

Abstract
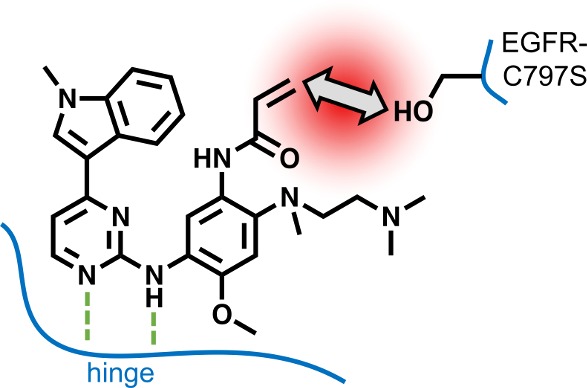
The first evidence of osimertinib resistance mediated by the epidermal growth factor receptor (EGFR) mutation C797S was reported three years ago. Since then, no major breakthroughs have been achieved to target the clinically relevant mutant variant that impedes covalent bond formation with irreversible EGFR inhibitors. Although several biochemically active compounds have been described, only a few inhibitors that potently act on the cellular level or in vivo have been introduced so far. Herein, we give an overview of current approaches in the field and highlight the challenges that need to be addressed in future research projects to overcome the C797S-mediated drug resistance.
EGFR-C797S Mutation Impedes Covalent Inhibitor Binding
In recent years, a variety of inhibitors for oncogenic and drug-resistant EGFR-mutations have been described. Most of these inhibitors, such as osimertinib (1), were designed to address mutant variants of EGFR in a covalent manner, exploiting a reactive cysteine in the binding site. However, a common mechanism of acquired drug resistance toward covalent inhibitors emerged, the C797S mutation of the covalent anchor point to a less reactive serine residue (Figure 1B).1
Figure 1.
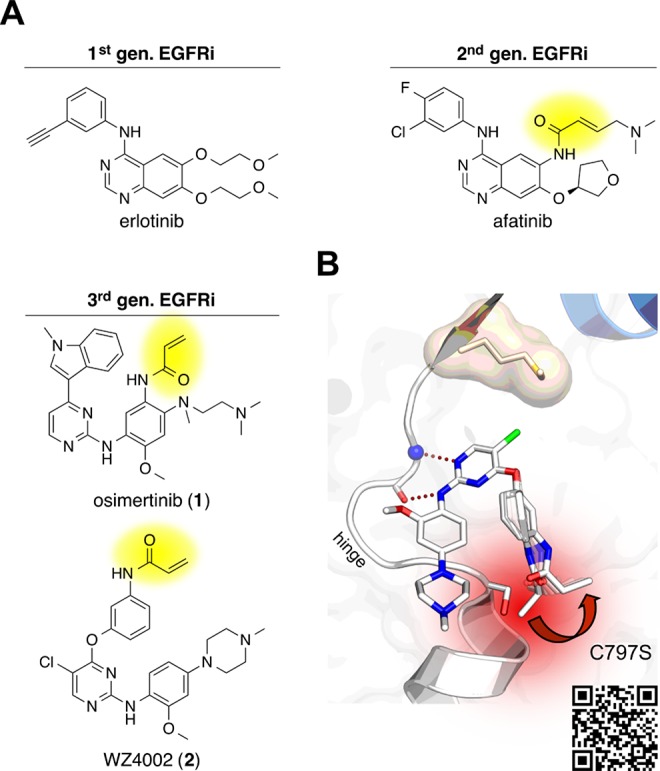
(A) Chemical structures of first, second, and third generation EGFR inhibitors. The acrylamide warhead is highlighted in yellow. (B) Structural analysis of EGFR-C797S that impedes covalent bond formation with irreversible inhibitors (model based on PDB IDs: 3IKA, 5X2K). The QR-code provides an augmented reality view of a 3D model of the binding site.
Must the Binding Affinity Be Improved To Target EGFR-C797S?
