

Abstract

We report the discovery of a novel series of 3-cinnoline carboxamides as highly potent and selective ataxia telangiectasia mutated (ATM) kinase inhibitors. Optimization of this series focusing on potency and physicochemical properties (especially permeability) led to the identification of compound 21, a highly potent ATM inhibitor (ATM cell IC50 0.0028 μM) with excellent kinase selectivity and favorable physicochemical and pharmacokinetics properties. In vivo, 21 in combination with irinotecan showed tumor regression in the SW620 colorectal tumor xenograft model, superior inhibition to irinotecan alone. Compound 21 was selected for preclinical evaluation alongside AZD0156.
Keywords: ATM, ataxia telangiectasia mutated kinase, PIKK, DDR
The ataxia telangiectasia mutated (ATM) kinase1 is involved in the DNA damage responses (DDR),2 specifically in the repair of DNA double strand breaks (DSB). Upon recruitment to sites with DNA DSB, which is mediated by the MRN (MRE11-RAD50-NBS1) complex, ATM is autophosphorylated on Ser1981 and signals activation of downstream DNA repair and cell cycle checkpoint.3 As DSBs can result from ionizing radiation (IR) or chemically induced DNA damage (e.g., DNA topoisomerase I inhibitors such as irinotecan4), the combination of ATM inhibition with clinically induced DNA damage in tumor cells could bring important benefits to cancer patients.5,6 In addition to its DNA damage signaling function, ATM has an additional role in signaling and protecting against reactive oxygen species (ROS). ATM has been reported as a redox sensor becoming directly activated by oxidation.7,8 This ROS-protecting function may be important for promoting neovascularization of tumors.9
ATM is a serine/threonine protein kinase belonging to the PIKK (phosphatidylinositol 3′-kinase [PI3K]-related kinase) family together with DNA-dependent protein kinase (DNAPK), ataxia telangiectasia mutated and RAD3-related (ATR) kinase, and mammalian target of rapamycin (mTOR). One of the first selective ATM inhibitors reported was Ku-55933.10 Although it was shown to be an ATM inhibitor with suitable PIKK selectivity, its poor physicochemical properties have precluded its use as an in vivo probe. Subsequent optimization led to the identification of Ku-60019, a more potent and selective ATM inhibitor.11 However, its physicochemical properties again still limit its use in vivo, especially for oral administration. We recently reported significant advances in the development of potent and selective ATM inhibitors with suitable physicochemical and pharmacokinetic properties for oral administration. Optimization of a series of quinoline carboxamides led to compounds 1–3 as highly selective and orally active ATM inhibitors (Figure 1).12 We subsequently disclosed the optimization of a series of imidazo[5,4-c]quinolin-2-ones leading to 4, a highly potent and selective ATM inhibitor with acceptable permeability and good bioavailability in multiple preclinical species. Compound 4 (also known as AZD0156) is currently being evaluated in Phase I clinical trials.13 We also reported the identification of another series based on an imidazopyrazine scaffold showing evidence of brain penetration.14
Figure 1.

Structure of selected ATM inhibitors (compounds 1–4) previously reported.
During the optimization of the quinoline carboxamide series, we noted that introduction of a basic side chain at C-6 at a suitable distance dramatically improved potency (e.g., compound 5, see Figure 2), but led to reduced permeability, precluding good oral bioavailability.13 This observation was subsequently integrated in the optimization of the imidazo[5,4-c]quinolin-2-one series. Starting from 7, which has much higher permeability than its equivalent quinoline carboxamide, exploration of various basic side chains at C-6 led to the identification of 4. As part of the modifications of the quinoline hinge binder, we identified that cinnoline carboxamides may be a suitable replacement for quinoline carboxamides. In this Letter, we report the optimization of this novel series as selective and orally active ATM inhibitors.
Figure 2.

Structure of compounds 5–7.
Although there is no reported crystal structure of ATM to aid design of inhibitors, the similarity of the binding mode of quinoline carboxamides (e.g., 6) and imidazo[5,4-c]quinolin-2-ones (e.g., 7) in PI3Kγ supports the parallel SAR observed at C-6 against ATM between the two series (see Figure 3). Both compounds exhibit key hydrogen-bonding interactions to Val882 and Asp964 and form a π-interaction with Tyr867. Although no crystal structure of cinnoline carboxamides bound to PI3Kγ was available, we hypothesized that they would have a similar binding mode as quinoline carboxamides.
