

Abstract
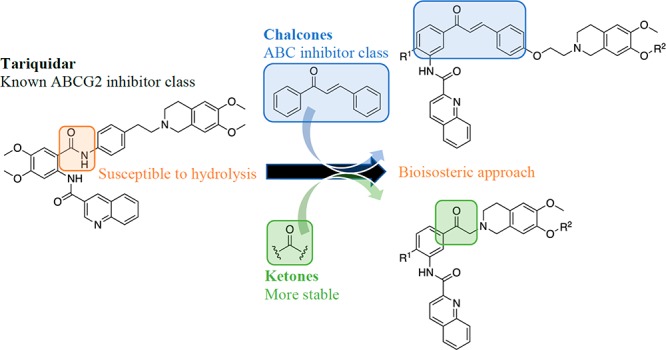
ABC transporters, including ABCG2, play a vital role in defending the human body against the vast range of xenobiotics. Even though this is beneficial for human health, these protein transporters have been implicated in the emerging resistance of cancer cells to a variety of structurally and functionally diverse anticancer drugs. In order to investigate their role in resistance, potent and selective ABCG2 modulators have been described in the literature. A leading class of modulators are the tariquidar analogues; however, their susceptibility to hydrolysis limits their applicable use. To overcome this, we synthesized a novel series of chalcone- and ketone-based compounds inspired by reported tariquidar analogues. Compounds were characterized and evaluated for their ABCG2 modulatory activity and ABC transporter selectivity. When compared to transporters ABCB1 and ABCC1, the chalcone-based compounds exhibited selectivity for ABCG2, while the ketone-based compounds showed only a slight preference for ABCG2. From the former series, chalcone 16d (UR-DP48) displayed similar activity to the reference fumitremorgin C, both producing comparable maximal effects. The compound exhibited marked antiproliferative activity, while cytotoxicity was less pronounced for the most active compound 17f from the ketone series. Chalcone-containing tariquidar analogues are promising modulators to aid in functional investigations of ABCG2 transporters.
Keywords: Tariquidar, chalcone, ketone, ABCG2, modulator
Cytotoxic drugs are well established in the treatment of many cancer types. In order to improve the effectiveness of treatment, multiple cytotoxic drugs with different mechanisms of action are administered simultaneously. However, emerging resistance of cells, known as multidrug resistance (MDR), has become an increasing issue for effective cancer treatment.1 A well-established mechanism of resistance is associated with an overexpression of ATP-binding cassette (ABC) transporters, including P-glycoprotein (P-gp/ABCB1), multidrug resistance protein 1 (MRP1/ABCC1), and breast cancer resistance protein (BCRP/ABCG2). Several anticancer drugs are substrates of these proteins and are exported from the tumor cells, which prevents the drugs from exerting their intracellular cytotoxic effects.2,3 Strategies to increase the efficacy of cancer chemotherapy involve the inhibition or modulation of such cellular efflux pumps. Even though the role of these pumps in drug resistance has been investigated for decades,4−6 ABCG2 transporters have only recently received increasing attention with a desire to further understand their structure,7,8 function,9,10 and molecular pharmacology.11
A class of compounds that are able to interact with ABC transporters are chalcones. These chemical scaffolds are present in various biomolecules and are synthetically accessible.12 Chalcone-based compounds exert a variety of biological effects including anticancer, anti-inflammatory, chemopreventive, antioxidant, and antimicrobial effects.13 Compounds, containing this scaffold, have been developed to inhibit the transport activity of ABCcassette proteins, such as P-gp. Further developments have resulted in compounds that selectively engage with ABCG2.14
Structural investigations of chalcones have shown that the methoxy and hydroxy substituents at positions 2′ and 4′ on ring A (see Figure 1; general chalcone structure and compound 2) are crucial for activity.12,13 Recent structure–activity relationship studies14 revealed that the 2′-OH-4′,6′-dimethoxyphenyl group (A-ring) could be replaced by either a 2′-naphthyl group or a 3′,4′-methylenedioxyphenyl moiety. It was found that at least two methoxy groups on the B-ring are necessary for optimal inhibition. Additionally, substitution at positions 3, 4, and 5 (B-ring) improved cytotoxicity, while the presence of a large O-benzyl substituent at position 4 and a 2′-naphthyl substituent (A-ring) decreased cytotoxicity.14 Recently published chalcone-based ABCG2 modulators are shown in Figure 1.15,16
Figure 1.

