

Abstract
Introduction:
Retinopathy remains as one of the most feared blinding complications of diabetes, and with the prevalence of this life-long disease escalating at an alarming rate, the incidence of retinopathy is also climbing. Although the cutting edge research has identified many molecular mechanisms associated with its development, the exact mechanism how diabetes damages the retina remains obscure, limiting therapeutic options for this devastating disease.
Areas covered:
This review focuses on the central role of mitochondrial dysfunction/damage in the pathogenesis of diabetic retinopathy, and how damaged mitochondria initiates a self-perpetuating vicious cycles of free radicals. We have also reviewed how mitochondria could serve as a therapeutic target, and the challenges associated with the complex double mitochondrial membranes and a well-defined blood-retinal barrier for optimal pharmacologic/molecular approach to improve mitochondrial function.
Expert opinion:
Mitochondrial dysfunction provides many therapeutic targets for ameliorating the development of diabetic retinopathy including their biogenesis, DNA damage and epigenetic modifications. New technology to enhance pharmaceuticals uptake inside the mitochondria, nanotechnology to deliver drugs to the retina, and maintenance of mitochondrial homeostasis via lifestyle changes and novel therapeutics to prevent epigenetic modifications, could serve as some of the welcoming avenues for a diabetic patient to target this sight-threatening disease.
Keywords: Diabetes, Diabetic retinopathy, Epigenetics, Mitochondria, Mitochondrial DNA, Reactive Oxygen Species, Retina
1. Introduction
Diabetes has now become as one of the main threats to the human health; number of people living with diabetes worldwide has escalated from 108 million in 1980 to a staggering 415 million in 2015, and by 2040 these numbers are expected to climb to 642 million. The disease has a major socioeconomic effect, it impacts physical and emotional well-being of a patient, and takes a heavy toll on national economy; the global annual cost of diabetes is now above 825 billion dollars (Harvard School of Public Health News Release, April 16, 2016). Sustained high circulating glucose, either due to lack of enough insulin production (type 1 diabetes), or poor use of the insulin produced by the body (type 2 diabetes), damages large (macrovascular) and small (microvascular) blood vessels, culminating in many chronic complications. Although the risk of most diabetes-related complications can be alleviated by maintaining good blood glucose, this becomes challenging for some diabetic patients for long durations, and some patients face many life-limiting and threatening complications including ischemic heart disease, retinopathy, nephropathy and neuropathy [1, 2].
Diabetes is considered as one of the major causes of vision loss, and it can affect the vision system in multiple ways. In diabetic retinopathy, damage to the blood vessels in the retina impairs retinal circulation, and if not controlled in the early stages, leads to retinal detachment and vision loss. Damage to the vasculature of the central retina, the macula, results in its swelling, leading to the loss of central vision. Diabetes also clouds the lens making the vision distorted, and damages the optic nerve increasing intraocular pressure [1]. Although all of these conditions threaten vision loss, diabetic retinopathy remains the leading cause of blindness in working-age adults. A staggering 28.5% diabetic patients aged 40 years and older have retinopathy; in 2010, 93 million people had diabetic retinopathy [3], and with the incidence of diabetes increasing at an alarming rate, 155 million are expected to have retinopathy in 2030. According to the World Health Organization, diabetic retinopathy accounts for 4.8% of the number of cases of blindness (37 million) worldwide. Within 5 years, 13% of type 1 and 25–40% type 2 diabetic patients have retinopathy, but after 15–20 years of diabetes (type 1 or type 2), ~85% patents end up showing some signs of retinopathy [1].
Diabetic retinopathy is a progressive disease, and the time of gnosis is considered as a pivotal moment in the patients’ lives. Clinically, it is associated with the presence of specific retinal microvascular signs, and is classified into two stages: during the early stages of the disease, the non-proliferative stage, microaneurysms and cotton-wool spots begin to appear, and due to weakening of the vasculature, they become leaky. As the disease progresses, the macula shows some swelling, and vascular damage in the macula results in loss of vision acuity. With the increase in duration and severity of non-proliferative retinopathy, ischemic regions begin to appear, and release of growth factors in these ischemic regions results in neovascularization. Leakage of blood from these new vessels into the vitreous clouds the vision, and if not controlled, disorganized neovascularization detaches the retina, leading to blindness [1]. Although the vasculature of the retina shows major histopathology, retinal neurodegeneration is also observed in diabetic patients, and both clinical and basic research has shown that retinal neurodegeneration precedes vascular pathology. Retinal electrophysiological dysfunction and the neuronal damage are seen in the early stage of the disease with significant thinning of the ganglion cell and outer nuclear layers [4]. Diabetic rodents and retinal cells in culture have also clearly documented that both neuronal and vascular cells of the retina undergo accelerated apoptosis before appearance of any clinical sign of diabetic retinopathy [5, 6].
Landmark studies have clearly identified hyperglycemia as the major factor in the development of diabetic retinopathy [7], but there are other systemic factors associated with this blinding disease including blood pressure and hyperlipidemia. Hypertension, via producing mechanical stretch, can dysfunction endothelial cells [8]. Though the role of hyperlipidemia in the development of diabetic retinopathy is still not clear, both animal (rats & mice) and in vitro (retinal cells) models have shown that hyperlipidemia exacerbates and accelerates retinal cell apoptosis and the development of diabetic retinopathy [9, 10], and dysregulation of sphingolipid metabolism is shown to play a major role in dyslipidemia-induced retinal damage in diabetes [11, 12]. Furthermore, increased neck and waist circumference is associated with both increased risk and higher severity of diabetic retinopathy, and physical activity provides beneficial effects [13]. Thus, overall systemic health of a diabetic patient has a major influence on the development/ progression of diabetic retinopathy.
The pathogenesis of a disease is also influenced by genetic factors; the risk of severe diabetic retinopathy is about 3-fold higher in siblings of affected individuals, but the severity of retinopathy, as evaluated by Early Treatment Diabetic Retinopathy Study Grading System, among diabetic patients with similar risk factors shows a varied range [14]. Many candidate genes including aldose reductase, vascular endothelial growth factor (VEGF), receptor for advanced glycation end products (RAGEs), endothelial nitric oxide synthase (eNOS) and paraoxonase1, have shown some association with the development of diabetic retinopathy. Polymorphisms at the regulatory regions of some of these genes are also evaluated as risk alleles for the susceptibility or progression of diabetic retinopathy [15]. A systematic meta-analysis of 20 candidate genes for diabetic retinopathy has presented the strongest evidence of association for the z-2 microsatellite and rs759853 on aldo-keto reductase family 1 member B, and some evidence for polymorphisms in NOS3, VEGF, intercellular adhesion molecule 1 genes. A follow up meta-analysis, however, failed to provide any reported loci after adjustment for multiple testing, raising the possibility of false positive results in the previous analyses [16]. Recent results from a Chinese type 2 diabetic cohort with proliferative diabetic retinopathy have shown the INSR gene as a possible susceptibility candidate for severe diabetic retinopathy, and have suggested the role of compensatory hyperinsulinemia in the pathogenesis of diabetic retinopathy [17], but the underpowered nature of the study fails to strengthen genetic associations. Thus, despite recent progress in defining the genetic basis, many challenges remain in understanding the genetic contribution to this sight-threatening complication.