Figure 3.
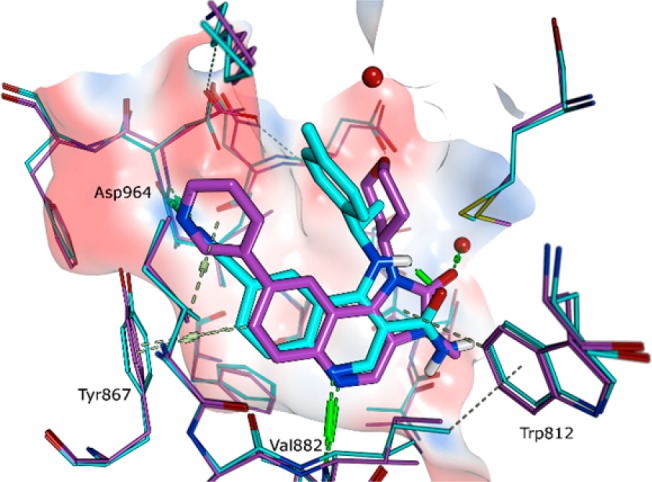
Overlay of crystal structures of compounds 6 (blue) and 7 (purple) in PI3Kγ; PDB codes 5G55 and 6GQ7, respectively. Hydrogen bonds are indicated in green, and π-interactions are indicated in gray.
Compounds 8–22 were made from a common intermediate (see synthetic section and details procedures in Supporting Information). The first cinnoline carboxamides 8, 9, and 10, having the same side chains at C-6 and C-4 as their quinoline carboxamide counterparts 1, 2, and 3, respectively, maintained a good level of ATM cellular potency, just slightly reduced compared to the corresponding quinoline carboxamides. However, 8 and 10 showed significantly higher permeability and lower efflux ratio in both Caco-2 and MDCK cell lines than their quinoline carboxamide counterparts 1 and 3. The reasons for this increased permeability/lower efflux may be a combination of lower basicity for cinnoline carboxamides vs quinoline carboxamides (measured pKa 5.3 vs 7.6 for the cinnoline group in 12 vs the quinoline group in 1) and a reduced number of effective H-bond donors (due to the potential intramolecular H-bond between amidic N–H and N2 of the cinnoline). The other physical and pharmacokinetic properties of 8–10 were considered as suitable for further optimization: moderate in vitro metabolic stability in rat hepatocytes and HLM for 8–10, and low clearance in vivo and moderate to good bioavailability for 8 (pharmacokinetic parameters in rats for 8 after iv and oral administration of 0.5 and 1 mg/kg respectively: CL 21 mL/min/kg; Vdss 0.7 L/kg; F 56%), although solubility was to be monitored (Table 1). Methylation of the carboxamide maintained similar potency (see compound 11 vs 8). This observation is not in line with what was observed in the quinoline carboxamide series, where loss of potency was observed based on one pair of compounds.12 The exact reason for this difference in SAR between the two series is not understood, as our initial assumption was that the SAR would be fully transferable from the quinoline carboxamide series to the cinnoline carboxamide series.
Table 1. Biological, Physicochemical, and Metabolic Data of Compounds 8-22 vs Previously Reported Compounds 1–3.

