

Abstract
Glutamate mediates fast excitatory neurotransmission via ionotropic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. The trafficking and gating properties of AMPA receptors (AMPARs) can be amplified by transmembrane AMPAR regulatory proteins (TARPs), which are often expressed in localized brain regions. Herein, we describe the discovery, lead optimization, and preclinical characterization of 5-arylbenzimidazolone and oxindole-based negative modulators of AMPARs associated with TARP γ-8, the primary TARP found in hippocampus. High-throughput screen lead 4 was optimized for potency and brain penetration to provide benzimidazolone 3, JNJ-55511118.1 Replacement of the benzimidazolone core in 3 with an oxindole mitigated reactive metabolite formation and led to the identification of 18 (GluA1/γ-8 pIC50 = 9.7). Following oral dosing in rats, 18 demonstrated robust target engagement in hippocampus as assessed by ex vivo autoradiography (ED50 = 0.6 mg/kg, plasma EC50 = 9 ng/mL).
Keywords: AMPA, α-amino-3-hydroxy-5-methy-4-isoxazolepropionic acid, AMPA, TARP γ-8, transmembrane AMPA receptor regulatory protein, auxiliary subunit, glutamate, hippocampus, epilepsy, glutathione, autoradiography
Fast excitatory transmission in the central nervous system (CNS) is mediated primarily by glutamate-gated ionotropic α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors. In the endoplasmic reticulum, AMPA receptors (AMPARs) are formed as tetrameric assemblies of subunits (GluA1–GluA4) and then trafficked to the synaptic membrane where they play an integral role in regulating synaptic strength.2 Although AMPARs are abundantly distributed throughout the brain, their activity can be modulated by auxiliary proteins that often have more discrete expression profiles. Specifically, transmembrane AMPAR regulatory proteins (TARPs), which are classified by sequence homology and function as Type I (γ-2, γ-3, γ-4, γ-8) or Type II (γ-5, γ-7), enhance AMPAR pharmacology by altering gating properties and amplifying receptor surface expression.3 In mammals, TARP γ-8 is predominantly expressed in forebrain regions and highly enriched in hippocampus.4 Consistent with this restricted expression profile, selective pharmacological disruption of AMPARs associated with γ-8 has been shown to preferentially attenuate excitatory neurotransmission in rat forebrain.1,5,6 Targeting this mechanism in human may ultimately provide improved therapies for temporal lobe epilepsy and other neurological disorders characterized by hyperexcitability within the hippocampus.7
The noncompetitive AMPAR antagonist 1 (perampanel, Fycompa; Figure 1) is approved by the FDA as an adjunctive treatment for partial-onset and generalized tonic-clonic seizures.8 However, like many antiepileptic drugs, perampanel administration is associated with CNS-related side effects such as dizziness, sedation, and falling.9 Perampanel is not a TARP-selective AMPAR antagonist, and motor impairment in patients may result from blockade of AMPARs dependent on stargazin (γ-2), the primary TARP expressed in cerebellum.10 Recently, Lilly reported the discovery of 2, LY3130481/CERC-611, an AMPAR negative modulator selective for γ-8.11 We have also disclosed a structurally distinct compound 3, JNJ-55511118, which exhibited comparable in vitro and in vivo pharmacology to 2.1 Notably, both γ-8 selective negative modulators were orally efficacious in rodent seizure models and did not impair motor function. Herein, we describe the medicinal chemistry efforts that resulted in the identification of 3, as well as a second-generation series of oxindole analogs with improved in vivo potency and reduced propensity for reactive metabolite formation.
Figure 1.

Perampanel and AMPAR/TARP γ-8 modulators.
A high-throughput screen (HTS) of the Janssen global screening deck was conducted to identify compounds that blocked glutamate-induced Ca2+ flux in HEK-293 cells expressing a fusion protein between γ-8 and the GluA1o “flop” splice variant.1 Confirmed hits were counter-screened against a similar cellular construct coexpressing GluA1o and γ-2. From this effort, 5-aryl oxindole 4 was identified as a preliminary lead due to its high ligand efficiency and selectivity over γ-2 (Figure 2). A subsequent methyl scan around the distal aromatic ring established that 2,6-disubstitution was preferred for γ-8 potency and provided a submicromolar analog 5 (pIC50 = 6.4). Replacement of the oxindole core with an isosteric benzimidazolone scaffold improved activity (6, pIC50 = 6.8) without sacrificing selectivity against γ-2. Furthermore, when administered to rats, 6 exhibited encouraging oral bioavailability (%F = 68), low i.v. clearance (9 mL/min/kg), and acceptable brain exposure (Kp u,u = 0.3).