2. Molecular mechanisms of diabetic retinopathy
As mentioned above, diabetic retinopathy is a slow progressing multifactorial complication, and hyperglycemia is the main initiator of its development. Retina is a complex structure with neuronal, glial and vascular components, and it receives blood supply form two different sources. While the outer 1/3rd retina receive blood from choroidal blood vessels, the inner 2/3rd receives from the inner retinal blood vessels. Sustained high circulating glucose initiates metabolic abnormalities, damaging the retina at cellular, structural and functional levels. Multiple cellular pathways and potential molecular mechanisms have been implicated in the development of diabetic retinopathy, including increased polyol pathway, increased advanced glycation end-products (AGEs) formation and hexosamine pathway, activation of protein kinase C (PKC) and increased oxidative stress [1, 2, 18]. However, despite extensive cutting-edge research worldwide, the exact mechanism responsible remains obscure.
Saturation of hexokinase by high glucose facilitates entry of excess glucose into the polyol pathway, and aldose reductase competes with glutathione reductase for conversion of nicotinamide adenine dinucleotide phosphate (NADPH) to NADP+, converting excess glucose to sorbitol. Sorbitol dehydrogenase then converts sorbitol to fructose and reduces NAD+ to NADH. Thus, activation of retinal polyol pathway decreases NAD+ and NADH, the cofactors important in redox reactions [19]. Despite experimental models and clinical samples documenting alterations in polyol pathway in diabetic retinopathy, clinical trials with structurally diverse aldose reductase inhibitors have been less than satisfactory [19], and have not solidified the role of polyol pathway in diabetic retinopathy.
Increased availability of glucose accelerates nonenzymatic reactions between reducing sugars and free amino groups of proteins/lipids/nucleic acids to exacerbate AGEs formation. AGEs, via interacting with cell-surface RAGEs, can activate many divergent cellular pathways, and increased AGEs are observed in the retinal microvasculature from diabetic patients and also from animal models [20]. Pentosidine, formed by the nonenzymatic reactions of pentose, is closely linked with increased vascular rigidity in diabetic retinopathy patients [21].
The hexosamine pathway, a relatively minor branch of glycolysis, usually accounts for only 2–5% of total glucose metabolism, but in diabetes, high circulating glucose increases flux through the hexosamine pathway, forming uridine diphosphate β-D-N-acetylglucosamine (UDP-GIcNAc), the donor for protein O-GIcNAcylation [18]. The O-GIcNAcylation process is a dynamic nutrient sensitive post-translational modification, which can affect proteins by altering their activity, cellular localization, signaling and transcription processes; in diabetes hexosamine pathway is activated in the retina [18]. In addition, increase in glucose flux through glycolysis also increases de novo synthesis of diacylglycerol (DAG), a key activator of PKC. Activation of PKC in the retina is associated in many abnormalities seen in diabetes including disturbances in hemodynamics and endothelial permeability, leukostasis and production of VEGF. PKC activation is also implicated in accelerated capillary cell apoptosis, and in reduction of the survival signaling pathway of platelet-derived growth factor [22]. Although an isoform-selective inhibitor of PKC-β, ruboxistaurin, which is not yet approved for diabetic retinopathy, has shown some promising results in reducing macular edema and sustained moderate visual loss [23], additional clinical trials are needed to investigate its effect on diabetic retinopathy.
Mitochondria utilize 95% of the available oxygen to produce ATP, but they are also the major endogenous source of free radicals. In normal conditions, only ~2% of oxygen enters the electron transport chain (ETC), which is subsequently oxidized to superoxide radicals [24]. In addition to mitochondria, cytosolic NADPH-oxidase (Nox) also generates reactive oxygen species (ROS), and in diabetes Nox2 is activated [25, 26], Hyperglycemia increases flux through ETC, hyperpolarizing the mitochondria generating free radicals [18]. The accumulation of ROS leads to oxidative stress, and free radicals, due to their unpaired electrons, readily react with the cellular molecules to damage structure and integrity of the cell. In diabetes, retinal mitochondria become swollen, their ultrastructure shows partial cristolysis, membrane integrity and function are impaired, cytochrome C leaks out into the cytosol, and the respiration and extracellular acidification are decreased [27, 28, 29, 30, 31]. In a normal situation, continuous generation of ROS is kept under control by endogenous antioxidants like glutathione (GSH), superoxide dismutase (Sod) and catalase [26, 32]. However, in diabetes, activated Nox2 and impaired antioxidant defense system continues to feed into excessive ROS accumulation [26, 32]. Hyperglycemia also induces nitric oxide synthase (iNOS), increasing nitric oxide production, and peroxynitrite, which is formed by a reaction of nitric oxide with superoxide radicals, damages cellular proteins and DNA [33]. In addition to increased production of ROS by autoxidation of glucose in hyperglycemic milieu, oxidative stress also links the damaging metabolic pathways induced by hyperglycemia in diabetic retinopathy; DNA strand breaks, induced by mitochondrial ROS, activate poly-(ADP-ribose)-polymerase (PARP), which inhibits glyceraldehyde 3-phosphate dehydrogenase (GAPDH), an enzyme converting glyceraldehyde 3-phosphate to D-glycerate 1,3-bisphosphate. GAPDH inhibition results in the accumulation glycolytic metabolites, and these metabolites, in turn, increase AGEs formation and activate PKC, polyol and hexosamine pathways [18]. Moreover, activation of PARP can also activate nuclear transcriptional factor-kB (NF-kB), which, via the toll-like receptor signaling pathway, could result in cell apoptosis [34].
Diabetic retinopathy is now also considered as a low grade inflammatory disease; hyperglycemia assists in production of inflammatory cytokines, and increased leukostasis is observed in retinal vasculature of experimental models before appearance of any histopathology [35]. Inflammatory events are also implicated in the early loss of capillary cells in diabetic retinopathy via activation of ATP-gated ion channels-purinergic P2X receptors (P2X7R), and release of ATP from activation of NLRP3 inflammasome, via activating P2X7R, could lead to cell necrosis/ apoptosis [36]. Thus, a number of interrelated molecular mechanisms could result in cell loss seen in the early stages of diabetic retinopathy.
3. Oxidative stress and diabetic retinopathy
The pathological state of imbalance in the cellular redox status due to increased free radicals creates oxidative stress, and unpaired electrons react with the cellular molecules to damage the structure and integrity of the cell. The continuous accumulation of ROS is kept under control by endogenous enzymatic (e.g., Sod and catalase) and nonenzymatic (glutathione) antioxidants. The retina is rich in fatty acid, making it highly susceptible to oxidative damage in diabetes. To add to that, oxidative stress connects the four major metabolic pathways that are considered major causative factors in the development of diabetic retinopathy such as activation of PKC, polyol and hexosamine pathways and formation of AGEs [1, 2, 18]. Consumption of NAD+ by polyol pathway leads to compromised antioxidant defense, and further increase in oxidative stress [19]. High glucose levels, and concomitant increase in fructose due to increased polyol pathway, results in the formation for AGEs, which act through their specific receptors to induce cellular dysfunction [20]. Thus, oxidative stress has a central place in the pathogenesis of diabetic retinopathy (Figure 1).