| Cpd | Core | R3 | R6 | ATM cell IC50a | ATR cell IC50a | LogD7.4b | Sol.c | Caco-2 Papp, A-B (ER)d | MDCK Papp, A-B (ER)d | HLM Clinte | Rat hep. kClintf |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.046 | >30 | 2.5 | 590 | 8.5 (3.2) | 4.8 (7.3) | 26 | <5.3 | |||
| 2 | 0.033 | >19 | 2.7 | 69 | 11 (3.1) | 15 (2.4) | 15 | 5.3 | |||
| 3 | 0.010 | >30 | 3.0 | 42 | 12 (1.2) | 13 (2.7) | 39 | 6.9 | |||
| 8 | A | H | CH2OMe | 0.14 | >30 | 2.7 | >1000 | 27 (0.7) | 19 (1.3) | 33 | 5.6 |
| 9 | B | H | CH2OMe | 0.26 | >30 | 3.0 | 76 | NA | NA | 14 | 4.9 |
| 10 | C | H | CH2OMe | 0.044 | >30 | 2.8 | 8 | 18 (0.9) | 27 (0.7) | 31 | 18 |
| 11 | A | Me | CH2OMe | 0.33 | >30 | 3.0 | 34 | 25 (0.5) | 20 (0.6) | 42 | 5.8 |
| 12 | A | H | OCH2CH2CH2NMe2 | 0.0019 | >30 | 1.9 | 870 | 7.5 (2.5) | 3.5 (9.0) | 28 | 3.4 |
| 13 | B | H | OCH2CH2CH2NMe2 | 0.0039 | >30 | 1.7 | 65 | NA | 2.9 (12) | 10 | 7.0 |
| 14 | C | H | OCH2CH2CH2NMe2 | 0.0009 | >30 | 2.1 | 290 | NA | 2.1 (16) | 27 | 12 |
| 15 | A | H | OCH2CH2CH2-pyrrolidine | 0.0018 | >30 | 1.9 | 620 | NA | 1.4 (24) | 23 | 1.9 |
| 16 | B | H | OCH2CH2CH2-pyrrolidine | 0.0011 | >30 | 1.8 | 490 | NA | 1.5 (23) | 10 | 4.6 |
| 17 | C | H | OCH2CH2CH2-pyrrolidine | 0.0005 | >30 | 2.1 | 510 | NA | 1.5 (20) | 40 | 8.1 |
| 18 | B | H | OCH2CH2NMe2 | 0.035 | >30 | 1.9 | 910 | NA | NA | 25 | 13 |
| 19 | A | Me | OCH2CH2NMe2 | 0.049 | >30 | 2.5 | 990 | NA | NA | 26 | 4.8 |
| 20 | A | H | NHCH2CH2NMe2 | 0.061 | >30 | 2.0 | >1000 | NA | NA | 18 | 4.2 |
| 21 | A | Me | OCH2CH2CH2NMe2 | 0.0028 | >30 | 2.3 | 880 | 5.0 (2.6) | 2.5 (6.6) | 22 | 5.7 |
| 22 | C | Me | OCH2CH2CH2NMe2 | 0.0039 | >30 | 2.3 | 620 | 4.0 (1.6) | 3.7 (3.2) | 33 | 6.7 |
Micromolar; unless stated otherwise, geometric means of at least two IC50 determinations per compound.
Measured using shake-flask methodology with a buffer/octanol volume ratio of 100:1.
Solubility in aqueous phosphate buffer, pH 7.4 after 24 h at 25 °C; μM.
Apparent permeability (Papp, A-B) in Caco-2 or MDCK-MDR1 cell line, A pH 7.4, B pH 7.4; 10–6 cm·s–1. Permeability was measured in both the apical to basolateral (A to B) and basolateral to apical (B to A) direction. Efflux ratio (ER) determined as the ratio of both permeabilities (B to A permeability divided by A to B permeability).
Human liver microsome intrinsic clearance (HLM Clint); μL.min–1.mg–1.
Intrinsic clearance measured from fresh rat hepatocytes, Clint; μL/min/106 cell. NA for not assessed.
We then explored the C-6 side chain. During optimization of the imidazo[5,4-c]quinolin-2-one series, introduction of a dimethylaminopropyloxy side chain dramatically increased potency against ATM, leading to the identification of 4. This observation was rationalized from a homology model of ATM based on the structure of the related protein mTOR. In this model, the dimethylamino group sits in a highly polar subpocket beyond the back pocket, surrounded by acidic residues Asp2725, Asp2720, and Asp2889.13 Indeed, introduction of this same chain again dramatically increased potency (ATM cellular potency 0.9 to 3.9 nM for compounds 12–14). Yet those compounds maintained acceptable permeability and limited efflux (measured in MDCK and/or Caco-2), high metabolic stability, and good solubility. Further modifications of the basic side chain were tolerated (e.g., pyrrolidines 15–17). As previously seen in the imidazo[5,4-c]quinolin-2-one series, the length of the linker was critical for maintaining the nM to sub-nM potency, the dimethylaminopropoxy side chain being significantly more active than the dimethylaminoethoxy side chain (e.g., 18 vs 13; 21 vs 19). It is worth noting that an amino link (e.g., 20) is also tolerated as well as an ether link. Finally with the observation that methylation of carboxamide at C-3 was tolerated (compound 11), we combined the methylcarboxamide at C-3 with the dimethylaminopropyloxy side chain at C-6 (compounds 21 and 22) with the expectation that permeability would be increased vs the carboxamide (compounds 12 and 14) because of a lower number of H-bond donors. Although no significant improvement was seen, compounds 21 and 22 maintained a similar degree of moderate to high permeability/low to moderate efflux.