In 2017, Uchibori et al. showed that the ALK-inhibitor brigatinib (3) could be repurposed to effectively reduce the viability of EGFR-C797S mutated cell lines. However, sufficient plasma levels to effectively inhibit EGFR-C797S in vivo could not be achieved, so a combination treatment with EGFR-targeted antibodies, such as cetuximab, was required to achieve marked effects in the mouse animal models.2 Based on the inhibitory effect of brigatinib and other ALK inhibitors against various EGFR mutants, the development of ALK/EGFR dual inhibitors was pursued. Chen et al. combined the features of the ALK inhibitor TAE684 (4) with an acrylamide warhead for covalent modification of Cys797 to gain the potent inhibitor 5. It displayed a good inhibition profile for several ALK and EGFR mutants but did not reveal convincing inhibition in multidrug-resistant EGFR-Ba/F3-cells.3 Kong et al. proposed a more hydrophilic character of the Ser797 that could lead to repulsion with the acrylamide moiety.4 Another compound series by Romu et al. was based on analogues of the aminopyrimidine WZ4002 (2). They discovered that the change from meta- to ortho-substituted amides 6 improved the inhibition against C797S-mutated EGFR.5 If we compare these series of compounds, it is striking that they all show a substitution in the ortho-position with phosphine oxides, sulfones, or amide moieties (Figure 2A, highlighted in yellow). Structural analysis revealed that these groups allow a strong interaction with the catalytic Lys745 side chain (Figure 2A). Thus, the interaction with the catalytic lysine and an increase in reversible binding affinity may be an important driver for inhibition of C797S-mutated EGFR and may provide a tool for the development of next-generation inhibitors. In accordance with this idea, Engel and Becker et al. showed that an increased degree of reversible binding is a crucial feature to target the C797S mutant variant.6 This factor was further exploited and validated by the work of Laufer and colleagues with inhibitors such as the pyridinyl imidazole derivatives 7–8, which bear alcohol moieties that form additional interactions within the phosphate binding site (Figure 2B, highlighted in orange). However, these compounds have only been characterized biochemically and, because of the bulky fluorophenyl moiety, these inhibitors possessed EGFR wild-type toxicity, which must be avoided in the clinical setting.7,8
Figure 2.

Binding modes and chemical structures of inhibitors targeting the C797S mutant EGFR. (A) Aminopyrimidine-based ALK and EGFR/ALK dual inhibitors 3–6. Interactions with the catalytic lysine are highlighted in yellow (PDB ID: 5GTZ). (B) Aminopyridinyl imidazole-based inhibitors 7–8. Interaction with the phosphate binding site is highlighted in orange (covalent docking results). (C) Aminopurine-based inhibitor 9. Interaction with the hydrophobic clamp motif is highlighted in green (PDB ID: 5X2C). (D) Phenol-based inhibitor 10 identified through virtual screening. Interaction with the Ser797 side chain is highlighted in red (docking results). (E) Allosteric inhibitor 11 occupies an allosteric binding site, highlighted in blue (PDB ID: 5D41).
The reversible binding of inhibitors was further approached through the exploitation of both the hydrophobic pocket adjacent to the gatekeeper residue and a hydrophobic clamp motif within the ATP-binding site. Therefore, Zhu et al. developed reversible purine derivatives that again showed structural similarity to the aminopyrimidine WZ4002 (2). Compound 9 was the lead compound with a cyclopentyl residue that interacts with Leu718, Val726, and Leu844, the aforementioned hydrophobic clamp (Figure 2C, highlighted in green), while the aniline moiety extends to the back pocket. Existing EGFR inhibitors do not fully exploit the clamp motif, which could provide another instrument to improve the binding affinity of reversible inhibitors. Of note, introduction of the C797S mutation reduced the activity of the reversible inhibitor 9 as well.9 We believe that the C797S mutant target kinase retains some biological secrets, and gaining a deeper understanding of its molecular features is crucial to develop the next generation of EGFR inhibitors.
Historically, the first EGFR inhibitors erlotinib and gefitinib showed cellular efficacy on the L858R mutant due to the decreased ATP affinity of the mutated kinase. One of the reasons contributing to resistance against these first generation inhibitors is the restored ATP-affinity with the acquired T790M gatekeeper drug-resistance mutation. This turn of events resulted in the development of covalent irreversible second and third generation inhibitors (Figure 1A) where the competition for the binding pocket can be shifted in favor of the inhibitor. Analysis of T790M/C797S mutated EGFR revealed a comparable ATP affinity as compared to the T790M mutant variant.4 This brings up the question whether an ATP-competitive inhibitor can provide sufficient potency via strong reversible binding and favorable pharmacologic properties when covalent bond formation with Cys797 is abolished.