Figure 1:
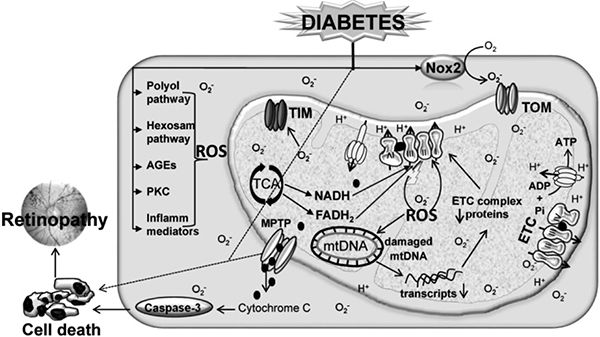
Circulating high glucose activates many metabolic pathways in the retina including polyol and hexosamine pathways, activates protein kinase C and Nox2, elevates inflammatory mediators, and damaged mitochondria. Increased formation of electron transport chain (ETC) substrates (NADH, FADH2) causes electron leakage from complex III, which results in the accumulation of mitochondrial ROS. Elevated mitochondrial ROS damage the mitochondrial transporters (TIM and TOM) and DNA (mtDNA), and the protein expressions of mtDNA-encoded genes, critical for ETC functioning, are decreased resulting in a vicious cycle of ROS. The damaged mitochondrial membranes, via mitochondrial permeability transition pores (MPTP), release cytochrome C in the cytosol, and this activates caspase-mediated cell death, and ultimately, results in the development of retinopathy. The dotted lines represent other possible mechanisms of cell death including necroptosis (via mitochondrial, or non-mitochondrial pathways).
3. 1. Production of ROS
Reactive oxygen species production by cellular respiration is a normal physiological process, and ROS can also be generated by enzymatic processes such as by cytosolic Nox complex and cytochrome p450 or xanthine oxidase. In diabetic environment, ROS are generated by both enzymatic and nonenzymatic pathways, and Nox and mitochondrial ETC pathways are their prominent sources [26, 37].
3. 1. 1. Cytosolic ROS production
The Nox family of enzymes is the main source of cytosolic ROS production and among these, Nox2 plays an active role in retinal ROS generation in diabetes. Nox2 is a highly regulated multi-protein membrane-associated enzyme which catalyzes the one-electron reduction of oxygen to superoxide anion by oxidizing cytosolic NADPH. The holoenzyme is comprised of cytosolic subunits-p47phox, p67phox and a small molecular G-protein Rac1, and the membrane-bound subunits p22phox and gp91phox [26]. Rac1 activation/deactivation is mediated by specific guanine exchange factors and GTPase-activating proteins. In diabetes, Rac1-Nox2 signaling axis in the retina and its capillary cells is activated in the initial stages of diabetes to increase intracellular ROS. Inhibition of guanine exchange factor, Tiam (T-lymphoma invasion and metastasis gene), attenuates Rac1 activation and ROS generation in the retina [26]. Activation of Rac1-Nox2-ROS is an initial event, which precedes mitochondrial damage. Although in the initial stages of the disease, concomitant increase in mitochondrial DNA (mtDNA) biogenesis/repair compensates for ROS-induced mitochondrial damage [26, 38], sustained hyperglycemia overwhelms the mtDNA biogenesis and repair mechanisms, and initiates a vicious cycle of ROS damaging the mitochondria [2, 39].
3. 1. 2. Mitochondrial ROS
As mentioned above, mitochondria are also the major endogenous source of free radicals, and generation of mitochondrial ROS mainly takes place during oxidative phosphorylation at the ETC. Electrons leakage from complex I and complex III results in partial reduction of oxygen, and superoxide thus formed is quickly dismutated to hydrogen peroxide. Low levels of ROS are important in intracellular signaling, but excessive production of ROS oxidatively modifies cellular lipids, proteins, or DNA. In the pathogenesis of diabetic retinopathy, high circulating glucose increases electron flux through the ETC and the activity of complex III is reduced, culminating in increased generation of ROS [39].
3. 2. Antioxidant response
Antioxidants safely interact with free radicals and terminate the chain reaction before vital molecules are damaged, and the antioxidant system consists of both enzymatic and nonenzymatic components. However, in diabetes, the antioxidant system is also compromised in the retina and the cells are exposed to reactive free radicals that subsequently damage biomolecules and affect vital cellular functions [40].
3. 2. 1. Enzymatic antioxidants
The cell has an efficient antioxidant defense enzyme system including Sod, which converts superoxide to H2O2. While copper-zinc Sod is a cytosolic dismutase, manganese Sod (Sod2), is a mitochondrial matrix enzyme. Catalase reduces hydrogen peroxide to water, and glutathione peroxidase reduces lipids and hydrogen peroxide. In diabetes, retinal antioxidant defense enzymes are also compromised, culminating in increased accumulation of free radicals [2, 40].
3. 2. 2. Non-enzymatic antioxidants
In addition to several antioxidant defense enzymes to scavenge free radicals, the cell also has intracellular antioxidants, and GSH is one of the major antioxidants which serves as a free radical scavenger and a detoxifying agent [41]. As a carrier of an active thiol group, it also directly interacts with reactive oxygen/nitrogen species. In the pathogenies of diabetic retinopathy, GSH levels are decreased in the retina and its vasculature [42]. Furthermore, nuclear factor-erythroid 2-related factor 2 (Nrf2) serves as a critical signaling pathway to regulate detoxifying enzymes important in elimination of ROS. Nrf2 serves as a transcription factor to regulate genes containing Antioxidant Response Element (ARE), such as genes encoding Sod, catalytic subunit of glutamate-cysteine ligase (GCLC) and thioredoxin reductase. In normal conditions, Kelch-like ECH-associated protein 1 (Keap1) anchors Nrf2 in the cytoplasm., and the cysteine residues in Keap1 act as oxidative stress sensors. Stress-induced oxidation of these cysteine residues dissociates Nrf2 to translocate in the nucleus, and initiate detoxifying gene transcription [43]. Biosynthesis of intracellular GSH is also intimately modulated by Nrf2 via its regulation of transcription of the catalytic subunit of the enzyme the rate limiting enzyme of GSH biosynthesis, GCLC, further adding to the increased oxidative stress [44]. In diabetic retinopathy, the binding of Nrf2 with Keap1 and the levels of Keap1 (protein and gene) are increased, and the DNA binding activity of Nrf2 is decreased [44]. As stated above, Nox2 is also a major source of ROS, and in diabetic retinopathy, it precedes mitochondrial damage; mouse model of oxygen-induced retinopathy has shown that activation of Nrf2 counteracts with Nox2, [45]. Furthermore, Rac1, an integral component of Nox2 activation, is also shown to activate Nrf2 via a Keap1 independent mechanism [46], suggesting that Nrf2 inactivation could also contribute to sustained Nox2-mediated ROS accumulation. Nrf2 also modulates mitochondrial membrane potential and biogenesis [47], via induction of hemeoxygenase-1 (HO-1) and NAD(P)H quinone oxidoreductase 1 [48]. Exposure of retinal pigment epithelium cells to high glucose is shown to regulate oxidative and metabolic cellular responses to the activators of Nrf2/HO-1 [49]. Thus, Nrf2 activation could have dual roles in decreasing free radical produced by both mitochondria and cytosolic free radicals.
4. Role of mitochondria in diabetic retinopathy
Mitochondria are integral part of cellular metabolic functions, and these double membrane organelles have an inter-membrane space within inner and outer membranes. The outer membrane is rich in lipids, and is freely permeable, but inner membrane acts as a tight diffusion barrier, and is permeable only to small molecules to help maintain mitochondrial membrane potential across the membrane. It has numerous cristae to increase the surface area of the inner membrane, important for high respiratory activity of the mitochondria [50].
Mitochondria produce >80% of all the energy that a cell needs to survive, and the number of mitochondria in a cell depends on its energy demand and can range from 200 to 2,000/cell. The ETC is a highly organized sequestered chain of four protein complex that allow a unilateral flow of electron from complex-I to complex-IV. During electron transport, the participating protein complexes push protons from the matrix into the intermembrane space, creating a proton gradient, and imperfectly coupled electron transport generates electrochemical potential difference leading to partial inhibition of the electron transport in complex III, generating ROS [51]. In diabetes, due to damaged retinal mtDNA, the genes encoding ETC proteins are subnormal, and the compromised ETC results in a loss of membrane potential, further generating free radicals [2].
