

Abstract

Antibody–drug conjugates (ADCs) are a novel modality that allows targeted delivery of potent therapeutic agents to the desired site. Herein we report our discovery of NAMPT inhibitors as a novel nonantimitotic payload for ADCs. The resulting anti-c-Kit conjugates (ADC-3 and ADC-4) demonstrated in vivo efficacy in the c-Kit positive gastrointestinal stromal tumor GIST-T1 xenograft model in a target-dependent manner.
Keywords: Antibody−drug conjugate (ADC), nicotinamide phosphoribosyltransferase (NAMPT), novel payload
Antibody–drug conjugates (ADCs) aim to deliver potent therapeutic agents to desired pathological sites while sparing healthy tissues from deleterious side effects.1 Of all conjugates currently undergoing clinical evaluation, those within the oncology space largely predominate. This includes four clinically approved ADCs Mylotarg,2 Adcetris,3 Kadcyla,4 and Besponsa.5 Despite this obvious bias, there are also efforts to extend the scope of this powerful technology toward targeting immune cells6 or treating bacterial infections.7 ADCs generally comprise three key components: an antibody, a linker, and a drug (payload). An antibody component, which serves as a targeting ligand, would typically recognize a receptor that is highly and selectively expressed on the cells of interest. Cleavable or noncleavable linkers represent the second key element of ADCs. Cleavable linkers are designed to release a free intact parent drug,8 which could exert a subsequent bystander killing, whereas noncleavable linkers result in formation of a charged adduct derived from the antibody catabolism in lysosomal compartments.9 To date, the choice of payload utilized in the ADC approach has been limited. Of the four approved ADCs, two (Adcetris and Kadcyla) employ tubulin-binding payloads (auristatines and maytansines) and the other two use the DNA double strand breaking compound calicheamicin. A literature survey reveals that only a few alternative mechanisms-of-action (MoAs) have been studied in the ADC context clinically. These include DNA minor groove binders pyrrolobenzodiazepines,10 a TOPO1 inhibitor camptothecin,11 and DNA alkylators such as indolinobenzodiazepines12 and duocarmycins.13
Screening of various potential MoAs in a select panel of cell lines, including c-Kit and HER2 expressing lines (NCI-H526, MDA-MB453, and NCI-N87) identified the NAMPT inhibitor 1 as being potent across multiple cell lines in the nanomolar range (Figure 1A, Table 1). Identification of cell lines both sensitive to the payload MoA and expressing the target receptor was a critical criterion in the selection of a new MoA for ADC delivery.
Figure 1.

(A) Novel NAMPT inhibitors as ADC payloads and (B) NAMPT inhibitors in clinical trials.
Table 1. Cellular Profiling of NAMPT Payloads.

The NAMPT enzyme converts nicotinamide to nicotinamide mononucleotide and is the rate-limiting enzyme that controls intracellular NAD+ concentration.14 The efficacy of NAMPT inhibitors has been demonstrated in various preclinical settings and several NAMPT inhibitors, e.g., APO866 (FK866),15GMX1778, and GMX1777,16 advanced into the clinic (Figure 1B).
Because clinical utility has been limited by on-target and dose-limiting toxicities such as thrombocytopenia and GI effects, we envisioned that vectorized, targeted delivery of NAMPT inhibitors into tumor tissues via an ADC route might significantly improve their therapeutic index.17
Compounds 1 and 2 originated from an internal research effort to discover novel NAMPT inhibitors.18 This gave us a solid platform with which we could begin our evaluation of NAMPT inhibitors as a novel ADC payload. We reasoned that 1 would be an excellent starting point for our optimization efforts due to the availability of X-ray cocrystal information with the NAMPT enzyme (Figure 2, PDB code 1gqh).18 This would help guide our design toward maximizing the payload potency and determine suitable exit vectors for our linkers.
Figure 2.
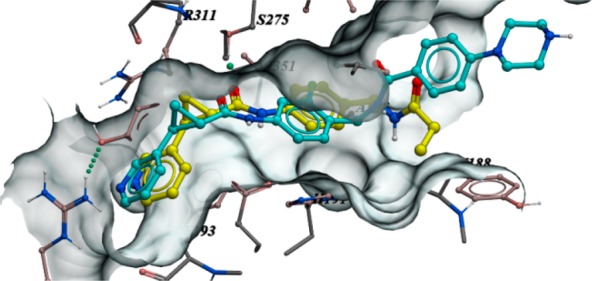
Overlay of the X-ray structure of 1 cocrystallized with the NAMPT enzyme (yellow, PDB code 1gqh) and 4 docked at the binding site (cyan).