Figure 2.

Evolution of benzimidazolone 6 from HTS lead 4.
Having established that benzimidazolone 6 was a CNS-penetrant drug-like lead, we sought to optimize this molecule into a tool compound suitable for evaluating AMPAR/γ-8 pharmacology in vivo. To this end, a library of ortho,ortho-disubstituted 5-arylbenzimidazolones was prepared, and promising compounds were initially profiled in a variety of in vitro assays (Table 1). Replacement of a single methyl group with a chlorine provided a 5-fold improvement in activity (compare 6 and 7), and consequently, this group was held constant while scanning the remaining ortho-position. Further gains in γ-8 potency were driven largely by lipophilicity, with relatively small structural changes yielding dramatic improvements (compare 7 and 8, as well as 3 and 13). The introduction of heterocycles (10) and polar substituents (11, 13) reduced potency, except for benzyl nitrile 12 (JNJ-56022486), which had the highest lipophilic ligand efficiency (LLE = 5.4). In addition to the encouraging potency and selectivity profile for 12, its auspicious physicochemical properties (i.e., high free fraction and solubility) prompted us to radiolabel the compound and establish binding and autoradiography assays in native tissue.12 As described previously, 12 was tritiated in the 6-position of the benzimidazolone, and the resulting radioligand displayed high specific binding to hippocampus in rodent and primate brain slices.1 Furthermore, the regional brain binding profile of [3H]-12 matched the γ-8 (CACNG8) mRNA expression pattern reported in the Allen Mouse Brain Atlas.13
Table 1. SAR and in Vitro ADME/DMPK Data for Disubstituted 5-Arylbenzimidazolones.
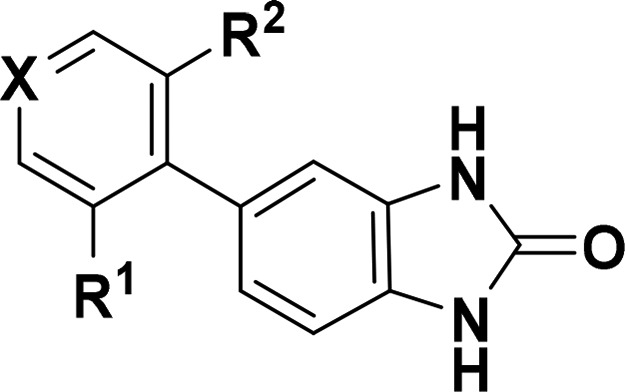
| Cmpd | X | R1 | R2 | GluA1/γ-8 pIC50a | GluA1/γ-2 pIC50b | HLM/RLM stabilityc | MDCK-MDR1d (B-A)/(A-B) × 10–6cm/s | Solubility (SGF/FaSSIF)e (μM) | PPB,fub (h/r)f | cLogP | LLE |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 6 | CH | CH3 | CH3 | 6.8 ± 0.1 | <5 | 0.3/0.5 | 34/42 | 5/276 | 2.1/3.7 | 3.4 | 3.4 |
| 7 | CH | Cl | CH3 | 7.3 ± 0.3 | <5 | <0.3/0.6 | 32/25 | <4/146 | 0.9/1.4 | 3.7 | 3.6 |
| 8 | CH | Cl | cPr | 8.2 ± 0.2 | <5 | <0.3/0.5 | 18/10 | <4/90 | 0.2/1.5 | 4.1 | 4.1 |
| 9 | CH | Cl | Cl | 7.3 ± 0.2 | <5 | <0.3/0.3 | 49/26 | <4/69 | 0.5/0.6 | 3.9 | 3.4 |
| 10 | N | Cl | Cl | 6.6 ± 0.2 | <5 | <0.3/<0.2 | 83/16 | 42/52 | 2.9/2.7 | 2.6 | 3.9 |
| 11 | CH | Cl | CN | 6.6 ± 0.3 | <5 | <0.3/<0.2 | 70/14 | <4/5 | 5.1/7.1 | 2.9 | 3.7 |
| 12 | CH | Cl | CH2CN | 8.0 ± 0.41 | <51 | <0.3/0.6 | 70/6.6 | 26/230 | 2.7/7.6 | 2.6 | 5.4 |
| 13 | CH | Cl | OCH3 | 6.9 ± 0.2 | <5 | 0.3/0.6 | 50/26 | 19/142 | 3.9/6.5 | 3.0 | 3.8 |
| 3 | CH | Cl | OCF3 | 8.3 ± 0.31 | <51 | <0.3/0.3 | 24/22 | <4/74 | 0.3/1.5 | 4.1 | 4.2 |
pIC50 measured in a FLIPR assay using HEK-293 cells expressing a human GluA1o-γ-8 fusion construct; all data are the result of at least three assays run in triplicate with the mean and standard deviation reported.
pIC50 measured in a FLIPR assay using HEK-293 cells expressing human GluA1-flop cotransfected with γ-2.
