 2008. The tip of the iceberg. American Scociety for Microbiology. http://schaechter.asmblog.org/schaechter/2008/06/the-tip-of-the.html.
- 72.Hopwood DA. 2013. Imaging mass spectrometry reveals highly specific interactions between actinomycetes to activate specialized metabolic gene clusters. mBio 4:e00612-13. doi: 10.1128/mBio.00612-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ichinose H, Suzuki R, Miyazaki T, Kimura K, Momma M, Suzuki N, Fujimoto Z, Kimura A, Funane K. 2017. Paenibacillus sp. 598K 6-α-glucosyltransferase is essential for cycloisomaltooligosaccharide synthesis from α-(1→4)-glucan. Appl Microbiol Biotechnol 101:4115–4128. doi: 10.1007/s00253-017-8174-z. [DOI] [PubMed] [Google Scholar]
- 74.Hosoya Y, Okamoto S, Muramatsu H, Ochi K. 1998. Acquisition of certain streptomycin-resistant (str) mutations enhances antibiotic production in bacteria. Antimicrob Agents Chemother 42:2041–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ochi K. 1987. Metabolic initiation of differentiation and secondary metabolism by Streptomyces griseus: significance of the stringent response (ppGpp) and GTP content in relation to A factor. J Bacteriol 169:3608–3616. doi: 10.1128/jb.169.8.3608-3616.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Inaoka T, Ochi K. 2011. Scandium stimulates the production of amylase and bacilysin in Bacillus subtilis. Appl Environ Microbiol 77:8181–8183. doi: 10.1128/AEM.06205-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Temuujin U, Chi WJ, Lee SY, Chang YK, Hong SK. 2011. Overexpression and biochemical characterization of DagA from Streptomyces coelicolor A3(2): an endo-type β-agarase producing neoagarotetraose and neoagarohexaose. Appl Microbiol Biotechnol 92:749–759. doi: 10.1007/s00253-011-3347-7. [DOI] [PubMed] [Google Scholar]
- 78.Natori Y, Nanamiya H, Akanuma G, Kosono S, Kudo T, Ochi K, Kawamura F. 2007. A fail-safe system for the ribosome under zinc-limiting conditions in Bacillus subtilis. Mol Microbiol 63:294–307. doi: 10.1111/j.1365-2958.2006.05513.x. [DOI] [PubMed] [Google Scholar]
- 79.Payne SH, Ames BN. 1982. A procedure for rapid extraction and high-pressure liquid chromatographic separation of the nucleotides and other small molecules from bacterial cells. Anal Biochem 123:151–161. doi: 10.1016/0003-2697(82)90636-4. [DOI] [PubMed] [Google Scholar]
- 80.Soga T, Heiger DN. 2000. Amino acid analysis by capillary electrophoresis electrospray ionization mass spectrometry. Anal Chem 72:1236–1241. doi: 10.1021/ac990976y. [DOI] [PubMed] [Google Scholar]
- 81.Soga T, Ohashi Y, Ueno Y, Naraoka H, Tomita M, Nishioka T. 2003. Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. J Proteome Res 2:488–494. doi: 10.1021/pr034020m. [DOI] [PubMed] [Google Scholar]
- 82.Sugimoto M, Wong DT, Hirayama A, Soga T, Tomita M. 2010. Capillary electrophoresis mass spectrometry-based saliva metabolomics identified oral, breast and pancreatic cancer-specific profiles. Metabolomics 6:78–95. doi: 10.1007/s11306-009-0178-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zerbino DR, Birney E. 2008. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. 2012. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 40:W445–W451. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.