Compounds 12 and 21 as representative compounds of this series were further evaluated in vitro and in vivo. The kinase selectivity of compounds 12 and 21 was assessed further. Compounds 12 and 21 did not show appreciable activity against other kinases of the PI3K or PIKK families (see Table 2: PI3Kα–δ, mTOR, and DNAPK enzyme assays; PI3Kα and PI3Kβ, ATR, and mTOR cellular assays). The excellent kinase selectivity was further confirmed in several kinase panels. Compound 12 did not show significant inhibition at 1 μM in a panel of 125 kinases (“Millipore panel”): no kinase with >70% inhibition, one kinase between 50 and 70% inhibition: CSF1R (56%). Compound 21 did not show significant inhibition at 1 μM in a panel of 272 kinases (“extended Millipore panel”): no kinase with >70% inhibition, three kinases between 50 and 70% inhibition: Flt-4 (63%), RET (59%), and NTRK2 (51%). The kinase selectivity of 21 was confirmed in the Ambit panel (KINOMEscan) against a panel of 392 kinases at 1 μM, with PI4KB being the only kinase showing less than 50% of control (PI4KB 17% of control).
Table 2. Biological Profile of Compounds 12 and 21.
| assay details | Cpd 12 IC50 (μM)a | Cpd 21 IC50 (μM)a | |
|---|---|---|---|
| enzyme inhibition | ATM | 0.00029 | 0.0007 |
| mTOR | 5.3 | 21 | |
| DNAPK | 1.8 | 2.8 | |
| PI3Kα | 2.2 | 3.8 | |
| PI3Kβ | 8.1 | 10.3 | |
| PI3Kγ | 2.9 | 3.0 | |
| PI3Kδ | 0.80 | 0.73 | |
| cellular inhibition | ATM | 0.0019 | 0.0028 |
| ATR | >30 | >30 | |
| PI3Kα | >30 | >30 | |
| PI3Kβ/mTOR | >15 | >19 |
Geometric means of multiple IC50 determinations.
On the basis of their high ATM potency, their excellent kinase selectivity and their overall suitable physicochemical and in vitro metabolic properties, in vivo pharmacokinetics of 12 and 21 were evaluated in rat and dogs (Table 3). Both compounds showed long half-lives in both rat and dogs, consistent with a high volume of distribution (as a consequence of the strong basicity of the dimethylaminopropoxy side chain), and acceptable oral bioavailability. Physical properties and safety properties of 12 and 21 were further investigated (see Table 3). Both 12 and 21 had excellent aqueous solubility: 870 and 880 μM in aqueous phosphate buffer (pH 7.4) from crystalline material. Mouse oral exposure was assessed for 21. At 10 mg/kg, compound 21 displayed free plasma exposure above the in vitro cellular ATM assay for at least 12 h (see Figure 4). Previous studies conducted in our laboratories had shown that monotherapy treatment with a selective ATM inhibitor did not result in significant efficacy in the SW620 model, but combination with topoisomerase I inhibitor irinotecan (acting as a DNA damaging agent, generating DSBs) resulted in significantly greater tumor growth inhibition compared to irinotecan alone. Indeed, in this same model, 21 was dosed in combination with irinotecan using the following schedule: (irinotecan, 50 mg/kg, i.p. once weekly; then 21, 50 mg/kg p.o. once daily for 3 days every week starting 24 h post-irinotecan dosing). The combination was well tolerated and resulted in excellent growth inhibition of the tumor (106%), while irinotecan alone resulted in only 74% tumor growth inhibition (see Figure 5). No single arm with 21 as monotherapy was present in the experiment as a control, based on our previous experience with other ATM selective inhibitors.