Stability in human and rat liver microsomes at 1 μM; data are reported as extraction ratios.
Apparent permeability in Madin–Darby canine kidney cells transfected with the MDR1 (P-gp) gene.
Thermodynamic solubility in simulated gastric fluid (pH = 1.6) and fasted state simulated intestinal fluid (pH = 6.5).
Human and rat plasma protein binding reported as fraction unbound (fub) at 1 μM. Details for all assay conditions are provided in the Supporting Information.
Although 12 proved to be a valuable compound for validating AMPAR/γ-8 selectivity in vitro, poor brain penetration limited its utility in vivo. However, trifluoromethoxy 3 and cyclopropyl 8 exhibited comparable potency and selectivity to 12, in addition to improved intrinsic permeability and efflux in a MDCK-MDR1 cellular assay. Since 3 and 8 achieved the best balance of potency and ADME/DMPK properties, both compounds were profiled extensively in vivo (Table 2). In rat pharmacokinetic studies, 3 and 8 displayed high bioavailability, as well as low to moderate i.v. clearance that correlated with the rank order predicted by in vitro liver microsomal stability. Furthermore, following oral dosing, both compounds reached full γ-8 receptor occupancy in rat hippocampus with plasma EC50s of 145 and 486 ng/mL for 3 and 8, respectively.
Table 2. Pharmacokinetic Parameters and ex Vivo GluA/γ-8 Receptor Occupancy in Rat Brain for Compounds 3 and 8.
| rat
PK parametersa |
rat ex vivo GluA/γ-8 receptor occupancyb |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cmpd | Vss (L/kg) | Cl (mL/min/kg) | i.v. t1/2 (h) | p.o. Cmax (ng/mL) | %F | ED50c (mg/kg) | EC50 [plasma] (ng/mL) | EC50 [brain] (ng/g) | Kpu,u (brain) |
| 3 | 3.3 ± 0.3 | 6.9 ± 1.2 | 5.8 ± 1.6 | 1027 ± 70 | 133 ± 31 | 0.51 | 1451 | 545 | 0.6 |
| 8 | 4.6 ± 0.2 | 21 ± 4.0 | 2.7 ± 0.6 | 502 ± 111 | 121 ± 17 | 3.2 | 486 | 1611 | 0.3 |
Compounds dosed as solutions in 20% HP-β-CD at 1 mg/kg (i.v.) and 5 mg/kg (p.o.) in Sprague–Dawley rats (n = 3/group).
GluA/γ-8 receptor occupancy in rat hippocampus at Tmax for each compound as measured by ex vivo autoradiography using [3H]12.
Compounds dosed p.o. in 0.5% HPMC (compound 3) or 20% HP-β-CD (compound 8) in Sprague–Dawley rats (8 doses, n = 3/dose group).
Due to its superior in vivo potency, long half-life, and promising anticonvulsant profile in rodents,1 compound 3 was advanced to a 14-day repeat dose rat toleration study. Although no CNS-related issues were noted at exposures that corresponded to full γ-8 receptor occupancy, subsequent histological analysis revealed hepatocellular necrosis in all dose groups (20 and 60 mg/kg/day), which precluded further development of 3.