General chalcone structure and chalcones, recently reported as ABCG2modulators.15−19
A new class of potent and selective ABCG2 inhibitors are the tariquidar analogues.17−19 These compounds were developed to investigate the functional role of ABCG2; however, their susceptibility to hydrolysis limits their use in biochemical and biological studies. In search for more stable analogues, we propose a bioisosteric approach for developing new tariquidar-like compounds. Two series of compounds were explored in this study–chalcones and ketones. The design of the former series involved incorporation of tariquidar’s key structural features necessary for ABCG2 inhibition. The latter series was designed to have a reduced molecular weight and more drug-like properties by replacing the 1,4-ethoxystyryl moiety by a methylene group. We hypothesized that this may reduce the compound toxicity. All compounds were evaluated for ABCG2, ABCB1, and ABCC1 modulatory activity by fluorescence based assays (an increase in cell-associated fluorescence is indicative of a decrease in transporter-mediated efflux) as well as for cytotoxicity.
As shown in Scheme 1, amines 12a–b were prepared from commercially available acetophenone via an electrophilic aromatic substitution, followed by a Bechamp reduction.20 The amides 9a–b were obtained by reaction with quinaldic acid, p-toluenesulfonyl chloride (Ts-Cl), and the amines (8a–b). Compound 10 was prepared according to known procedures.21
Scheme 1. Synthesis of Compounds 16–17a–f.

Reagents and conditions: (i) Quinaldic acid, Ts-Cl, TEA, 50 °C, overnight; (ii) NaOH 30%, rt, 24 h; (iii) NBS, methanol, reflux, 2 h; (iv) (a) 2-methoxyethyl 4-methylbenzenesulfonate or 2-(2-(2-methoxyethoxy)ethoxy)ethyl 4-methylbenzenesulfonate, KOH, THF, N2, 6 h; (b) DCM, trifluoroacetic acid, rt, overnight; (v) 15a–b or 16a–b, K2CO3, acetonitrile, 90 °C, overnight.
A Claisen–Schmidt condensation of the ketones 9a–b with the aldehyde 10 resulted in the desired protected chalcones 11a–b, while compounds 12a–-b were obtained by a bromination with N-bromosuccinimide. Tetrahydroisoquinoline 14 was obtained commercially as a hydrochloride salt. The Boc-protected tetrahydroisoquinoline 13 was synthesized as previously described.18 This compound was reacted with 2-methoxyethyl 4-methylbenzenesulfonate or 2-(2-(2-methoxyethoxy)ethoxy)ethyl 4-methylbenzenesulfonate and subsequently deprotected to produce the desired compounds as trifluoroacetic salts (15a–b). Finally, a nucleophilic substitution between the tosylated compounds (11a–b) or brominated compounds (12a–b) and the respective tetrahydroisoquinolines (14, 15a–b) led to the target compounds (16a–f and 17a–f, Scheme 1) in moderate to good yields (54–92%, Scheme 1). All synthesized compounds were characterized by nuclear magnetic resonance (NMR), infrared spectroscopy (IR), and high resolution mass spectrometry (HR-MS), and the purity was confirmed by analytical high performance liquid chromatography (HPLC).
The synthesized compounds 16a–f and 17a–f and the reference compounds fumitremorgin C (FTC) and tariquidar were investigated in a Hoechst 33342 microplate assay22 using ABCG2-overexpressing MCF-7/Topo cells. Relative IC50 values,23 i.e. the concentration corresponding to the response midway between the lower and the upper plateau of a full concentration–response curve, relative to the maximal response achieved by each individual compound, were calculated according to a four parameter curve fit (GraphPad Prism 7). Relative IC50 values are summarized in Table 1. Concentration response curves of selected compounds obtained in the Hoechst 33342 assay are shown in Figure 2.