Table 3. Physicochemical, Pharmacokinetics, and Other Properties of Compounds 12 and 21.
| properties | Cpd 12 | Cpd 21 |
|---|---|---|
| hu/rat/dog/mo ppb; fu (%)a | 13; 6; 44; 46 | 4; 2; 30; 21 |
| rat/dog/hu hepatocyte Clint (μL/min/106 cell)b | 3.4; 5.1; 2.8 | 5.7; 8.8; 4.1 |
| rat/dog pharmacokinetic parameters | ||
| clearance: Cl (mL/min/kg)c | 25; 49 | 7.8; 48 |
| Vd (L/kg)c | 5.5; 20 | 2.8; 14 |
| bioavailability: F (%)c | 26; 43 | 41; 35 |
| terminal half-life (h) | 4.1; 6.2 | 5.6; 4.7 |
| aqueous solubility (μM)d | 870 | 880 |
| intrinsic permeability (10–6 cm.s–1)e | 2.8 | 2.0 |
| CYP inhibition IC50 (μM)f | >30 (all) | >30 (all) |
| hERG IC50 (μM)g | 24 | 15 |
Plasma protein binding (ppb) in human, rat, dog, and mouse plasma, fraction unbound fu; determined from DMSO stock solution by equilibrium dialysis in 10% plasma.
Intrinsic clearance measured from fresh rat/dog hepatocytes and cryopreserved human hepatocytes, Clint.
From plasma concentrations in male Han Wistar rats and male Beagle dogs (at least n = 2), compound administered at 1 mg/kg i.v. and 2 mg/kg p.o. as a formulation in 0.5% (w/v) HPMC and 0.1% (w/v) Tween 80 in water for the oral arm and as a solution formulation in 5% DMSO and 95% SBE-β-cyclodextrin (30% w/v in water) for the iv arm. Intravenous parameters (including half-life) calculated from an intravenous bolus; bioavailability, from oral and i.v. AUC.
Solubility in aqueous phosphate buffer, pH 7.4 after 24 h at 25 °C.
Intrinsic permeability measured in Caco-2 cell line, A pH 6.5, B pH 7.4, measured in the apical-to-basolateral (A to B) direction in the presence of an efflux inhibitor.
Inhibition of cytochrome P450, IC50 (1A2, 2C9, 2C19, 2D6, 3A4).
Inhibition of the hERG tail current was measured using a plate-based planar patch clamp system (IonWorks).
Figure 4.
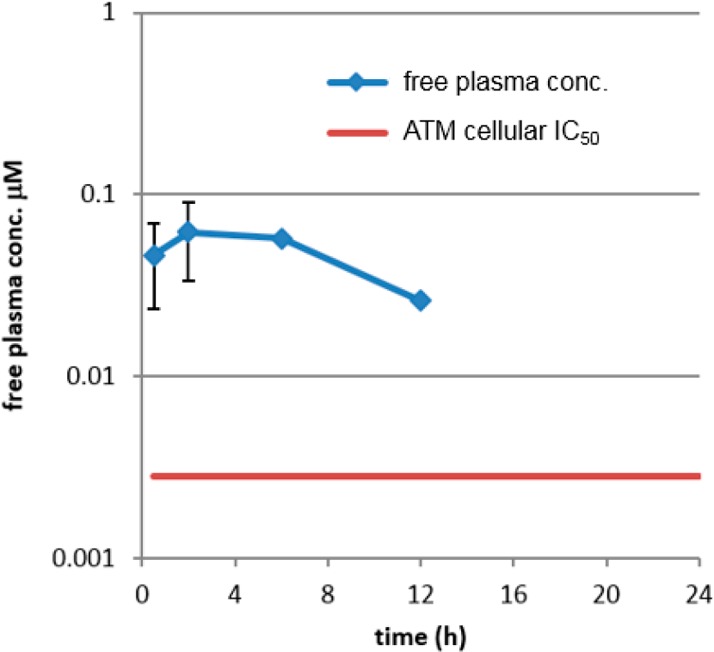
Mouse free plasma exposure of compound 21 after oral administration (10 mg/kg).
Figure 5.