4.1. Mitochondrial damage
As mentioned above, in the pathogenesis of diabetic retinopathy, Nox activation precedes mitochondrial damage, and sustained increase in cytosolic ROS damages and dysfunctions mitochondria [2, 38], but how hyperglycemic milieu damages mitochondria remains elusive. The cell possesses matrix metalloproteinases (MMPs), a family of extracellular matrix degrading proteins that are implicated in angiogenesis, vascular remodeling and apoptosis [52]. Although MMPs are mainly cytosolic enzymes, increase in ROS within cardiac mitochondria or in the cytoplasm is shown to activate them, resulting in matrix metabolizing and impaired cardiac function [53]. In the etiology of diabetic retinopathy, activation of retinal MMP-9 and MMP-2 is an early event, and is seen before mitochondrial damage. Although MMPs are cytosolic, in the pathogenesis of diabetic retinopathy, they are transported inside the mitochondria with the help of heat shock protein (Hsp70), and once inside, damage mitochondrial integrity by modulating Hsp60 and connexin 43 [54, 55]. Mitochondrial membrane loses their potential, and allows cytochrome c to leak out, activating the apoptotic machinery [31] (Figure 2). Thus, MMP-2 and MMP-9 play dual roles in the development of diabetic retinopathy: in the early stages of the disease (pre-neovascularization) they activate apoptotic machinery [31], and during the later stages, help in neovascularization [56].
Figure 2:
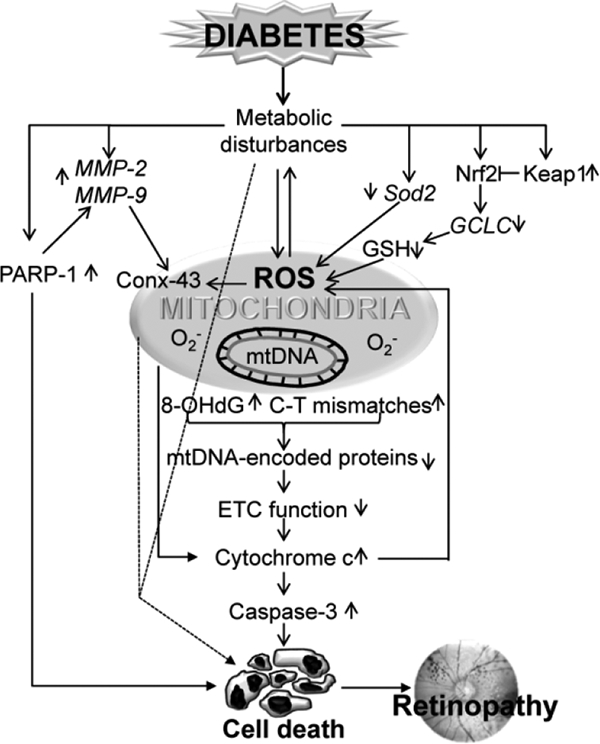
Diabetic environment brings in many metabolic disturbances, and induces oxidative stress. The gene expressions of proteins involved in the cellular antioxidant signaling (Sod2, Keap1-Nrf2, GCLC, GSH) are altered. Activation of retinal MMPs (MMP-2/MMP-9) damage mitochondrial membrane, and damage of connexin 43 (conx43) releases cytochrome C in the cytosol, activating caspase-mediated cell death. Diabetes also increases PARP-1, which enhances retinal MMPs. Sustained increase in ROS and decrease in antioxidant responsive genes, further adds to mitochondrial dysfunction and increase in ROS, subsequently damaging mtDNA (oxidation of guanine base and formation of 8-OHdG and base mismatches). Suppression of transcription of mtDNA-encoded genes, dysfunctions ETC, resulting in continuous free radical accumulation. Other possible mitochondrial and non-mitochondrial cell death routes are represented by dotted lines.
4. 2. Mitochondrial DNA
Mitochondria have multiple copies of small (16.5kb) circular DNA, which lacks protective histones, and their close proximity to ROS makes them vulnerable to free radical damage. The DNA has a 1.1 kb non-coding region, the displacement-loop (D-loop), which contains initiation sites for transcription and replication by the mitochondria-specific polymerase-gamma (POLG) [57]. Mitochondrial DNA encodes 37 genes, including 13 ETC complex proteins, two ribosomal RNAs (12S and 16S) and 22 tRNAs. The remaining hundreds of proteins required for mitochondrial metabolism and maintenance are synthesized in the cytosol, and are imported into the mitochondria [58]. In diabetes, retinal mtDNA is damaged with elevated levels of oxidized nucleoside, 8-hydroxy-2-deoxy-guanosine (8-OHdG), and increased sequence variants [28]. The damage is more extensive at the D-loop region compared to other regions, and mtDNA transcription is impaired, further compromising the ETC complex [28, 59].
4. 2. 1. DNA repair
To repair the damaged DNA, mitochondria has an efficient DNA repair systems including enzymes proteins to repair oxidatively modified bases, base excisions and mismatches. 8-OHdG is not recognized by DNA polymerase, and can lead to DNA mutation, but the cell has an efficient base excision repair (BER) enzyme, 8-oxoguanine glycosylase 1 (OGG1), which removes 8-oxo-deoxyguanine moiety. To remove uncomplimentary base pairs and the insertion and/or deletion loops that are formed during DNA replication, mitochondria possess a mismatch repair system; while Msh2 of MutS family recognizes mismatches, Mlh1 of MutL family cuts the mismatches [60]. In diabetes, although the expression of retinal OGG1 and other BER enzymes are not decreased, their translocation into the mitochondria is impaired, compromising the repair mechanism [39]. Furthermore, mismatch repair system is also compromised with reduced levels of Mlh1 in the mitochondria, culminating in increased sequence variants [28].
4. 2. 2. Biogenesis
Biogenesis of mitochondria is a complex process, which involves synthesis of the inner and outer mitochondrial membranes and mitochondrial encoded proteins, import of nuclear encoded proteins, and replication of mtDNA. Peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC1) is the master regulator of mitochondrial biogenesis, it interacts with nuclear transcription factors, nuclear respiratory factor 1 and 2 (NRF1 and NRF2). NRF1/NRF2 activate mitochondrial transcription factor A (TFAM), and TFAM helps maintain mitochondrial copy numbers by binding at the D-loop to initiate transcription [58]. Since the protein-coding capacity of mtDNA is limited to subunits of the ETC, all of the other gene products necessary for the mitochondrial biogenesis are derived from nuclear DNA [61]. In diabetes, mitochondria copy numbers are decreased, and to overcome mitochondrial dysfunction, the retina begins to increase the transcripts of PGC1, NRF1 and TFAM. But, because of a dysfunctional transport mechanism and posttranslational modification of TFAM, mtDNA biogenesis remains impaired [59], further adding to mitochondrial dysfunction.
The replication of mtDNA is critical for the mitochondria to function properly, and mitochondria have excellent replication machinery [62]. POLG holoenzyme is a heterotrimer consisting of a single 140kDa catalytic subunit, POLG1, and a 55kD accessory subunit, POLG2, and the holoenzyme functions in conjunction with the mtDNA helicase. In the pathogenesis of diabetic retinopathy, the gene transcripts of POLG1, POLG2 and Twinkle are decreased, and their translocation into the mitochondria is also attenuated [63].