There Is an Urgent Need for Novel Scaffolds and Approaches
Thus far, we have learned that drug resistant mutants are not just mutant variants of the target protein but should be considered as novel targets, and to effectively address these targets, innovative approaches and new chemical entities must be developed. In light of the strategies that increase the reversible binding character of the described inhibitors 3–6, which are based on aminopyrimidines, and 7–9, which are more or less derived from this scaffold, we propose the development of novel scaffolds that inhibit EGFR-C797S, exhibit novel features, and provide a highly reversible binding affinity toward the binding site.
One example for the development of new scaffolds is the work of Park et al. A virtual de novo design approach was used to identify 2-aryl-4-aminoquinazolines as a potent and mutant-selective scaffold for del19/T790M/C797S mutated EGFR. The most potent inhibitor 10 showed a novel hinge-binding motif exploiting a phenolic hydroxyl group to form two hydrogen bonds. The aminomethylpyrazine adds affinity by forming two hydrogen bonds with Ser797 (Figure 2A, highlighted in red), which was analogously observed with similar compounds crystallized with the Pim-1 kinase (PDB ID: 3UIX). To date, these scaffolds lack the proof of cellular activity;10 nonetheless, the described features might stimulate further research and the development of new inhibitors for C797S-mutated EGFR.
An innovative approach was introduced by Jia et al., who reported the first allosteric inhibitor EAI045 (11) that overcame the EGFR-C797S. The inhibitor exploited an allosteric pocket (Figure 2E, highlighted in blue) emerging with the inactive kinase conformation and exhibited low nanomolar activity with selectivity over wild-type EGFR in biochemical assays. However, an effect at the cellular level or in vivo is achievable only in combination with the antibody cetuximab.11 Recently, the widespread PROTAC/dTAG system has been utilized in this context by Gray and his group, who patented bifunctional hybrid compounds consisting of EAI045 and a thalidomide tag, which induced proteasomal degradation of the kinase. These compounds show promising cellular efficacy as single drugs; however, the transfer of the PROTAC/dTAG system into clinical applications has yet to be performed. Furthermore, the EAI045-based approaches are limited to the L858R-activating mutation and show no activity on del19-mutated EGFR,12 which is the predominant activating mutation found in osimertinib-resistant patients.
There Is Still a Long Journey Ahead
Drug resistance represents a major challenge in targeted cancer therapy and has been foreseen to arise with third generation inhibitors. The success of covalent inhibitors targeting the gatekeeper mutant EGFR-T790M gave way to the advent of acquired resistance toward these drugs, which was a great misfortune for affected patients. Even worse, despite some interesting approaches to address the C797S mutation that have been followed over the last years, there is still an unmet medical need for an efficacious inhibitor to treat osimertinib-resistant tumors. Several approaches have focused on improving the reversible binding affinity of established aminopyrimidine-based inhibitors, but because of the restored competition with cellular ATP for the binding site, these compounds have thus far failed as single agents in cellular contexts or in vivo. Moreover, several reports lacked the proof of cellular efficacy, which is perplexing to scientists in the field and also highlights the absence of general access to meaningful cellular model systems. However, the emergence of T790M-mediated resistance a decade ago taught us that simply improving existing inhibitors did not counteract the acquired drug resistance. If the C797S mutant variant is druggable at all, we are certain that novel chemical structures must be developed and that innovative approaches are needed to yield next-generation EGFR inhibitors. Here, we have presented features that increase the reversible binding character, novel scaffolds that incorporate the mutant Ser797 side chain into the interaction pattern, and allosteric inhibitors that might be effectively combined with PROTACs. However, all of these approaches exhibited their limitations, and intensive research will be required to develop efficient drugs and to unravel the molecular and biochemical phenomena around the multimutated EGFR kinase. We will be excited to learn who is first off the mark—the patients count on us!
Glossary
ABBREVIATIONS
- ALK
anaplastic lymphoma kinase
- EGFR
epidermal growth factor receptor
- EGFRi
EGFR inhibitors
- PROTAC
protein-targeting chimeric molecules
- dTAG
degradation tag
Author Contributions
‡ These authors contributed equally. The manuscript was written through contributions of all authors.
Views expressed in this editorial are those of the authors and not necessarily the views of the ACS.
The authors declare no competing financial interest.