Table 1. ABCG2 Inhibitory Activity in Comparison to Reference Compounds, Chalcones 16a–f, and Ketones 17a–f.
| ABCG2a |
|||
|---|---|---|---|
| compd | IC50 (μM)b | Imax (%)c | clogPd |
| FTC | 0.731 ± 0.092 | 100 | 1.23 |
| Tariquidar | 0.526 ± 0.085 | 69 ± 5 | 6.38 |
| 16a | 4.08 ± 0.29 | 79 ± 4 | 6.51 |
| 16b | 2.13 ± 0.47 | 72 ± 8 | 6.18 |
| 16c | 2.83 ± 0.30 | 81 ± 5 | 5.46 |
| 16d | 0.88 ± 0.02 | 86 ± 10 | 6.97 |
| 16e | 1.55 ± 0.36 | 97 ± 3 | 6.64 |
| 16f | 2.72 ± 0.54 | 111 ± 4 | 5.92 |
| 17a | 6.73 ± 1.02 | 77 ± 1 | 4.45 |
| 17b | 14.39 ± 11.8 | 55 ± 2 | 4.12 |
| 17c | 1.13 ± 0.27 | 47 ± 1 | 3.40 |
| 17d | 8.76 ± 1.59 | 77 ± 1 | 4.91 |
| 17e | 23.24 ± 4.36 | 79 ± 1 | 4.58 |
| 17f | 6.65 ± 1.27 | 86 ± 2 | 3.86 |
Hoechst 33342 microplate assay using ABCG2-overexpressing MCF-7/Topo cells.
Relative IC50 value23calculated using GraphPad Prism 7 four parameter curve fitting and presented as mean values ± SEM from three independent experiments performed in triplicate.
Maximal inhibitory effects (%) are expressed as inhibition caused by the highest concentration (100 μM) of the compound, relative to the inhibitory effect caused by 10 μM FTC (100% inhibition).
Calculated values using ACD/Laboratories I-Lab 2.0 ilab.acdlabs, Algorithm Version: v5.0.0.184.
Figure 2.

Concentration dependent inhibition of ABCG2 mediated Hoechst 33342 efflux by chalcones 16a–f (A) and ketones 17a–f (B) using MCF-7/Topo cells; inhibition relative to 10 μM FTC (100%).
All chalcones (16a–f) exhibited maximal inhibitory activity between 72 and 111% (Table 1), indicating an ABCG2 related decrease in Hoechst 33342 efflux. Compounds with a methyl substituent at the amino chalcone core (16d–f) were superior in potency and maximal inhibitory effect than compounds bearing a hydrogen atom (16a–c). This suggests an importance in the substitution pattern at this particular position. Compound 16d (UR-DP48) showed similar potency to the reference compound FTC (Figure 2). Moreover, the maximal inhibitory effects of these compounds (16d–f, 86%, 97%, and 111%, respectively) were higher than the effects of tariquidar and even surpassed that of FTC. The insertion of ethylene and triethylene glycol chains (R2 in the structure, Scheme 1) at the tetrahydroisoquinoline moiety in compounds 16a–c had no effect on the IC50 or Imax values (Table 1). In contrast, the maximal inhibitory effect achieved by the compounds 16d–f increased from 86 to 111% (Table 1).
The partition coefficient (log P) is a measure of the differential solubility of a compound in a hydrophobic solvent (octanol) and a hydrophilic solvent (water). The logarithm of these two values enables compounds to be ranked in terms of hydrophilicity (or hydrophobicity).24 As shown in Table 1, compounds bearing a methyl substituent at the amino chalcone core (16d–f) exhibited a higher calculated partition coefficient, and so, were more lipophilic, in comparison to analogs with hydrogen as a substituent at the same position (16a–c). These compounds also had the highest values for the maximal inhibition of Hoechst 33342 efflux. Comparing compounds 16a and 16d, the maximal inhibition for the most lipophilic compound 16d (86%) was higher and the IC50 value was almost 5 times lower (0.88 μM, Table 1). Chalcones 16e and 16f showed a considerable increase in maximal inhibitory effect (97% and 111%) in comparison to compound 16b and 16c (72% and 81%). With the highest partition coefficient (log P) among all synthesized compounds, chalcone 16d (UR-DP48) was the most potent inhibitor in the series, with similar activity to the reference compound (FTC).