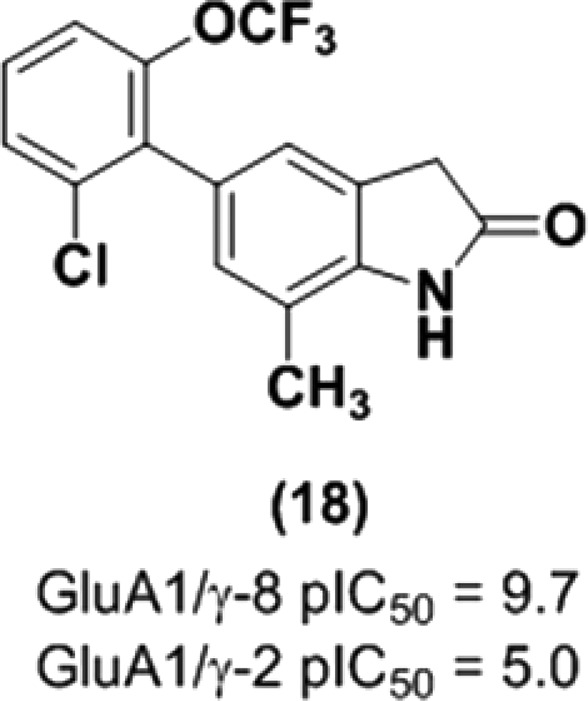
From metabolite identification studies in hepatocytes with 3, multiple thiol adducts were observed by LC–MS/MS analysis as the primary metabolites in all species except monkey, which suggested that the liver toxicity in rat may be attributed to reactive metabolites (Figure 3). M3, a glutathione (GSH) adduct formed through oxidative dechlorination in mouse, implicated the 5-aryl ring as the site of bioactivation. However, modifications to this region of the molecule did not prevent glutathione trapping when compounds were incubated in human liver microsomes supplemented with GSH and NADPH (data not shown). Fortunately, a scan of isosteric replacements for the benzimidazolone scaffold ultimately mitigated this key issue (Table 3). While benzoxazolone 14 (pIC50 = 5.6) and benzothiazolone 15 (pIC50 = 8.6) differed greatly in γ-8 potency, both compounds trapped glutathione following metabolic activation. In contrast, oxindole analogs 16–18 did not form GSH adducts under identical conditions. Although oxindole 16 (pIC50 = 7.8) was 5-fold less active than the corresponding benzimidazolone 3 (pIC50 = 8.3), the introduction of small lipophilic substituents in the 7-position improved γ-8 potency dramatically. Notably, the subnanomolar 7-methyl derivative 18 (pIC50 = 9.7) was 25-times more potent than 3.
Figure 3.
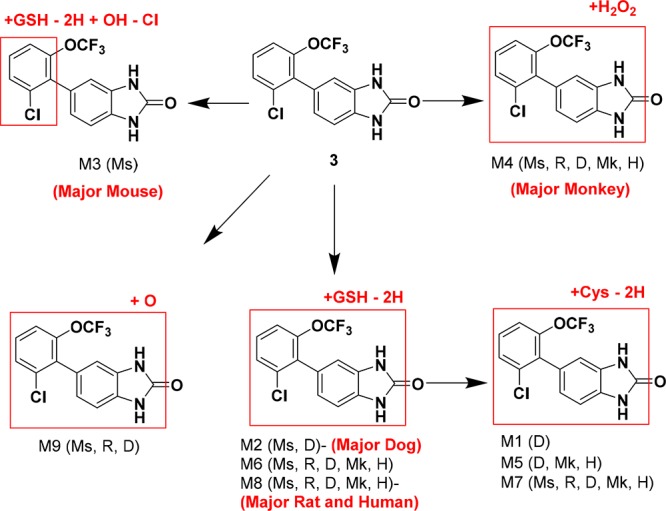
Cross-species hepatocyte metabolism for 3.
Table 3. Reactive Metabolite SAR for Analogs of Compound 3.
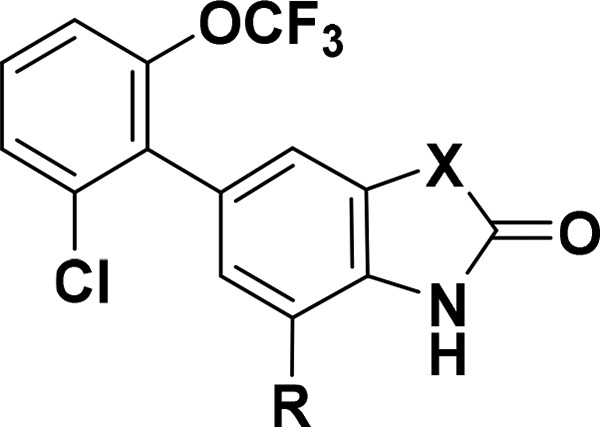
| Cmpd | X | R | GluA1/γ-8 pIC50a | GSH adductb | cLogP |
|---|---|---|---|---|---|
| 3 | NH | H | 8.3 ± 0.31 | yes | 4.1 |
| 14 | O | H | 5.6 (n = 1) | yes | 4.1 |
| 15 | S | H | 8.6 ± 0.3 | yes | 4.7 |
| 16 | CH2 | H | 7.8 ± 0.2 | no | 3.6 |
| 17 | CH2 | F | 8.6 ± 0.3 | no | 4.1 |
| 18 | CH2 | CH3 | 9.7 ± 0.3 | no | 4.2 |
pIC50 measured in a FLIPR assay using HEK-293 cells expressing a human GluA1o-γ-8 fusion construct; unless noted, all data are the result of at least three assays run in triplicate with the mean value and standard deviation reported.