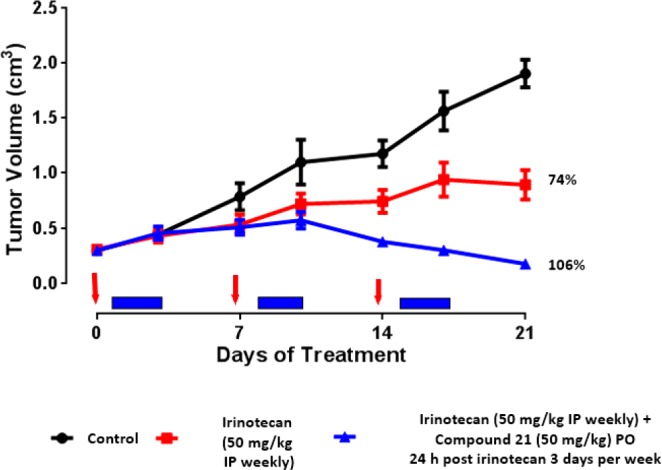
Tumor growth inhibition in the SW620 xenograft model of 21 in combination with irinotecan vs irinotecan alone.
In conclusion, we have discovered a series of cinnoline carboxamides as highly potent and selective ATM inhibitors. Optimization of this series focusing on potency and physical properties (especially permeability) led to the identification of compound 21, a highly potent ATM inhibitor (ATM cell IC50 0.0028 μM) with excellent kinase selectivity and favorable physicochemical and pharmacokinetic properties. In vivo, 21 in combination with irinotecan showed tumor regression in the SW620 colorectal tumor xenograft model, superior efficacy to irinotecan alone. Compound 21 was selected for preclinical evaluation alongside AZD0156.
Acknowledgments
We acknowledge chemists at Pharmaron, Beijing for the synthesis of some compounds; Paul Davey and Eva Lenz for the assistance with characterization of compounds; Rebecca Morris for mouse pharmacokinetic data; Ian Barrett and Caroline Truman for their input into assay design and data generation; Andrew Thomason for leading the in vitro biological evaluation of ATM inhibitors; and Philip Jewsbury for helpful discussions regarding the manuscript.
Glossary
ABBREVIATIONS
- 2nd generation X-Phos precatalyst
chloro(2-dicyclohexylphosphino-2′,4′,6′-triisopropyl-1,1′-biphenyl)[2-(2′-amino-1,1′-biphenyl)]palladium(II)
- ATM
ataxia telangiectasia mutated
- ATR
ataxia telangiectasia Rad3-related
- BPin
pinacolatoboryl
- Clint
intrinsic clearance
- DIPEA
N-ethyl-N-isopropylpropan-2-amine
- CSF1R
colony stimulating factor 1 receptor kinase
- DDR
DNA damage repair
- DNAPK
DNA-dependent protein kinase
- DSB
double strand break
- Flt-4
Fms-related tyrosine kinase 4
- HLM
human liver microsome
- MDCK
Madin–Darby canine kidney epithelial cell line
- mTOR
mammalian target of rapamycin
- NTRK2
neurotrophic tyrosine receptor kinase type 2
- Papp
apparent permeability
- PI4K
phosphatidylinositol 4-kinase
- PIKK
PI3K-related kinase
- PTEN
phosphatase and tensin homologue
- RET
“rearranged during transfection” kinase
- SAR
structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00200.
Details of the full synthesis and spectroscopic characterization of all compounds and intermediates; crystallographic information; all protocols for in vitro and in vivo experiments and kinase selectivity (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Savitsky K.; Bar-Shira A.; Gilad S.; Rotman G.; Ziv Y.; Vanagaite L.; Tagle D. A.; Smith S.; Uziel T.; Sfez S.; Ashkenazi M.; Pecker I.; Frydman M.; Harnik R.; Patanjali S. R.; Simmons A.; Clines G. A.; Sartiel A.; Gatti R. A.; Chessa L.; Sanal O.; Lavin M. F.; Jaspers N. G.; Taylor A. M.; Arlett C. F.; Miki T.; Weissman S. M.; Lovett M.; Collins F. S.; Shiloh Y. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–53. 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Kurz E. U.; Lees-Miller S. DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair 2004, 3, 889–900. 10.1016/j.dnarep.2004.03.029. [DOI] [PubMed] [Google Scholar]
- Abraham R. T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- Wu J.; Yin M.; Hapke G.; Toth K.; Rustum Y. M. Induction of biphasic DNA double strand breaks and activation of multiple repair protein complexes by DNA topoisomerase I drug 7-ethyl-10-hydroxy-camptothecin. Mol. Pharmacol. 2002, 61, 742–748. 10.1124/mol.61.4.742. [DOI] [PubMed] [Google Scholar]