By contrast, all ketones (17a–f) exhibited lower modulatory activities (47–86% maximal inhibition of Hoechst 33342 efflux) in comparison to the chalcone series (16a–f). The synthesized ketones were found to have lower partition coefficients compared to the chalcones (3.40–4.91 vs 5.46–6.97), which could explain the reduction in ABCG2 modulation (Table 1). As ABCG2 is located in the cell membrane, compound lipophilicity may be essential for modulation. In addition, recently published cryo-EM structural studies suggest that compounds can access one binding pocket of ABCG2 via a hydrophobic membrane entrance from the lipid bilayer.8 This reflects the importance of lipophilicity, which should be considered when designing ABCG2 modulators. In accordance to this hypothesis, the two most hydrophilic compounds in the ketone series (17b and 17c) showed the lowest maximal inhibitory effect. Interestingly, compound 17f, which also had a low calculated log P value, showed the highest maximal inhibition in the Hoechst 33342 accumulation assay. This may emphasize the importance of a methyl group at the R1 position in the central phenyl core.
In summary, the results of the Hoechst assay illustrated that the synthesized chalcones are more potent and efficacious in ABCG2 modulation than the ketones. Additionally, similar potencies were observed for chalcone 16d (IC50 0.880 μM) and the previously reported ABCG2 modulators 2 (IC50 0.75 μM), 4 (IC50 1.4 μM), 5 (IC50 0.94 μM), and 6 (IC50 0.501 μM) described in Figure 1.
Compound modulatory activities for ABCB1 and ABCC1 were screened in a calcein accumulation assay, using tariquidar (10 μM) and reversan (30 μM) as reference compounds. Tariquidar has been reported as a potent and specific inhibitor of ABCB1 (P-gp); therefore, it is commonly used as a reference compound for this transporter inhibition assay.25,26 Reversan is a potent ABCC1 modulator.27−29 Screening was conducted at a fixed concentration of 1 μM and 10 μM using Kb-V1 (ABCB1) and MDCKII (ABCC1) cell lines (Figure 3). Concentration–response curves were not constructed, as none of the compounds showed more than 25% inhibition of calcein-AM efflux (ABCB1 and ABCC1) at 10 μM. The results from the calcein accumulation assay suggested that the chalcones (16a–f) and ketones (17a–f) had a low inhibitory activity against ABCB1 and ABCC1 (values below 25%, Figure 3A, red and green bar respectively).
Figure 3.

Comparison of ABC transporter mediated efflux inhibition by 10 μM of 16a–f (A) and 17a–f (B) on MCF-7/Topo cells (ABCG2; blue), KbV1 cells (ABCB1; red), and MDCKII/MRP1 cell (ABCC1; green); the accumulation of Hoechst 33342 (ABCG2) and calcein (ABCB1 and ABCC1) was measured relative to 10 μM FTC (ABCG2), 10 μM tariquidar (ABCB1), and 30 μM reversan, respectively (100%).
To evaluate the chemical stability, all synthesized chalcones and ketones were incubated in assay medium (DMEM supplemented with 10% fetal calf serum) at 37 °C. Aliquots were then analyzed by HPLC over a period of 24 h. Products of cleavage and degradation of compounds were not detected (see the Supporting Information, pp 62–63), indicating that all compounds were not affected by the components present in the assay medium.
Finally, the effect of the modulators 16d and 17f on the proliferation of ABCG2-overexpressing MCF-7/Topo cells was investigated by means of a kinetic chemosensitivity assay (Figure 4).30 The cytostatic drug vinblastine was used as a positive control.
Figure 4.
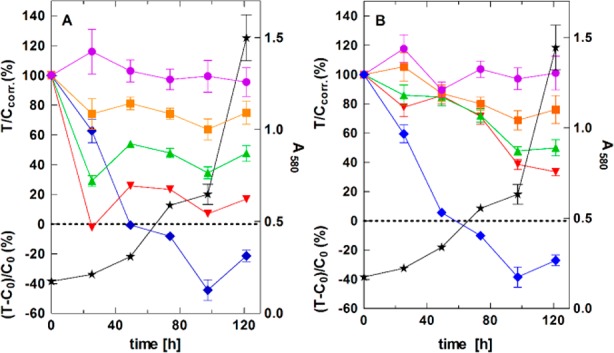
Concentration dependent effect of 16d (UR-DP48) (A) and 17f (B) on proliferating MCF-7/Topo cells upon long-term exposure; 100 nM (purple circles); 1 μM (orange squares); 3 μM (green triangles); 10 μM (red inverted triangles); positive control: 100 nM vinblastine (blue diamonds); growth curve of vehicle (DMSO) treated cells (negative control) (black stars).
Compound 16d showed a concentration dependent cytostatic effect on proliferating MCF-7/Topo cells, which may be due to the reactive α–β-unsaturated ketone core.31 In accordance with the working hypothesis, cytotoxicity was reduced in the case of ketone 17f, which is lacking the Michael acceptor system.