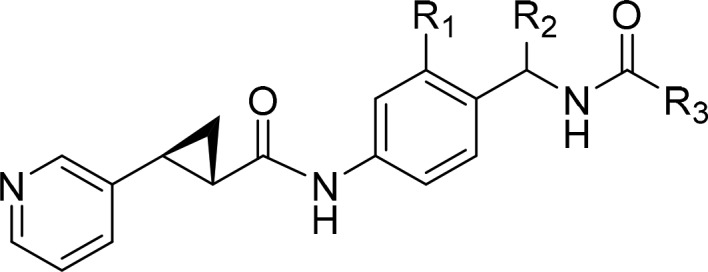
Before embarking on our exploration, we had to take into account a key aspect of the NAMPT inhibitor. The pyridine nitrogen is known to undergo ribophosphorylation by the NAMPT enzyme and is thus an essential part of the pharmacophore. Furthermore, we reported that the whole 3-pyridine (S,S) cyclopropyl carboxamide motif is crucial to achieve the highest level of cellular activity; therefore, we did not pursue further optimization of this portion of the molecule. Instead, we performed a focused SAR exploration in the R3 region, which points toward a solvent exposed area. As shown previously, various substituents are tolerated at this position. In particular, the ethyl substituent can be replaced by a more rigid unsubstituted phenyl ring (2, Table 1). This substitution resulted in a comparable cellular potency when tested on the NAMPT sensitive A2780 and CORL23 cell lines. As our ultimate goal was to generate conjugates, we also included the cell lines NCI-H526, MDA-MB453, and NCI-N87 in our panel. NCI-H526 expresses high levels of the receptor tyrosine kinase c-Kit, whereas MDA-MB453 and NCI-N87 cell lines overexpress the receptor tyrosine kinase HER2, both suitable antigens for ADC targeting.
Molecular docking suggested that the phenyl ring can be further derivatized at the para-position providing an optimal vector for linker attachment. Next, we needed to introduce an appropriate functional group that could be utilized for linker attachment. Although many functional groups have been employed in the context of ADCs, secondary or primary amines offer the highest flexibility and can be modified with both cleavable and noncleavable linkers. Therefore, our goal was to append a primary or secondary amine to the phenyl ring while retaining or improving the payload potency. Among various groups that have been tried, a piperazine substituent installed at the para-position of the phenyl ring turned out to be well tolerated (3, Table 1). Furthermore, molecular docking suggested that the free secondary amine on the piperazine group points into a solvent-exposed area and can be utilized as a handle for linker attachment to allow subsequent conjugation to an antibody. Phenylpiperazine became our preferred substituent at position R3. Finally, keeping R3 constant, we revisited the SAR in the central portion of the payload. A quick evaluation of R1 and R2 substituents resulted in a slightly improved analogue 4 (the payload synthesis and characterization is described in the Supporting Information).
Payload 4 displayed single digit nanomolar potency on c-Kit and HER2 expressing cell lines and was, therefore, selected for a subsequent modification with a focused set of cleavable (L1) and noncleavable (L2–L5) linkers (Table 2). All linker-payloads were initially conjugated to an anti-c-Kit monoclonal antibody selected for its ability to efficiently internalize in c-Kit expressing cells. Maleimide chemistry was used to conjugate the linker-payloads to interchain disulfides of the antibodies (the linker-payload synthesis and their characterization and bioconjugation methods are described in the Supporting Information). The cathepsin-cleavable ValCit linker L1, which has been widely used in the literature and utilized in the approved ADC Adcetris,3 was our first choice, as it should furnish the unmodified payload 4 upon intracellular release. Despite the promising in vitro potency of the conjugate ADC-1, the utility of this linker is compromised by the resulting high aggregation of 29% (Table 2). Therefore, we turned our attention to the noncleavable linkers (L2–L5). It is worth mentioning that, while linkers L2–L4 turn the secondary amine of the payload into an amide, the L5 linker was designed to retain the basic tertiary amine, which we thought would be the closest version of payload 4. Interestingly, linker L5 along with linker L2 resulted in conjugates with high aggregation. In contrast, linkers L3 and L4 reduced aggregation to the acceptable level of below 10% and afforded drug-to-antibody ratio (DAR) in the range of 2–4. All conjugates were tested for in vitro cytotoxicity on c-Kit expressing cell lines such as GIST-T1 and NCI-H526. The anti-cKIT conjugates ADC-3 and ADC-4 turned out to be not only low aggregating but also possessed the best potency (IC50 of 9 and 40 pM, respectively) on the NCI-H526 line, representing a lower antigen density compared to GIST-T1. As a consequence, two linker-payloads (LP3 and LP4) emerged as our leads. This led to conjugation of LP4 to an anti-HER2 trastuzumab (ADC-5) and a nonbinding isotype control antibody (ADC-6). The target-dependent cellular activity was confirmed (Table 2), with the nonbinding IgG isotype control ADC-6 being significantly less active.