References
- Lu X.; Yu L.; Zhang Z.; Ren X.; Smaill J. B.; Ding K. Targeting EGFR(L858R/T790M) and EGFR(L858R/T790M/C797S) resistance mutations in NSCLC: Current developments in medicinal chemistry. Med. Res. Rev. 2018, 10.1002/med.21488. [DOI] [PubMed] [Google Scholar]
- Uchibori K.; Inase N.; Araki M.; Kamada M.; Sato S.; Okuno Y.; Fujita N.; Katayama R. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat. Commun. 2017, 8, 14768. 10.1038/ncomms14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Wu J.; Wang A.; Qi Z.; Jiang T.; Chen C.; Zou F.; Hu C.; Wang W.; Wu H.; Hu Z.; Wang W.; Wang B.; Wang L.; Ren T.; Zhang S.; Liu Q.; Liu J. Discovery of N-(5-((5-chloro-4-((2-(isopropylsulfonyl)phenyl)amino)pyrimidin-2-yl)amino)-4-met hoxy-2-(4-methyl-1,4-diazepan-1-yl)phenyl)acrylamide (CHMFL-ALK/EGFR-050) as a potent ALK/EGFR dual kinase inhibitor capable of overcoming a variety of ALK/EGFR associated drug resistant mutants in NSCLC. Eur. J. Med. Chem. 2017, 139, 674–697. 10.1016/j.ejmech.2017.08.035. [DOI] [PubMed] [Google Scholar]
- Kong L. L.; Ma R.; Yao M. Y.; Yan X. E.; Zhu S. J.; Zhao P.; Yun C. H. Structural pharmacological studies on EGFR T790M/C797S. Biochem. Biophys. Res. Commun. 2017, 488, 266–272. 10.1016/j.bbrc.2017.04.138. [DOI] [PubMed] [Google Scholar]
- Romu A. A.; Lei Z.; Zhou B.; Chen Z. S.; Korlipara V. Design, synthesis and biological evaluation of WZ4002 analogues as EGFR inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 4832–4837. 10.1016/j.bmcl.2017.09.048. [DOI] [PubMed] [Google Scholar]
- Engel J.; Becker C.; Lategahn J.; Keul M.; Ketzer J.; Mühlenberg T.; Kollipara L.; Schultz-Fademrecht C.; Zahedi R. P.; Bauer S.; Rauh D. Insight into the Inhibition of Drug-Resistant Mutants of the Receptor Tyrosine Kinase EGFR. Angew. Chem., Int. Ed. 2016, 55, 10909–10912. 10.1002/anie.201605011. [DOI] [PubMed] [Google Scholar]
- Günther M.; Juchum M.; Kelter G.; Fiebig H.; Laufer S. Lung Cancer: EGFR Inhibitors with Low Nanomolar Activity against a Therapy-Resistant L858R/T790M/C797S Mutant. Angew. Chem., Int. Ed. 2016, 55, 10890–10894. 10.1002/anie.201603736. [DOI] [PubMed] [Google Scholar]
- Günther M.; Lategahn J.; Juchum M.; Döring E.; Keul M.; Engel J.; Tumbrink H. L.; Rauh D.; Laufer S. Trisubstituted Pyridinylimidazoles as Potent Inhibitors of the Clinically Resistant L858R/T790M/C797S EGFR Mutant: Targeting of Both Hydrophobic Regions and the Phosphate Binding Site. J. Med. Chem. 2017, 60, 5613–5637. 10.1021/acs.jmedchem.7b00316. [DOI] [PubMed] [Google Scholar]
- Zhu S.-J.; Zhao P.; Yang J.; Ma R.; Yan X.-E.; Yang S.-Y.; Yang J.-W.; Yun C.-H. Structural insights into drug development strategy targeting EGFR T790M/C797S. Oncotarget 2018, 9, 13652–13665. 10.18632/oncotarget.24113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H.; Jung H. Y.; Mah S.; Hong S. Discovery of EGF Receptor Inhibitors That Are Selective for the d746–750/T790M/C797S Mutant through Structure-Based de Novo Design. Angew. Chem., Int. Ed. 2017, 56, 7634–7638. 10.1002/anie.201703389. [DOI] [PubMed] [Google Scholar]
- Jia Y.; Yun C. H.; Park E.; Ercan D.; Manuia M.; Juarez J.; Xu C.; Rhee K.; Chen T.; Zhang H.; Palakurthi S.; Jang J.; Lelais G.; DiDonato M.; Bursulaya B.; Michellys P. Y.; Epple R.; Marsilje T. H.; McNeill M.; Lu W.; Harris J.; Bender S.; Wong K. K.; Jänne P. A.; Eck M. J. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray N.; Jang J.; De-Clercq D.; Eck M.. Bifunctional Molecules For Degradation of EGFR and Methods of Use. WO2017185036A1, 2017.