4. 3. Fusion-Fission
Mitochondria are very dynamic in structure, and to ensure efficient content exchange between mitochondria and the proper mitochondrial morphology, they undergo fusion and fission [64]. Fusion-fission maintains shape and also controls physiological processes including mitochondrial membrane potential, respiration and genome integrity. Although the loss of mitochondrial fission has no deleterious effect, defects in fusion are associated with accumulation of mtDNA mutations and deletions and compromised ETC [65]. Mitochondria dynamics are regulated by GTPases; fission is controlled by dynamin-related protein1 (Drp1), and fusion by mitofusin 1 and 2 (Mfn1, Mfn2), controlling the outer membrane fusion. The optic atrophy 1 (Opa1) helps in the inner membrane fusion [64]. In diabetes, mitochondria are swollen in the retina and its capillary cells, while Mfn2 expression is decreased in the damaged mitochondria, that of Drp1 is increased, suggesting mitochondrial fragmentation [66, 67].
4. 4. Mitophagy
Since damaged mitochondria induce catastrophic consequences, the quality of mitochondria is essential, and mitochondrial fusion by OPA1/Mfn1/2 is a protective phenomenon of fusing damaged membrane with intact membrane to minimizing the damage. However, the damaged mitochondria that fail to fuse, become potential targets of sequestration by autophagosomes, and the mitophagy is initiated before apoptosis or necrosis can be triggered. Before undergoing mitophagy, the damaged mitochondria undergo Drpl-mediated fission, and due to impaired membrane potential, PTEN-induced putative kinase protein 1 (PINK1), a voltage-sensitive kinase, accumulates in the outer membrane [68]. PINK1 accumulation facilitates the recruitment of E3 ubiquitin ligase, Parkin, to the mitochondrial surface, and ubiquitination signals mitophagy, targeting the removal of damaged mitochondria by autophagosomes by interacting with autophagy adaptor p62. Rat Müller cell line, exposed to high glucose, have increased mitochondrial autophagosome marker Microtubule-associated protein 1A/1 B-light chain 3 BII and lysosomal-associated membrane protein 2A [67], however, the role of mitophagy in diabetic retinopathy, is still in its incipient stages.
4. 5. Apoptosis
Permeabilization of the outer mitochondrial membrane via BCL2-associated X protein (BAX) and BCL2-antagonistic/killer (BAK) releases pro-apoptotic proteins cytochrome c/apoptosis-inducing factor to activate apoptosis [69]. In diabetes, the release of Bax from cytosol into the retinal mitochondria, and cytochrome C from mitochondria into the cytosol is increased [70]. Furthermore, calcium ion is also a major trigger of apoptosis; intracellular Ca2+ signaling involving activation of Ca2+/Calmodulin-dependent protein kinase II (CaMKII), catalyzes the phosphorylation of proteins that translate these Ca2+ signals into cellular responses, is an important mediator of retinal cell apoptosis in diabetes [71]. CaMKII, via the death receptor Fas, plays an essential role in endoplasmic reticulum stress-induced apoptosis, and via c-Jun N-terminal kinase (JNK)-mediated, in mitochondrial apoptosis [72]. Furthermore, mitochondrial damage, under compromised mitophagy or alternative repair phenomenon, also leads to cellular apoptosis by increasing mitochondrial membrane permeabilization [73]. As mentioned above, apoptosis of retinal capillary cells precedes the development of histopathology characteristic of diabetic retinopathy [5, 74], suggesting that the mitochondria occupy central place in the development of diabetic retinopathy (Figure 3). Mitochondrial damage can also increase necroptosis directly by breaking down DNA [75], and caspase-independent necroptotic cell death is shown in ischemia/reperfusion-induced retinal damage [75, 76]; the possibility of a similar mitochondrial-dependent or independent necroptotic phenomenon in the development of diabetic retinopathy needs further investigation.
Figure 3:
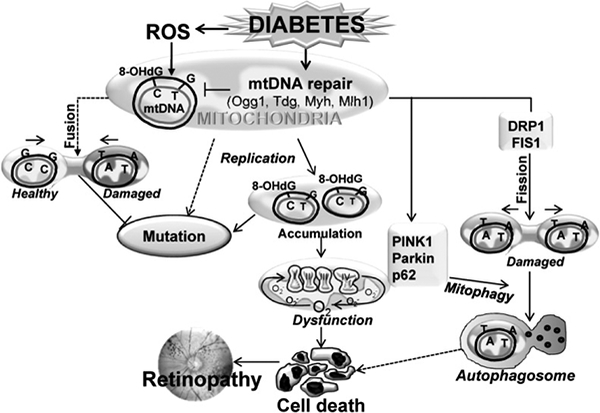
Increased production of cytosolic ROS damages the mitochondria, increasing mitochondrial ROS, and subsequently damaging mtDNA. Due to compromised mtDNA repair system (Ogg1, Tdg, Myh, Mlh1), repair of modified mtDNA bases is impaired. This leads to accumulation of base-mismatches and mutations, and results in sustained mitochondrial dysfunction. Replication of damaged mtDNA, and/or decreased mitochondrial fusion (protective), further enhances the propagation of damaged mtDNA. Dysfunctional mitochondria signals cell death, or via recruitment of fission proteins (PINK1, Parkin, p62, DRP1 and FIS1), forms autophagosomes for lysosomal degradation.
5. Epigenetics and mitochondrial damage
Diabetes and its complications are life-long, and expression of many genes associated with the metabolic, structural and functional abnormalities are altered. Recent evidence suggests that both genetic predisposition and environmental factors are crucial in the development of diabetic complications, and epigenetic modifications, the functionally relevant modifications to the genome without involving any changes in the nucleotide sequence, play a significant role in the pathogenesis of diabetic retinopathy [2, 37]. Different mechanisms such as DNA methylation, histone modifications, and RNA-associated silencing by small non-coding RNAs, regulate gene expression [2, 77]. Covalent modifications of histone and DNA bases in the regulatory regions of genes, alternatively, regulate their expression; e.g., histone acetylases add acetyl group on lysine residue of histones and DNA methyl transferases (Dnmts) add a methyl group on the cytosine base pair forming methyl cytosine (5mC). To keep a balance, histone deacetylases remove acetyl group and Ten eleven translocases (Tets) hydroxymethylate 5mC to form 5-hydroxymethyl cytosine [78]. The ~21-mer noncoding RNAs, microRNAs (miRNAs), also regulate the gene expression by binding to the 3’-untranslated regions (UTRs) of specific mRNAs. About 1000–30,000 copies/cell of these miRNAs, can interact with specific mRNAs through complementary base-pairing, and modulate expression of a gene [79]. Recent work has shown an important role of epigenetic modifications in the etiology of diabetic retinopathy; 349 CpG sites for DNA methylation, associated with 233 unique genes including the genes that are routinely altered in the patients with proliferative diabetic retinopathy, including the gene encoding tumor necrosis factor, have been identified [80]. Both in vitro and in vivo models of diabetic retinopathy have identified that the retinal mtDNA is hypermethylated, and the levels of Dnmts are increased in the mitochondria [81]. Furthermore, there appears to be a close crosstalk between mtDNA hypermethylation and increase in sequence variants in the retina in diabetes, and deamination of 5mC to thymine is increased, making mtDNA more vulnerable for sequence variants. The promoter DNA of POLG, a gene associated with mtDNA replication, is hypermethylated [82], impairing mitochondrial biogenesis, and contributing to decreased mitochondrial copy numbers. Due to dynamic DNA methylation of the gene encoding MMP-9, an enzyme implicating in retinal mitochondrial damage in diabetes, the promoter is hypomethylated and gene transcription activated [83]. Demethylation of lysine 9 at histone 3 (H3K9me2) at MMP-9 promoter acetylates lysine 9, facilitating the recruitment of the NF-kB, and upregulation of its gene transcription [84]. Activation of histone methylation enzyme Enhancer of Zeste homolog 2 (Ezh2), which catalyzes methylation of histone 3 lysine 27 (H3K27), in diabetes increases H3K27me3 at the MMP-9 promoter, allowing recruitment of the DNA methylation-hydroxymethylation enzymes, and regulation of its DNA methylation status [85]. Diabetes also epigenetically modifies genes responsible for protecting mitochondrial damage; e.g. tri-methylation of lysine 20 of histone 4 (H4K20me3) at the promoter/enhancer of Sod2 suppress its expression [86], and epigenetic modifications of thioredoxin interacting protein, an endogenous inhibitor of antioxidant thioredoxin, regulates its transcription in diabetes [87]. Due to increased H3K4me1 of Keap1 promoter, binding of transcriptional factors is increased, resulting in its transcriptional activation, and inhibition of Nrf2 nuclear localization [88]. Nrf2 also regulates GSH biosynthesis by regulating GCLC; histone modifications at the GCLC-ARE4 decrease GCLC transcription and results in subnormal GSH biosynthesis [89]. Retina from human donors with established diabetic retinopathy have further confirmed the role of epigenetics in its development [81, 89].