- Weber A. M.; Ryan A. J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. 10.1016/j.pharmthera.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Teng P.; Bateman N. W.; Darcy K. M.; Hamilton C. A.; Maxwell G. L.; Bakkenist C. J.; Conrads T. P. Pharmacologic inhibition of ATR and ATM offers clinically important distinctions to enhancing platinum or radiation response in ovarian, endometrial, and cervical cancer cells. Gynecol. Oncol. 2015, 136, 554–561. 10.1016/j.ygyno.2014.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z.; Kozlov S.; Lavin M. F.; Person M. D.; Paull T. T. ATM activation by oxidative stress. Science 2010, 330, 517–521. 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- Alexander A.; Cai S. L.; Kim J.; Nanez A.; Sahin M.; Mac Lean K. H.; Inoki K.; Guan K. L.; Shen J.; Person M. D.; Kusewitt D.; Mills G. B.; Kastan M. B.; Walker C. L. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 4153–4158. 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno Y.; Nakamura-Ishizu A.; Otsu K.; Suda T.; Kubota Y. Pathological neoangiogenesis depends on oxidative stress regulation by ATM. Nat. Med. 2012, 18, 1208–1216. 10.1038/nm.2846. [DOI] [PubMed] [Google Scholar]
- Hickson I.; Zhao Y.; Richardson C. J.; Green S. J.; Martin N. M. B.; Orr A. I.; Reaper P. M.; Jackson S. P.; Curtin N. J.; Smith G. C. M. Identification and Characterization of a Novel and Specific Inhibitor of the Ataxia-Telangiectasia Mutated Kinase ATM. Cancer Res. 2004, 64, 9152–9159. 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- Golding S. E.; Rosenberg E.; Valerie N.; Hussaini I.; Frigerio M.; Cockcroft X. F.; Chong W. Y.; Hummersone M.; Rigoreau L.; Menear K. A.; O’Connor M. J.; Povirk L. F.; van Meter T.; Valerie K. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 2009, 8, 2894–2902. 10.1158/1535-7163.MCT-09-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degorce S. L.; Barlaam B.; Cadogan E.; Dishington A.; Ducray R.; Glossop S. C.; Hassall L. A.; Lach F.; Lau A.; McGuire T. M.; Nowak T.; Ouvry G.; Pike K. G.; Thomason A. G. Discovery of Novel 3-Quinoline Carboxamides as Potent, Selective and Orally Bioavailable Inhibitors of Ataxia Telangiectasia Mutated (ATM) Kinase. J. Med. Chem. 2016, 56, 6281–6292. 10.1021/acs.jmedchem.6b00519. [DOI] [PubMed] [Google Scholar]
- Pike K. G.; Barlaam B.; Cadogan E.; Campbell A.; Chen Y.; Colclough N.; Davies N. L.; de-Almeida C.; Degorce S. L.; Didelot M.; Dishington A.; Ducray R.; Durant S. T.; Hassall L. A.; Holmes J.; Hughes G. D.; MacFaul P. A.; Mulholland K. R.; McGuire T. M.; Ouvry G.; Pass M.; Robb G.; Stratton N.; Wang Z.; Wilson J.; Zhai B.; Zhao K.; Al-Huniti N. The Identification of Potent, Selective, and Orally Available Inhibitors of Ataxia Telangiectasia Mutated (ATM) Kinase: The Discovery of AZD0156 (8-{6-[3-(Dimethylamino)propoxy]pyridin-3-yl}-3-methyl-1-(tetrahydro-2H-pyran-4-yl)-1,3-dihydro-2H-imidazo[4,5-c]quinolin-2-one). J. Med. Chem. 2018, 61, 3823–3841. 10.1021/acs.jmedchem.7b01896. [DOI] [PubMed] [Google Scholar]
- Karlin J.; Allen J.; Odedra R.; Hughes G.; Farrington P.; Ducray R.; Ouvry G.; Degorce S.; Wilson J.; Smith A.; Patel B.; Thomason A.; Vincent J.; Colclough N.; Ahmad S. F.; Beckta J. M.; Tokarz M.; Mukhopadhyay N. D.; Barlaam B.; Pike K. G.; Cadogan E.; Pass M.; Valerie K.; Durant S. Blood-brain barrier penetrating ATM inhibitor radio-sensitizes intracranial gliomas in mice. Cancer Res. 2016, 76, 3041. 10.1158/1538-7445.AM2016-3041. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.