Compounds were incubated for 1 h in human liver microsomes supplemented with glutathione (GSH) and NADPH.
To offset the increased lipophilicity introduced in the 7-position of the oxindole, pyridines were investigated as phenyl replacements in the 5-position (Table 4). Consistent with the SAR established for the benzimidazolone core, 4-pyridyl 19 (pIC50 = 8.6) was an order of magnitude less potent than the corresponding phenyl analog 18 (pIC50 = 9.7), but had comparable lipophilic ligand efficiency. The reduced hydrophobicity translated into improved physicochemical properties as pyridines 19–20 displayed lower plasma protein and brain tissue binding, as well as higher aqueous solubility at acidic pH compared to 18. Difluoromethoxypyridine 20, the compound with the lowest cLogP, was especially impressive in this regard. Furthermore, both pyridines exhibited minimal turnover in human microsomes, high intrinsic permeability, and low efflux. This contrasts with the large efflux ratios observed for related derivatives from the benzimidazolone series (i.e., see 10Table 1), presumably due to the differing number of hydrogen bond donors between the two cores. In addition to their favorable in vitro ADME profiles, oxindoles 18–20 retained exquisite selectivity for γ-8 over γ-2 (>1000×) and did not form glutathione adducts in the presence of human liver microsomes.
Table 4. SAR and in Vitro ADME/DMPK Data for Disubstituted 5-Aryloxindoles.
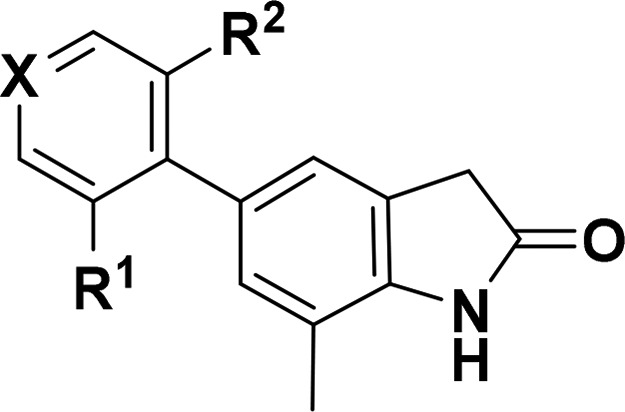
| Cmpd | X | R1 | R2 | GluA1/γ-8 pIC50a | GluA1/γ-2 pIC50b | GSH adductc | HLM/RLM stabilityd | MDCK-MDR1e (B-A)/(A-B) | solubility (SGF/FaSSIF)f | PPB,fub (h/r)g | BTB,fub rath | cLogP | LLE |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 18 | CH | Cl | OCF3 | 9.7 ± 0.3 | 5.0i | No | 0.5/0.4 | NDj | <4/55 | 0.3/0.6 | 0.4 | 4.2 | 5.5 |
| 19 | N | Cl | OCF3 | 8.6 ± 0.3 | <5 | No | <0.3/0.5 | 51/44 | 11/45 | 4.4/6.0 | 1.4 | 3.2 | 5.4 |
| 20 | N | Cl | OCF2H | 8.1 ± 0.2 | <5 | No | <0.3/0.4 | 66/34 | 140/64 | 11/8.6 | 4.8 | 2.6 | 5.4 |
pIC50 measured in FLIPR assay using HEK-293 cells expressing a human GluA1o-γ-8 fusion construct; all data are the result of at least three assays run in triplicate with the mean value and standard deviation reported.
pIC50 measured in a FLIPR assay using HEK-293 cells transiently expressing human GluA1o cotransfected with γ-2 (n = 1, run in triplicate).
Compounds were incubated for 1 h in human liver microsomes supplemented with glutathione (GSH) and NADPH.
Stability in human and rat liver microsomes at 1 μM; data are reported as extraction ratios, where the predicted hepatic clearance is divided by the species-specific hepatic blood flow.
Apparent permeability (× 10–6cm/s) in Madin–Darby canine kidney cells transfected with the MDR1 (P-gp) gene.
Thermodynamic solubility (μM) in simulated gastric fluid (pH = 1.6) and fasted state simulated intestinal fluid (pH = 6.5).
Human and rat plasma protein binding reported as fraction unbound (fub) at 1 μM.
Rat brain tissue binding reported as fraction unbound (fub) at 5 μM.
Calcium-flux data for 18 with related GluA/TARP constructs are shown in Supplementary Table S1.