In addition, miRNAs are also implicated in diabetic retinopathy [90], and could play a significant role in regulating mitochondrial homeostasis. Although miRNAs are encoded in the nuclear genome, some miRNAs are now being considered to have mitochondrial origin (i.e., miR-4485–3p, miR-1973, miR-4284) [91]. Since majority of proteins essential for mitochondrial homeostasis are nuclear DNA-encoded, genes encoding those proteins, including Sod2, can also be regulated by miRNAs. Thus, the role of epigenetics in diabetic retinopathy is gaining strength, and opens up the possibility of using epigenetic markers as biomarkers, and/or therapeutic targets to combat this blinding disease.
6. Therapeutic implications
Although many therapeutic options including dexamethasone and anti-VEGF strategies are now becoming available for the treatment of diabetic macular edema, another common vision threatening problem faced by many patients [92], despite extensive cutting edge research in the field, strict control of glycemia and laser photocoagulation remains the conventional management options for a diabetic patient to control the progression of diabetic retinopathy. Maintenance of strict glycemic control for long durations, however, can be challenging for some diabetic patients, and laser photocoagulation is associated with many risks including possibility of unwanted loss of central vision due to inadvertent laser application to the foveal and parafoveal regions and reduction in accommodative power. Thus, additional therapies are being sought actively for this blinding disease. Based on the central role of mitochondria in diabetic retinopathy and their damage being associated with many inter-related metabolic abnormalities [2, 18], strategies to maintain mitochondrial homeostasis by targeting their dysfunction, or the agents to enhance mitochondrial bioenergetics, appear promising for diabetic retinopathy.
Majority of the data generated from experimental models have clearly demonstrated beneficial effects of therapies ameliorating mitochondrial damage on the molecular mechanisms implicated in the development of diabetic retinopathy, and also on the histopathology characteristic of diabetic retinopathy. For example, overexpression of Sod2 in mouse model prevents diabetes-induced increase in mitochondrial ROS, membrane permeability and mtDNA damage, and these mice are also protected from the development of diabetic retinopathy [93]. Overexpression of Sirt1, a Class III histone deacetylase important is regulating many cellular functions including cell proliferation and apoptosis, in mice protects their retinal vasculature from diabetes-induced mitochondrial damage, and prevents increase in apoptotic cells and formation of degenerative capillaries [94]. Furthermore, repression of MMP-9 in mice prevents diabetes-induced retinal mitochondrial damage and the development of retinopathy [31]. In rats, administration of lipoic acid, a natural thiol antioxidant, or multi-antioxidants mixture containing vitamins, N-acetyl cysteine, β-carotene and selenium, or antioxidant mixture employed in the Age-Related Eye Disease Study, soon after induction of diabetes, attenuates retinal oxidative stress and 8-OHdG formation, and also prevents the formation of degenerative capillaries [42, 95]. Benfotiamine, a fat-soluble synthetic derivative of vitamin B1, inhibits the major metabolic pathways, and ameliorate the severity of retinopathy in diabetic rats [96]. In in vitro models, addition of N-acetyl cysteine or a MnSOD mimic, MnTBAP, or a Sirt1 activator, resveratrol, protects glucose-induced increase in ROS and mitochondrial damage in retinal endothelial cells, and also prevents their accelerated apoptosis [97]. Furthermore, inhibition of Tiam1-Rac1 mediated Nox2 activation inhibits glucose-induced mitochondrial dysfunction and apoptosis [26].
Despite a good body of positive experimental data, clinical trials using antioxidants, in contrast, have not been conclusive. Analysis of clinical, functional and morphological changes in the retina in type 2 diabetic patients with early retinopathy has demonstrated beneficial effects of treatment with antioxidant and angioprotective agents [98]. A double masked, placebo controlled clinical trial on limited number of diabetic patients (<70 patients) has shown some meaningful improvements in the visual function using a multi-component nutritional formula consisting of vitamins C, D3, and E, zinc oxide, eicosapentoenoic acid, docosahexaenoic acid, alpha-lipoic acid, coenzyme Q10, zeaxanthin, lutein, benfotiamine, N-acetyl cysteine, resveratrol, turmeric root extract, green tea leaf [99]. Diabetic patients have lower plasma carotenoids, and potential benefits of lutein and zeaxanthin intake has been reported in diabetic patients with non-proliferative diabetic retinopathy with decrease in foveal thickness [100]. In a double blind randomized, phase II, clinical trial, administration of coenzyme Q10 or antioxidant mixture consisting of lutein, astaxanthin, zeaxanthin, vitamin C, vitamin E, zinc, and copper is shown to improving the oxidative stress in patients with non-proliferative diabetic retinopathy [101]. Furthermore, oral administration of a mixture of pycnogenol, Vitamin E and coenzyme Q10 is shown to reduce ROS levels and retinal thickness in non-proliferative diabetic retinopathy patients [102]. Administration of Mito Q, a mixture of ubiquinol and ubiquinone, which mimics the activity of CoQ antioxidant coenzyme, has been shown to decrease ischemia-reperfusion cardiac injury in rats [103], but its role in protecting retinal damage needs further evaluation. Despite some beneficial effects by these antioxidant supplementation, the duration of these trials is relatively short (6–12 months) for a progressing disease, which is asymptomatic in its early stages and takes decades to manifest. A retrospective study in type 2 diabetic patients, based on a single 24-hour diet recall, failed to show any association between antioxidant supplementation (vitamins C and E, and β-carotene) and decrease in the severity of retinopathy [104], making the association between diabetic retinopathy and dietary antioxidant therapy remains elusive. Some of the possible reasons for failure to find a strong association between antioxidants and oxidative stress, and the development of diabetic retinopathy, could include the ease of antioxidants to pass through an orchestrated blood-retinal barrier to the reach to the retina [105], and their availability inside the mitochondria, due to highly impermeable cardiolipin-rich inner membrane [106]. As documented by DCCT/EDIC studies that although maintenance of tight glycemic control impedes the development of diabetic retinopathy, prior damage plays a major role in the outcome of the tight glycemic control [107]; the severity of hyperglycemia, and the duration are also some of other key factors that could influence therapeutic outcome for this slow progressing disease.