Table 2. In Vitro Cytotoxicity of NAMPT ADCs.

| ADC | linker-payload | linker (L) | target antigen | Aggr. (%) | DAR | GIST-T1 (c-Kit+, nM) | NCI-H526 (c-Kit+, nM) | MDA-MB453 (HER2+, nM) | NCI-N87 (HER2+, nM) |
|---|---|---|---|---|---|---|---|---|---|
| ADC-1 | LP1 | L1 | c-Kit | 29 | 2.8 | 0.014 | >0.720 | not tested | not tested |
| ADC-2 | LP2 | L2 | c-Kit | 23 | 4.0 | <0.003 | 0.047 | >33 | >33 |
| ADC-3 | LP3 | L3 | c-Kit | 6.1 | 3.9 | <0.003 | 0.009 | 2.13 | 4.05 |
| ADC-4 | LP4 | L4 | c-Kit | 5.3 | 2.3 | <0.007 | 0.040 | 6 | 12 |
| ADC-5 | LP4 | L4 | HER2 | 6.2 | 2.9 | 18.7 | 71.5 | 0.006 | 0.017 |
| ADC-6 | LP4 | L4 | IgG | 10 | 4.4 | 0.1274 | 12.5 | 1.42 | 2.76 |
| ADC-7 | LP5 | L5 | c-Kit | 23 | 3.9 | <0.003 | 0.352 | >33 | >33 |
Next, we confirmed the mechanism-dependent pharmacodynamic (PD) modulation by looking at the NAD+/NADH levels upon exposure of the GIST-T1 cell line to our anti-c-Kit conjugate ADC-4 in comparison to the nonbinding control ADC-6 (Figure 3). To this end we employed a cellular assay system that uses a luminescent substrate to quantify cellular NAD+/NADH levels and compared the activity of ADC-4 to that of the nonbinding ADC-6, demonstrating that only the c-Kit-targeting ADC-4 resulted in a concentration-dependent inhibition of signal.
Figure 3.
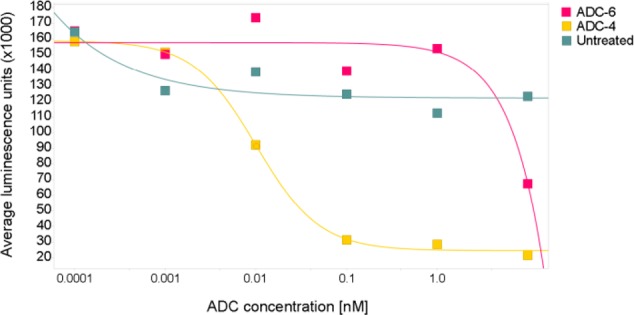
Mechanism-dependent PD modulation: NAD+/NADH levels in GIST-T1 cells at 24 h post-dosing.
In addition, the nonantimitotic MoA of our lead conjugate was confirmed by analyzing the cell-cycle profiles of GIST-T1 cells exposed to either s-methyl-DM1 (s-me-DM1), a cell-permeable maytansine analog,19 or ADC-4.
Treatment with s-me-DM1 resulted in accumulation of cells in G2/M phase, consistent with inducing cell cycle arrest at the G2/M transition. This was due to microtubule depolymerization. In contrast, treatment of GIST-T1 cells with ADC-4 inhibited cell proliferation (Figure 4B) without overtly affecting the cell cycle (Figure 4A).