Not determined due to low compound recovery. Details of all assay conditions are provided in the Supporting Information.
Whereas compounds 3–15 were synthesized using Suzuki reactions between commercially available or readily accessible aryl halides and boronic acids/esters, convergent multistep routes were often required to construct oxindoles 16–20. As a representative example, the reaction sequences used to prepare perfluoroalkoxy analogs 16–20 are shown in Scheme 1. Final compounds were assembled from Pd-catalyzed couplings between functionalized phenyl or pyridyl iodides 25–27 and oxindole boronic esters 31–33, which were either commercially available or derived from a sequential bromination/borylation sequence starting with 7-substituted oxindoles (i.e., 28, R2 = F). Intermediate 25 (X = CH) was prepared in one pot through a regioselective lithiation/iodination of 3-chloro-5-trifluoromethoxybenzene 21, while the perfluoroalkoxy groups in pyridines 26 (X = N, R1 = H) and 27 (X = N, R1 = F) were installed after the iodination step. Specifically, alkylation of hydroxypyridine 24 with sodium chlorodifluoroacetate14 provided difluoromethoxy 26, whereas trifluoromethoxy 27 was assembled in two steps from 24 via fluoride displacement of a bromodifluoroalkoxypyridine intermediate.15
Scheme 1. Synthesis of Compounds 16–20 as a Representative Route to 5-Aryl Oxindoles/Benzimidazolones.

Reagents and conditions: (a) nBuLi, THF, −78 °C, then I2, 85%; (b) MOMCl, KOtBu, THF, 0 °C to rt, 65%; (c) nBuLi, THF, −78 °C, then I2, 47%; (d) 4 N HCl, dioxane, 93%; (e) Na(CO2CF2Cl), Cs2CO3, DMF/H2O, 100 °C, 27%; (f) CBr2F2, NaH, KOtBu, DMF, 80 °C, 15%; (g) AgBF4, DCM, −78 °C to rt, 62%; (h) NBS, MeCN, 0 °C, 72%; (i) B2(Pin)2, KOAc, cat Pd(dppf)Cl2–CH2Cl2, dioxane, 85 °C, 54–75%; (j) K3PO4, cat Pd(dppf)Cl2CH2Cl2 or Pd(dtbpf)Cl2, dioxane/H2O, 95 °C, 37–62%.
Based on their promising in vitro ADME/DMPK profiles, oxindoles 18–20 were further evaluated in vivo (Table 5). Although 20 exhibited the lowest plasma clearance, this compound also had the shortest i.v. half-life due to its reduced volume of distribution relative to 18 and 19. To assess target engagement in rat hippocampus, compounds were dosed orally at 3 mg/kg (for 18) or 10 mg/kg (for 19–20), and receptor occupancy was determined by ex vivo autoradiography. All three compounds reached micromolar brain concentrations, which resulted in robust target engagement (>70% GluA/γ-8 receptor occupancy).
Table 5. PK/PD Profiles of Oxindoles 18–20 in Rat.
| PK parametersa |
ex vivo ARGb |
||||
|---|---|---|---|---|---|
| Cmpd | Vss (L/kg) | Cl (mL/min/kg) | t1/2 (h) | GluA/γ-8 R.O. (%)c | [brain]/[plasma]d |
| 18 | 3.5 ± 1.6 | 28 ± 0.5 | 1.9 ± 0.4 | 82 ± 10 | 524/135 |
| 19 | 2.7 ± 0.6 | 37 ± 4.0 | 1.4 ± 0.1 | 86 ± 3 | 983/788 |
| 20 | 1.9 ± 0.1 | 19 ± 2.0 | 1.2 ± 0.1 | 72 ± 6 | 2276/2367 |
Compounds dosed as solutions in 1:1 PEG400/H2O at 1 mg/kg (i.v.) in Sprague–Dawley rats (n = 3/group).
Compounds dosed p.o. as suspensions in 0.5% HPMC at 3 mg/kg (compound 18) or 10 mg/kg (compounds 19–21) in Sprague–Dawley rats (n = 2/dose group).
GluA/γ-8 receptor occupancy in rat hippocampus at Tmax for each compound as measured by ex vivo autoradiography using [3H]12.
Brain and plasma concentrations in ng/g and ng/mL, respectively, at Tmax.