Although free radicals are considered as harmful, their production is strongly regulated processes and they also act as secondary messengers to control many biological processes including calcium homeostasis, enzymatic functions, gene expression, apoptosis and cell growth [108]. Conjugation to a lipophilic cation such as triphenylphosphonium (TPP), makes a small molecule compound available for its uptake into the mitochondrial matrix; conjugation of TPP+ with quinone analogues, mitoquinone, mitovitamin E, mitophenyltertbutyline, and Sod mimetic, Mito-CP, increases their accumulation within the mitochondrial matrix by several-fold higher compared to their accumulation within the cytosolic compartment. Experimental results using such strategy have been very encouraging, and some of these are in phase I & II clinical trials for other chronic diseases [109], but their use to potentially inhibit/retard diabetic retinopathy is not yet known. Since a vast majority of proteins required for mitochondrial homeostasis are nuclear in origin, and are transported into the mitochondria, this opens up the possibility for the use of compounds that are not targeted to mitochondria, but act by binding to specific targets, for future therapeutic options.
As detailed above, MMPs play a significant role in damaging retinal mitochondria in diabetes, and MMP-9 knockout diabetic mice have normal mitochondria in their retinal endothelium and are protected from the development of retinal histopathology [31, 55], targeting MMPs can be an option to prevent mitochondrial damage in diabetic retinopathy. MMP inhibitors, such as synthetic bisphosphonates, collagen peptidemimetics and nonpeptidomimetic, and other zinc chelators are some of the good options, however, clinical trials with MMP inhibitors for chronic diseases have not been very encouraging [110]. Also, since recent research continues to implicate the role of epigenetic modifications in diabetic retinopathy [2, 90], regulation of the enzymes responsible for dynamic epigenetic modifications, such as Dnmts and histone modifying enzymes, and miRNAs have potential to serve as good therapeutic targets to prevent mitochondrial damage; Dnmt inhibitors, 5-azacytidine and 5-aza-2’-deoxycitidine are already approved by US Food and Drug Administration (FDA) for the treatment of myeloid cancers and cutaneous T cell lymphoma [111, 112]. Moreover, synthetic DNA methylation inhibitors such as hydralazine and procainamide are intensively evaluated in multiple clinical trial, and these inhibitors are relatively safe and do not pose many adverse side effects [113]. HDAC inhibitors vorinostat 1 (Zolinza®; Merck) and belinostat 3 (Beleodaq®; Spectrum Pharmaceuticals) are being used for the treatment of peripheral T-cell and refractory cutaneous T-cell lymphomas [114]. An Ezh2 inhibitor Tazemetostat is now in phase I & II clinical trials for lymphomas and sarcomas [115]. Targeting epigenetic modifications via chemical inhibitors, however, comes with clinical and ethical challenges including resistance to epigenetic therapies and less than desirable drug specificity of the Dnmt and HDAC inhibitors. Interestingly, dietary flavonoids and polyphenols such as resveratrol and catechins also directly inhibit Dnmts, or indirectly suppress the methyl donor S-adenosyl methionine. Furthermore, polyphenols like curcumin (diferuloylmethane) and resveratrol can also modulate histone modifications by regulating histone acetylation/ deacetylation [113]. The possibility of using such inhibitors (chemical or natural) in future for the treatment of retinopathy would be a welcome sign for diabetic patients.
In addition to the challenge of relatively impermeable inner mitochondrial membrane to translate discoveries into therapeutic interventions, the retina is protected by blood retinal barrier, making the accessibility of macromolecules to the back of the eye and their stability in the biological environment, more challenging. However, with the recent advancements in conjugating small molecule to lipophilic cations to increase availability to the mitochondrial matrix, nanotechnology-based delivery encapsulation technology, and advancement in injectable hydrogels, which can also serve as implants for controlled delivery and filling purposes, new avenues are being explored for targeting this blinding disease.
7. Conclusions
Retinopathy is one of the most feared complications of diabetes, and the severity of hyperglycemia is a good predictor of this blinding disease. In the early stages of this progressive disease, the patient is asymptomatic, but, with the extending duration of diabetes and leaving it untreated, this can lead to vision loss. Although great advances in technology have facilitated diagnosis of diabetic retinopathy, and cutting-edge research going on all across the globe has identified many molecular mechanisms associated with diabetic retinopathy, due to the multifactorial nature of this slow progressing disease, the exact mechanism remains unclear. Mitochondrial dysfunction occupies a central place in the pathogenesis of this blinding disease as it responds to many metabolic signals by undergoing functional and morphological changes, and these changes, in turn, generate signals influencing various cellular and molecular functions contributing to disease complexity. Mitochondria provide multiple targets for therapeutics, but the optimal pharmacologic or molecular approach to improve mitochondrial homeostasis remains to be better defined, making such options challenging. With the ongoing advancement in drug encapsulation and delivery techniques, the future holds a great promise to treat this blinding disease, which carries a major socioeconomic effect on a diabetic patient.
8. Expert opinion
As discussed in this review, diabetes damages mitochondria including damage to mitochondrial function and structure, compromising of the ETC activity and increased formation of ROS, and mitochondrial dysfunction has a central role in the development of this devastating disease. Since many pathways feed into mitochondrial damage, and damaged mitochondria, in turn, propagate multiple metabolic abnormalities, targeting mitochondria to inhibit the development of diabetic retinopathy appears promising. With the emerge of technology to increase the uptake of pharmaceuticals inside the mitochondria by conjugating them a lipophilic cation to stimulate mitochondrial biogenesis and/or quench superoxide radicals, and by encapsulating the drugs with nanoparticles for their delivery into the retina, new avenues are being explored to target this devastating disease. The new research has also documented the role of epigenetic modifications in the mitochondrial damage-the development of diabetic retinopathy; and lifestyle changes and novel therapeutics to prevent epigenetic modifications appear very promising. Thus, the future for preventing/ slowing down this sight-threatening disease looks very hopeful for diabetic patients.
Acknowledgement
The authors thank their laboratory associates, especially Dr. Arul J. Duraisamy, for some of the initial literature research.
Funding
This article is supported in part by the grants to RAK from the National Institutes of Health (EY014370, EY017313 and EY022230), Juvenile Diabetes Research Foundation (1–2008764 and 5–2012-13) and the Thomas Foundation, and an unrestricted grant to the Ophthalmology Department from Research to Prevent Blindness.