In summary, new types of tariquidar-related chalcone and ketone derivatives were synthesized and investigated for their inhibitory effect and selectivity toward ABCG2. All chalcones were selective for ABCG2 inhibition. Compounds bearing a methyl substituent at the A ring (16d–f) were most active and potent (86%, 97%, and 111%, respectively). Compound 16d showed similar potency to the reference compound (FTC) with an IC50 value of 0.88 μM in the Hoechst assay and a maximal inhibitory effect of 86%. The compounds of the ketone series showed none or only very small preference for ABCG2 over ABCB1 and ABCC1. Compounds 17d–f surpassed the maximal inhibitory effect of tariquidar although requiring higher concentrations. The difference in ABCG2 modulation and selectivity when comparing the two series of compounds indicated that the lipophilicity plays a critical role. The chemosensitivity assay demonstrated antiproliferative activity of the synthesized chalcones, whereas cytotoxicity was less pronounced in the ketone series. With further optimization, chalcone 16d, displaying pronounced ABCG2 modulating potential with a submicromolar IC50 value, is a promising lead to aid in future investigations of ABCG2 to further understand it́s role in cancer drug resistance.
Acknowledgments
D.P.-S thanks COLCIENCIAS for a graduate fellowship, and C.O.-P thanks COLCIENCIAS, the German Academic Exchange Service (DAAD), and Universidad Nacional de Colombia for providing financial support. We thank Ms. Ranit Lahmy for proof reading the manuscript.
Glossary
Abbreviations
- ABCB1
ATP-binding cassette transporter, subfamily B, member 1
- ABCC1
ATP-binding cassette transporter, subfamily C, member 1
- ABCG2
ATP-binding cassette transporter, subfamily G, member 2
- BCRP
breast cancer resistance protein (= ABCG2)
- IC50
concentration of inhibitor required to give 50% inhibition of activity
- MDR
multidrug resistance
- MRP1
multidrug resistance associated protein 1 (= ABCC1)
- p-gp
pglycoprotein (= ABCB1)
- SEM
standard error of the mean.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00289.
Chemical synthesis, characterization of target compounds; and protocols of biological assays (PDF)
The authors declare no competing financial interest.
Author Status
∥ Died July 18, 2017.
Supplementary Material
References
- Horsey A. J.; Cox M. H.; Sarwat S.; Kerr I. D. The multidrug transporter ABCG2: still more questions than answers. Biochem. Soc. Trans. 2016, 44 (3), 824–830. 10.1042/BST20160014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhuri S.; Klaassen C. D. Structure, Function, Epression, Genomic Organization, and Single Nucleotide Polymorphisms of Human ABCB1 (MDR1), ABCC (MRP), and ABCG2 (BCRP) Efflux Transporters. Int. J. Toxicol. 2006, 25 (4), 231–259. 10.1080/10915810600746023. [DOI] [PubMed] [Google Scholar]
- Staud F.; Pavek P. Breast Cancer Resistance Protein (BCRP/ABCG2). Int. J. Biochem. Cell Biol. 2005, 37, 720–725. 10.1016/j.biocel.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Pleban K.; Ecker G. F. Inhibitors of P-Glycoprotein - Lead Identification and Optimisation. Mini-Rev. Med. Chem. 2005, 5, 153–163. 10.2174/1389557053402729. [DOI] [PubMed] [Google Scholar]
- Boumendjel A.; Baubichon-Cortay H.; Trompier D.; Perrotton T.; Di Pietro A. Anticancer multidrug resistance mediated by MRP1: Recent advances in the discovery of reversal agents. Med. Res. Rev. 2005, 25 (4), 453–472. 10.1002/med.20032. [DOI] [PubMed] [Google Scholar]