Since oxindoles 18 and 19 had the lowest efficacious plasma concentrations in the rat ex vivo autoradiography screen, full dose responses were obtained (Figure 4). Whereas 18 displayed dose-proportional plasma and brain exposures, the plasma levels for pyridyl 19 were below the lower limit of quantification at low doses (<0.3 mg/kg) and increased in a nonlinear manner with ascending doses (i.e., 87 ng/mL at 3 mg/kg and 2049 ng/mL at 10 mg/kg). Consequently, the ED50 for 19 (5.4 mg/kg) was significantly right-shifted relative to 18 (0.6 mg/kg). The corresponding plasma and brain EC50s for 18 were 9 ng/mL and 71 ng/g, respectively, which represent a 16-fold improvement in efficacious plasma concentration relative to benzimidazolone 3 (see Table 2).
Figure 4.
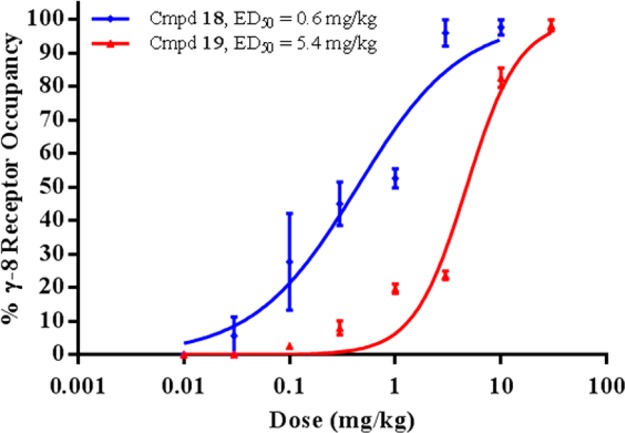
Ex vivo GluA/γ-8 receptor occupancy in rat hippocampus for compounds 18 and 19: dose dependency following p.o. administration (n = 3/dose ± SEM). Receptor occupancy was measured at Tmax for each compound using [3H]-12 as the radiotracer.
In summary, we have described the discovery and optimization of a series of 5-arylbenzimidazolone- and oxindole-based AMPAR modulators selective for TARP γ-8. Starting from a micromolar benzimidazolone lead 4, systematic modification of the 5-aryl region to optimize potency and brain penetration afforded a single-digit nanomolar tool compound 3. Although hepatotoxicity in rats prevented development of 3, this compound established that selective negative modulation of AMPAR/γ-8 can provide robust seizure protection in rodents without causing sedation or motor impairment. A benzimidazolone to oxindole scaffold switch mitigated the reactive metabolite liabilities associated with 3, and the introduction of small lipophilic groups in the 7-position of the oxindole dramatically improved potency. Accordingly, in rats, the lead oxindole 18 exhibited an order of magnitude lower efficacious plasma concentration than benzimidazolone 3. Related series with favorable PK profiles in higher species have been identified and will be disclosed in due course.
Acknowledgments
The authors wish to thank Moravek, Inc. (Brea, CA) for the synthesis of [3H]12, NovAliX (Strasbourg, France) and WuXi AppTec (Shanghai, China) for the resynthesis of compounds 3 and 8, and BioBlocks, Inc. (San Diego, CA) for the resynthesis of compound 20.
Glossary
ABBREVIATIONS
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- TARP
transmembrane AMPA receptor regulatory protein
- CNS
central nervous system
- FDA
Food and Drug Administration
- LE
ligand efficiency
- LLE
ligand-lipophilicity efficiency
- ADME
absorption, distribution, metabolism, excretion
- DMPK
drug metabolism and pharmacokinetics
- SAR
structure–activity relationship
- CYP
cytochrome P450
- MW
molecular weight
- FLIPR
fluorescence imaging plate reader
- p.o.
per os
- i.v.
intravenous
- PK
pharmacokinetics
- CL
clearance
- Vss
volume of distribution at steady state
- t1/2
half-life
- Cmax
maximum concentration
- F
bioavailability
- RLM
rat liver microsome
- HLM
human liver microsome
- PPB
plasma protein binding
- fub
fraction unbound
- MDR1
multidrug resistance gene
- MDCK-MDR1
Madin–Darby canine kidney cell line transfected with MDR1
- SGF
simulated gastric fluid
- FaSSIF
fasted state simulated intestinal fluid
- GSH
glutathione
- NADPH
nicotinamide adenine dinculeotide phosphate
- RO
receptor occupancy
- HPMC
hydroxypropyl methylcellulose
- PEG
polyethylene glycol
- SEM
standard error of the mean
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00215.
Experimental procedures, characterization, and assay conditions (PDF)
Author Present Address
† Sanford Burnham Prebys Medical Discovery Institute, 10901 North Torrey Pines Road, La Jolla, California 92037, United States.