Figure 4.
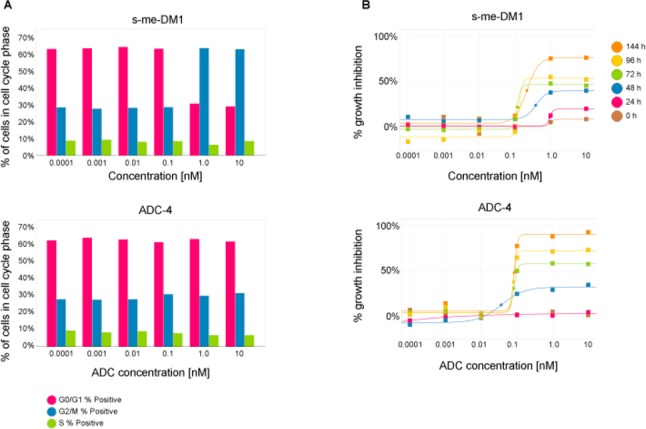
(A) Cell cycle and (B) kinetic viability analysis in GIST-T1 cells treated with either s-me-DM1 or ADC-4.
Encouraged by the in vitro activity, the anti-c-Kit ADC-3 and ADC-4 were assessed for in vivo efficacy in the c-Kit positive gastrointestinal stromal tumor GIST-T1 xenograft model in mice. A single 20 mg/kg administration of either anti-cKit ADCs induced tumor stasis in the GIST-T1 model, with a clear differential from the nonbinding isotype control ADC-6 treated group (Figure 5A).
Figure 5.
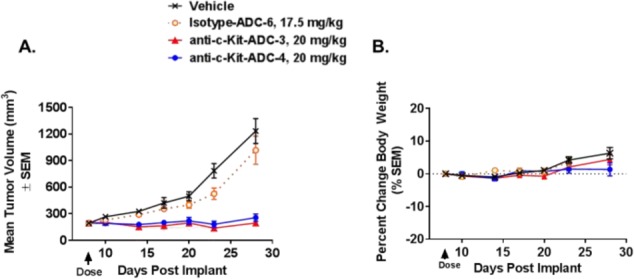
(A) Antitumor efficacy after a single intravenous administration of vehicle, isotype control ADC-6, or anti-c-Kit ADC-3 or ADC-4 at indicated dose levels in the GIST-T1 xenograft model in SCID bg mice and (B) effect on the body weight.
The conjugates were well tolerated at this dose as indicated by the stable body weight (Figure 5B).
In summary, we designed and synthesized a series of antibody–drug conjugates employing payloads with a novel NAMPT inhibitor mechanism of action. The two noncleavable linker formats afforded ADCs that were generally low-aggregating with highly promising in vitro profiles. Furthermore, the conjugates were well tolerated and demonstrated target-dependent efficacy in vivo. The conjugates are currently undergoing further evaluation across additional in vivo models.
Acknowledgments
We are grateful to Christophe Bury, Jeff Hewett, Laryssa Tierney, and Thomas Wolf for their excellent technical assistance.
Glossary
ABBREVIATIONS
- ADC
antibody–drug conjugate
- Aggr
aggregation
- DAR
drug-to-antibody ratio
- MoA
mechanism-of-action
- NAD+
nicotinamide adenine dinucleotide (oxidized)
- NADH
nicotinamide adenine dinucleotide (reduced)
- NAMPT
nicotinamide phosphoribosyltransferase
- NMN
nicotinamide mononucleotide
- RNA
ribonucleic acid
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00254.
Complete experimental procedures and analytical characterization of novel compounds, bioconjugation methods, in vitro cytotoxicity, and in vivo efficacy data (PDF)
Author Present Address
∥ (M.D.) Ideaya Biosciences, 7000 Shoreline Court, Suite 350, South San Francisco, California 94080, United States.
Author Present Address
⊥ (G.P.) Merck Exploratory Science Center, 320 Bent Street, Cambridge, Massachusetts, United States.