- Sandor V.; Fojo T.; Bates S. E. Future perspectives for the development of P-glycoprotein modulators. Drug Resist. Updates 1998, 1 (3), 190–200. 10.1016/S1368-7646(98)80039-3. [DOI] [PubMed] [Google Scholar]
- Taylor N. M. I.; Manolaridis I.; Jackson S. M.; Kowal J.; Stahlberg H.; Locher K. P. Structure of the human multidrug transporter ABCG2. Nature 2017, 546, 504. 10.1038/nature22345. [DOI] [PubMed] [Google Scholar]
- Jackson S. M.; Manolaridis I.; Kowal J.; Zechner M.; Taylor N. M. I.; Bause M.; Bauer S.; Bartholomaeus R.; Bernhardt G.; Koenig B.; Buschauer A.; Stahlberg H.; Altmann K.-H.; Locher K. P. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25 (4), 333–340. 10.1038/s41594-018-0049-1. [DOI] [PubMed] [Google Scholar]
- Polgar O.; Robey R. W.; Bates S. E. ABCG2: structure, function and role in drug response. Expert Opin. Drug Metab. Toxicol. 2008, 4 (1), 1–15. 10.1517/17425255.4.1.1. [DOI] [PubMed] [Google Scholar]
- Ferreira R. J.; Bonito C. A.; Cordeiro M. N. D. S.; Ferreira M.-J. U.; dos Santos D. J. V. A. Structure-function relationships in ABCG2: insights from molecular dynamics simulations and molecular docking studies. Sci. Rep. 2017, 7 (1), 15534. 10.1038/s41598-017-15452-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacy A. E.; Jansson P. J.; Richardson D. R. Molecular pharmacology of ABCG2 and its role in chemoresistance. Mol. Pharmacol. 2013, 84 (5), 655–669. 10.1124/mol.113.088609. [DOI] [PubMed] [Google Scholar]
- Valdameri G.; Gauthier C.; Terreux R. l.; Kachadourian R. m.; Day B. J.; Winnischofer S. M.; Rocha M. E.; Frachet V. r.; Ronot X.; Di Pietro A. Investigation of chalcones as selective inhibitors of the breast cancer resistance protein: critical role of methoxylation in both inhibition potency and cytotoxicity. J. Med. Chem. 2012, 55 (7), 3193–3200. 10.1021/jm2016528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juvale K.; Pape V. F.; Wiese M. Investigation of chalcones and benzochalcones as inhibitors of breast cancer resistance protein. Bioorg. Med. Chem. 2012, 20 (1), 346–355. 10.1016/j.bmc.2011.10.074. [DOI] [PubMed] [Google Scholar]
- Rangel L. P.; Winter E.; Gauthier C.; Terreux R.; Chiaradia-Delatorre L. D.; Mascarello A.; Nunes R. J.; Yunes R. A.; Creczynski-Pasa T. B.; Macalou S. New structure–activity relationships of chalcone inhibitors of breast cancer resistance protein: polyspecificity toward inhibition and critical substitutions against cytotoxicity. Drug Des., Dev. Ther. 2013, 7, 1043. 10.2147/DDDT.S46983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter E.; Gozzi G. J.; Chiaradia-Delatorre L. D.; Daflon-Yunes N.; Terreux R.; Gauthier C.; Mascarello A.; Leal P. C.; Cadena S. M.; Yunes R. A. Quinoxaline-substituted chalcones as new inhibitors of breast cancer resistance protein ABCG2: polyspecificity at B-ring position. Drug Des., Dev. Ther. 2014, 8, 609. 10.2147/DDDT.S56625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraege S.; Köhler S. C.; Wiese M. Acryloylphenylcarboxamides: A New Class of Breast Cancer Resistance Protein (ABCG2) Modulators. ChemMedChem 2016, 11 (21), 2422–2435. 10.1002/cmdc.201600341. [DOI] [PubMed] [Google Scholar]
- Puentes C. O.; Höcherl P.; Kühnle M.; Bauer S.; Bürger K.; Bernhardt G.; Buschauer A.; König B. Solid phase synthesis of tariquidar-related modulators of ABC transporters preferring breast cancer resistance protein (ABCG2). Bioorg. Med. Chem. Lett. 2011, 21 (12), 3654–3657. 10.1016/j.bmcl.2011.04.094. [DOI] [PubMed] [Google Scholar]