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Maher M. P.; Wu N.; Ravula S.; Ameriks M. K.; Savall B. M.; Liu C.; Lord B.; Wyatt R. M.; Matta J. A.; Dugovic C.; et al. Discovery and characterization of AMPA receptor modulators selective for TARP-γ8. J. Pharmacol. Exp. Ther. 2016, 357, 394–414. 10.1124/jpet.115.231712. [DOI] [PubMed] [Google Scholar]
- Huganir R. L.; Nicoll R. A. AMPARs and synaptic plasticity: the last 25 years. Neuron 2013, 80, 704–717. 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson A. C.; Nicoll R. A. The expanding social network of ionotropic glutamate receptors: TARPs and other transmembrane auxiliary subunits. Neuron 2011, 70, 178–199. 10.1016/j.neuron.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita S.; Chen L.; Kawasaki Y.; Petralia R. S.; Wenthold R. J.; Nicoll R. A.; Bredt D. S. Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J. Cell Biol. 2003, 161 (4), 805–815. 10.1083/jcb.200212116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher M. P.; Matta J. A.; Gu S.; Seierstad M.; Bredt D. S. Getting a handle on neuropharmacology by targeting receptor-associated proteins. Neuron 2017, 96, 989–998. 10.1016/j.neuron.2017.10.001. [DOI] [PubMed] [Google Scholar]
- Kato A. S.; Burris K. D.; Gardinier K. M.; Gernert D. L.; Porter W. J.; Reel J.; Ding C.; Tu Y.; Schober D. A.; Lee M. R.; et al. Forebrain-selective AMPA-receptor antagonism guided by TARP γ-8 as an antiepileptic mechanism. Nat. Med. 2016, 22, 1496–1501. 10.1038/nm.4221. [DOI] [PubMed] [Google Scholar]
- Small S. A.; Schobel S. A.; Buxton R. B.; Witter M. P.; Barnes C. A. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat. Rev. Neurosci. 2011, 12, 585–601. 10.1038/nrn3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibi S.; Ueno K.; Nagato S.; Kawano K.; Ito K.; Norimine Y.; Takenaka O.; Hanada T.; Yonaga M. Discovery of 2-(2-oxo-1-phenyl-5-pyridin-2-yl-1,2-dihydropyridin-3-yl)benzonitrile (perampanel): a novel, noncompetitive α-amino-3-hydroxy-5-methyl-4-isoxazolepropanoic acid (AMPA) receptor antagonist. J. Med. Chem. 2012, 55, 10584–10600. 10.1021/jm301268u. [DOI] [PubMed] [Google Scholar]
- Greenwood J.; Valdes J. Perampanel (Fycompa) a review of clinical efficacy and safety in epilepsy. P T 2016, 41 (11), 683–688. [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Chetkovich D. M.; Petralia R. S.; Sweeney N. T.; Kawasaki Y.; Wenthold R. J.; Bredt D. S.; Nicoll R. A. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 2000, 408, 936–943. 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- Gardinier K. M.; Gernert D. L.; Porter W. J.; Reel J. K.; Ornstein P. L.; Spinazze P.; Stevens F. C.; Hahn P.; Hollinshead S. P.; Mayhugh D.; et al. Discovery of the first α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonist dependent upon transmembrane AMPA receptor regulatory protein (TARP) γ-8. J. Med. Chem. 2016, 59, 4753–4768. 10.1021/acs.jmedchem.6b00125. [DOI] [PubMed] [Google Scholar]
- Langlois X.; te Riele P.; Wintmolders C.; Leysen J. E.; Jurzak M. Use of the β-imager for rapid ex vivo autoradiography exemplified with central nervous system penetrating neurokinin 3 antagonists. J. Pharmacol. Exp. Ther. 2001, 299, 712–717. [PubMed] [Google Scholar]
- Lein E. S.; Hawrylycz M. J.; Ao N.; Ayres M.; Bensinger A.; Bernard A.; Boe A. F.; Boguski M. S.; Brockway K. S.; Byrnes E. J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Sperry J. B.; Sutherland K. A safe and practical procedure for difluoromethylation of methyl 4-hydroxy-3-iodobenzoate. Org. Process Res. Dev. 2011, 15, 721–725. 10.1021/op200052z. [DOI] [Google Scholar]
- a Li X.; Pan H.; Jiang X. Spontaneous reactions of potassium phenoxide with dibromoperfluoroalkanes, first evidence for bromophilic attack on C-Br bonds by the phenoxide anion. Tetrahedron Lett. 1984, 25, 4937–4940. 10.1016/S0040-4039(01)91263-8. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.