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Chari R. V. J. Expanding the Reach of Antibody-Drug Conjugates. ACS Med. Chem. Lett. 2016, 7, 974–976. 10.1021/acsmedchemlett.6b00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jen E. Y.; Ko C.-W.; Lee J. E.; Del Valle P. L.; Aydanian A.; Jewell C.; Norsworthy K. J.; Przepiorka D.; Nie L.; Liu J.; Sheth C. M.; Shapiro M.; Farrell A. T.; Pazdur R. FDA Approval: Gemtuzumab Ozogamicin for the Treatment of Adults with Newly-diagnosed CD33-positive Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 10.1158/1078-0432.CCR-17-3179. [DOI] [PubMed] [Google Scholar]
- Senter P. D.; Sievers E. L. The Discovery and Development of Brentuximab Vedotin for Use in Relapsed Hodgkin Lymphoma and Systemic Anaplastic Large Cell Lymphoma. Nat. Biotechnol. 2012, 30, 631–637. 10.1038/nbt.2289. [DOI] [PubMed] [Google Scholar]
- Lambert J. M.; Chari R. V. J. Ado-trastuzumab Emtansine (T-DM1): An Antibody-Drug Conjugate (ADC) for HER2-Positive Breast Cancer. J. Med. Chem. 2014, 57, 6949–6964. 10.1021/jm500766w. [DOI] [PubMed] [Google Scholar]
- Yilmaz M.; Richard S.; Jabbour E. The Clinical Potential of Inotuzumab Ozogamicin in Relapsed and Refractory Acute Lymphocytic Leukemia. Ther. Adv. Hematol. 2015, 6, 253–261. 10.1177/2040620715596715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern J. C.; Cancilla M.; Dooney D.; Kwasnjuk K.; Zhang R.; Beaumont M.; Figueroa I.; Hsieh S.; Liang L.; Tomazela D.; Zhang J.; Brandish P. E.; Palmieri A.; Stivers P.; Cheng M.; Feng G.; Geda P.; Shah S.; Beck A.; Bresson D.; Firdos J.; Gately D.; Knudsen N.; Manibusan A.; Schultz P. G.; Sun Y.; Garbaccio R. M. Discovery of Pyrophosphate Diesters as Tunable, Soluble, and Bioorthogonal Linkers for Site-Specific Antibody-Drug Conjugates. J. Am. Chem. Soc. 2016, 138, 1430–1445. 10.1021/jacs.5b12547. [DOI] [PubMed] [Google Scholar]
- Lehar S. M.; Pillow T.; Xu M.; Staben L.; Kajihara K. K.; Vandlen R.; DePalatis L.; Raab H.; Hazenbos W. L.; Morisaki J. H.; Kim J.; Park S.; Darwish M.; Lee B.-C.; Hernandez H.; Loyet K. M.; Lupardus P.; Fong R.; Yan D.; Chalouni C.; Luis E.; Khalfin Y.; Plise E.; Cheong J.; Lyssikatos J. P.; Strandh M.; Koefoed K.; Andersen P. S.; Flygare J. A.; Tan M. W.; Brown E. J.; Mariathasan S. Novel Antibody-Antibiotic Conjugate Eliminates Intracellular S. aureus. Nature 2015, 527, 323–328. 10.1038/nature16057. [DOI] [PubMed] [Google Scholar]
- Okeley N. M.; Miyamoto J. B.; Zhang X.; Sanderson R. J.; Benjamin D. R.; Sievers E. L.; Senter P. D.; Alley S. C. Intracellular Activation of SGN-35, a Potent Anti-CD30 Antibody-Drug Conjugate. Clin. Cancer Res. 2010, 16, 888–897. 10.1158/1078-0432.CCR-09-2069. [DOI] [PubMed] [Google Scholar]
- Erickson H. K.; Park P. U.; Widdison W. C.; Kovtun Y. V.; Garrett L. M.; Hoffman K.; Lutz R. J.; Goldmacher V. S.; Blättler W. A. Antibody-Maytansinoid Conjugates Are Activated in Targeted Cancer Cells by Lysosomal Degradation and Linker-Dependent Intracellular Processing. Cancer Res. 2006, 66, 4426–4433. 10.1158/0008-5472.CAN-05-4489. [DOI] [PubMed] [Google Scholar]