- Ochoa-Puentes C.; Bauer S.; Kühnle M.; Bernhardt G. n.; Buschauer A.; König B. Benzanilide–Biphenyl Replacement: A Bioisosteric Approach to Quinoline Carboxamide-Type ABCG2Modulators. ACS Med. Chem. Lett. 2013, 4 (4), 393–396. 10.1021/ml4000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer S.; Ochoa-Puentes C.; Sun Q.; Bause M.; Bernhardt G.; König B.; Buschauer A.. Quinoline Carboxamide-Type ABCG2Modulators: Indole and Quinoline Moieties as Anilide Replacements. ChemMedChem 2013, 8, 1773. 10.1002/cmdc.201300319 [DOI] [PubMed] [Google Scholar]
- Gamble A. B.; Garner J.; Gordon C. P.; O’Conner S. M. J.; Keller P. A. Aryl Nitro Reduction with Iron Powder or Stannous Chloride under Ultrasonic Irradiation. Synth. Commun. 2007, 37 (16), 2777–2786. 10.1080/00397910701481195. [DOI] [Google Scholar]
- Watanabe H.; Ono M.; Kimura H.; Kagawa S.; Nishii R.; Fuchigami T.; Haratake M.; Nakayama M.; Saji H. A dual fluorinated and iodinated radiotracer for PET and SPECT imaging of β-amyloid plaques in the brain. Bioorg. Med. Chem. Lett. 2011, 21 (21), 6519–6522. 10.1016/j.bmcl.2011.08.063. [DOI] [PubMed] [Google Scholar]
- Kühnle M.Institut für Pharmazie, Universität Regensburg, 2010. [Google Scholar]
- Sebaugh J. L. Guidelines for accurate EC50/IC50 estimation. Pharmaceutical Statistics 2011, 10 (2), 128–134. 10.1002/pst.426. [DOI] [PubMed] [Google Scholar]
- Savjani K. T.; Gajjar A. K.; Savjani J. K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 195727. 10.5402/2012/195727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry P.; Stewart A. J.; Dangerfield W.; Okiji S.; Liddle C.; Bootle D.; Plumb J. A.; Templeton D.; Charlton P. In Vitro and in Vivo Reversal of P-Glycoprotein-mediated Multidrug Resistance by a Novel Potent Modulator, XR9576. Cancer Res. 2001, 61, 749–758. [PubMed] [Google Scholar]
- Kuehnle M.; Egger M.; Mueller C.; Mahringer A.; Bernhardt G.; Fricker G.; Koenig B.; Buschauer A., Potent and Selective Inhibitors of Breast Cancer Resistance Protein (ABCG2) Derived from the p-Glycoprotein (ABCB1) Modulator Tariquidar. J. Med. Chem. 2009, 52, 1190–1197. 10.1021/jm8013822 [DOI] [PubMed] [Google Scholar]
- Obreque-Balboa J. E.; Sun Q.; Bernhardt G.; König B.; Buschauer A. Flavonoid derivatives as selective ABCC1 modulators: Synthesis and functional characterization. Eur. J. Med. Chem. 2016, 109, 124–133. 10.1016/j.ejmech.2015.12.010. [DOI] [PubMed] [Google Scholar]
- Burkhart C.; Murray J.; Isachenko N.; Pajic M.; Watt F.; Flemming C.; Smith J.; Gurova K.; Marshall G.; Norris M.; Gudkov A.; Haber M. Reversan, a novel inhibitor of MRP1, increases the therapeutic index of conventional chemotherapeutic agents. Cancer Res. 2008, 68 (9 Supplement), 3242–3242. [Google Scholar]
- Burkhart C. A.; Watt F.; Murray J.; Pajic M.; Prokvolit A.; Xue C.; Flemming C.; Smith J.; Purmal A.; Isachenko N.; Komarov P. G.; Gurova K. V.; Sartorelli A. C.; Marshall G. M.; Norris M. D.; Gudkov A. V.; Haber M. Small Molecule MRP1 Inhibitor Reversan Increases the Therapeutic Index of Chemotherapy in Mouse Model of Neuroblastoma. Cancer Res. 2009, 69 (16), 6573–6580. 10.1158/0008-5472.CAN-09-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt G.; Reile H.; Birnböck H.; Spruß T.; Schönenberger H. Standardized kinetic microassay to quantify differential chemosensitivity on the basis of proliferative activity. J. Cancer Res. Clin. Oncol. 1992, 118 (1), 35–43. 10.1007/BF01192309. [DOI] [PubMed] [Google Scholar]
- Zhuang C.; Zhang W.; Sheng C.; Zhang W.; Xing C.; Miao Z. Chalcone: a privileged structure in medicinal chemistry. Chem. Rev. 2017, 117 (12), 7762–7810. 10.1021/acs.chemrev.7b00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.