- Tiberghien A. C.; Levy J.-N.; Masterson L. A.; Patel N. V.; Adams L. R.; Corbett S.; Williams D. G.; Hartley J. A.; Howard P. W. Design and Synthesis of Tesirine, a Clinical Antibody-Drug Conjugate Pyrrolobenzodiazepine Dimer Payload. ACS Med. Chem. Lett. 2016, 7, 983–987. 10.1021/acsmedchemlett.6b00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogitani Y.; Aida T.; Hagihara K.; Yamaguchi J.; Ishii C.; Harada N.; Soma M.; Okamoto H.; Oitate M.; Arakawa S.; Hirai T.; Atsumi R.; Nakada T.; Hayakawa I.; Abe Y.; Agatsuma T. DS-8201a, a Novel HER2-targeting ADC with a Novel DNA Topoisomerase I inhibitor, Demonstrates a Promising Anti-tumor Efficacy with Differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097. 10.1158/1078-0432.CCR-15-2822. [DOI] [PubMed] [Google Scholar]
- Miller M. L.; Fishkin N. E.; Li W.; Whiteman K. R.; Kovtun Y.; Reid E. E.; Archer K. E.; Maloney E. K.; Audette C. A.; Mayo M. F.; Wilhelm A.; Modafferi H. A.; Singh R.; Pinkas J.; Goldmacher V.; Lambert J. M.; Chari R. V. J. A New Class of Antibody–Drug Conjugates with Potent DNA Alkylating Activity. Mol. Cancer Ther. 2016, 15, 1870–1878. 10.1158/1535-7163.MCT-16-0184. [DOI] [PubMed] [Google Scholar]
- Van der Lee M. M. C.; Groothuis P. G.; Ubink R.; Van der Vleuten M. A. J.; Van Achterberg T. A.; Loosveld E. M.; Damming D.; Jacobs D. C. H.; Rouwette M.; Egging D. F.; Van den Dobbelsteen D.; Beusker P. H.; Goedings P.; Verheijden G. F. M.; Lemmens J. M.; Timmers M.; Dokter W. H. A. The Preclinical Profile of the Duocarmycin-Based HER2-Targeting ADC SYD985 Predicts for Clinical Benefit in Low HER2-Expressing Breast Cancers. Mol. Cancer Ther. 2015, 14, 692–703. 10.1158/1535-7163.MCT-14-0881-T. [DOI] [PubMed] [Google Scholar]
- Roulston A.; Shore G. C. New Strategies to Maximize Therapeutic Opportunities for NAMPT Inhibitors in Oncology. Molecular & Cellular Oncology. 2016, 3, e1052180. 10.1080/23723556.2015.1052180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holen K.; Saltz L. B.; Hollywood E.; Burk K.; Hanauske A.-R. The Pharmacokinetics, Toxicities, and Biologic Effects of FK866, a Nicotinamide Adenine Dinucleotide Biosynthesis inhibitor. Invest. New Drugs 2008, 26, 45–51. 10.1007/s10637-007-9083-2. [DOI] [PubMed] [Google Scholar]
- Von Heideman A.; Berglund A.; Larsson R.; Nygren P. Safety and Efficacy of NAD Depleting Cancer Drugs: Results of a Phase I Clinical Trial of CHS 828 and Overview of Published Data. Cancer Chemother. Pharmacol. 2010, 65, 1165–1172. 10.1007/s00280-009-1125-3. [DOI] [PubMed] [Google Scholar]
- During the review of this manuscript it was pointed out that the use of NAMPT inhibitors as payloads for the ADC approach was disclosed in a recent patent application: Neumann C. S.; Olivas K.. Targeted Delivery of Nicotinamide Adenine Dinucleotide Salvage Pathway Inhibitors. WO 2018/075600 A1.
- Palacios D. S.; Meredith E.; Kawanami T.; Adams C.; Chen X.; Darsigny V.; Geno E.; Palermo M.; Baird D.; Boynton G.; Busby S. A.; George E. L.; Guy C.; Hewett J.; Tierney L.; Thigale S. Structure Based Design of Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors from a Phenotypic Screen. Bioorg. Med. Chem. Lett. 2018, 28, 365–370. 10.1016/j.bmcl.2017.12.037. [DOI] [PubMed] [Google Scholar]
- Sun X.; Widdison W.; Mayo M.; Wilhelm S.; Leece B.; Chari R.; Singh R.; Erickson H. Design of Antibody-maytansinoid Conjugates Allows for Efficient Detoxification via Liver Metabolism. Bioconjugate Chem. 2011, 22, 728–735. 10.1021/bc100498q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.