Footnotes
Declaration of Interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Frank RN. Diabetic Retinopathy. N Engl J Med. 2004;350:48–58. •• A comprehensive review of diabetic retinopathy-clinical aspects [DOI] [PubMed] [Google Scholar]
- 2.Kowluru RA, Kowluru A, Mishra M, Kumar B. Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog Retin Eye Res. 2015;48:40–61. •• A comprehensive review of diabetic retinopathy-basic science [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yau JW, Rogers SL KR, Lamoureux EL, et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care. 2012;35:556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Dijk HW, Verbraak FD, Kok PH, et al. Decreased retinal ganglion cell layer thickness in patients with type 1 diabetes. Invest Ophthalmol Vis Sci. 2010;51:3660–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizutani M, Kern TS, Lorenzi M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest. 1996;97:2883–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barber AJ, Lieth E, Khin SA, et al. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest 1998;102:783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DCCT. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329:977–86. [DOI] [PubMed] [Google Scholar]
- 8.Sena CM, Pereira AM, Seica R. Endothelial dysfunction - a major mediator of diabetic vascular disease. Biochim Biophys Acta. 2013;1832:2216–31. [DOI] [PubMed] [Google Scholar]
- 9.Kowluru RA, Mishra M, Kowluru A, Kumar B. Hyperlipidemia and the development of diabetic retinopathy: Comparison between type 1 and type 2 animal models. Metabolism. 2016;65:1570–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar B, Kowluru A, Kowluru RA. Lipotoxicity augments glucotoxicity-induced mitochondrial damage in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2015;56:2985–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Opreanu M, Tikhonenko M, Bozack S, et al. The unconventional role of acid sphingomyelinase in regulation of retinal microangiopathy in diabetic human and animal models. Diabetes. 2011;60:2370–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakravarthy H, Navitskaya S, O’Reilly S, et al. Role of Acid Sphingomyelinase in Shifting the Balance Between Proinflammatory and Reparative Bone Marrow Cells in Diabetic Retinopathy. Stem cells. 2016;34:972–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loprinzi PD, Brodowicz GR, Sengupta S, et al. Accelerometer-assessed physical activity and diabetic retinopathy in United States. JAMA Ophthalmol. 2014;13:1017–9. [DOI] [PubMed] [Google Scholar]
- 14.Looker HC, Nelson RG, Chew E, et al. Genome-wide linkage analyses to identify Loci for diabetic retinopathy. Diabetes. 2007;56:1160–66. [DOI] [PubMed] [Google Scholar]
- 15.Cilensek I, Mankoc S, Globocnik Petrovic M, Petrovic D. The 4a/4a genotype of the VNTR polymorphism for endothelial nitric oxide synthase (eNOS) gene predicts risk for proliferative diabetic retinopathy in Slovenian patients (Caucasians) with type 2 diabetes mellitus. Mol Biol Rep. 2012;39:7061–67. [DOI] [PubMed] [Google Scholar]
- 16.Dahlstrom E, Sandholm N. Progress in Defining the Genetic Basis of Diabetic Complications. Curr Diab Rep. 2017;17:80. [DOI] [PubMed] [Google Scholar]
- 17.Cheung CY, Hui EY, Lee CH, et al. Impact of genetic loci identified in genome-wide association studies on diabetic retinopathy in Chinese Patients With Type 2 Diabetes. Invest Ophthalmol Vis Sci. 2016;57:5518–24. [DOI] [PubMed] [Google Scholar]
- 18.Brownlee M The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–25. •• Mitochondria and diabetic complications [DOI] [PubMed] [Google Scholar]
- 19.Kador PF, Wyman M, Oates PJ. Aldose reductase, ocular diabetic complications and the development of topical Kinostat(R). Prog Retin Eye Res. 2016;54:1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stitt AW. AGEs and diabetic retinopathy. Invest Ophthalmol Vis Sci. 2010;51:4867–74. [DOI] [PubMed] [Google Scholar]
- 21.Sato E, Nagaoka T, Yokota H, et al. Correlation between plasma pentosidine concentrations and retinal hemodynamics in patients with type 2 diabetes. Am J Ophthalmol. 2012;153:903–09 e1. [DOI] [PubMed] [Google Scholar]
- 22.Geraldes P, Hiraoka-Yamamoto J, Matsumoto M, et al. Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat Med. 2009;15:1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheetz MJ, Aiello LP, Shahri N, et al. Effect of ruboxistaurin (RBX) On visual acuity decline over a 6-year period with cessation and reinstitution of therapy: results of an open-label extension of the Protein Kinase C Diabetic Retinopathy Study 2 (PKC-DRS2). Retina. 2011;31:1053–59. [DOI] [PubMed] [Google Scholar]
- 24.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757:509–17. [DOI] [PubMed] [Google Scholar]
- 25.Al-Shabrawey M, Rojas M, Sanders T, et al. Role of NADPH oxidase in retinal vascular inflammation. Invest Ophthalmol Vis Sci. 2008;49:3239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kowluru RA, Kowluru A, Veluthakal R, et al. TIAM1-RAC1 signalling axis-mediated activation of NADPH oxidase-2 initiates mitochondrial damage in the development of diabetic retinopathy. Diabetologia. 2014;57:1047–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tien T, Zhang J, Muto T, et al. High glucose induces mitochondrial dysfunction in reyinal Muller cells: Implications for diabetic retinopathy. Invest Ophthalmol Vis Sci. 2017;58:2915–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mishra M, Kowluru RA. Retinal mitochondrial DNA mismatch repair in the development of diabetic retinopathy, and its continued progression after termination of hyperglycemia. Invest Ophthalmol Vis Sci. 2014;55:6960–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masser DR, Otalora L, Clark NW, et al. Functional changes in the neural retina occur in the absence of mitochondrial dysfunction in a rodent model of diabetic retinopathy. J Neurochem. 2017;143:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han WH, Gotzmann J, Kuny S, et al. Modifications in retinal mitochondrial respiration precede Type 2 diabetes and protracted microvascular retinopathy. Invest Ophthalmol Vis Sci. 2017;58:3826–39. [DOI] [PubMed] [Google Scholar]
- 31.Kowluru RA, Mohammad G, dos Santos JM, et al. Abrogation of MMP-9 gene protects against the development of retinopathy in diabetic mice by preventing mitochondrial damage. Diabetes. 2011;60:3023–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finkel T Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–54. [DOI] [PubMed] [Google Scholar]
- 33.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–37. [DOI] [PubMed] [Google Scholar]
- 34.Madonna R, Balistreri CR, Geng YJ, et al. Diabetic microangiopathy: Pathogenetic insights and novel therapeutic approaches. Vasc Pharmacol. 2017;90:1–7. [DOI] [PubMed] [Google Scholar]
- 35.Joussen AM, Poulaki V, Le ML, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18:1450–62. [DOI] [PubMed] [Google Scholar]
- 36.Platania CBM, Giurdanella G, Di Paola L, et al. P2X7 receptor antagonism: Implications in diabetic retinopathy. Biochem Pharmacol. 2017;138:130–39. [DOI] [PubMed] [Google Scholar]
- 37.Kowluru RA, Mishra M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim Biophys Acta. 2015;1852:2474–83. •• Mitochondria and diabetic retinopathy [DOI] [PubMed] [Google Scholar]
- 38.Santos JM, Tewari S, Kowluru RA. A compensatory mechanism protects retinal mitochondria from initial insult in diabetic retinopathy. Free Rad Biol Med. 2012;53:1729–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Madsen-Bouterse SA, Mohammad G, Kanwar M, et al. Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid Redox Signal. 2010;13:797–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kowluru RA, Kern TS, Engerman RL. Abnormalities of retinal metabolism in diabetes or experimental galactosemia. IV. Antioxidant defense system. Free Radic Biol Med. 1997;22:587–92. [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Gimenez JL, Ibanez-Cabellos JS, Seco-Cervera M, et al. Glutathione and cellular redox control in epigenetic regulation. Free Radic Biol Med. 2014;75:S34. [DOI] [PubMed] [Google Scholar]
- 42.Kowluru RA, Odenbach S. Effect of long-term administration of alpha lipoic acid on retinal capillary cell death and the development of retinopathy in diabetic rats. Diabetes. 2004;53:3233–38. [DOI] [PubMed] [Google Scholar]
- 43.Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16:123–140. [DOI] [PubMed] [Google Scholar]
- 44.Zhong Q, Mishra M, Kowluru RA. Transcription factor Nrf2-mediated antioxidant defernse system in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2013;54:3941–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei Y, Gong J, Xu Z, et al. Nrf2 promotes reparative angiogenesis through regulation of NADPH oxidase-2 in oxygen-induced retinopathy. Free Radic Biol Med. 2016;99:234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cuadrado A, Martin-Moldes Z, Ye J, et al. Transcription factors NRF2 and NF-kappaB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation [J Biol Chem. 2014;289:15244–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med. 2015;88:179–88. • Mitochondrial function and Nrf2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strom J, Xu B, Tian X, et al. Nrf2 protects mitochondrial decay by oxidative stress. FASEB J. 2016;30:66–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Foresti R, Bucolo C, Platania CM, et al. Nrf2 activators modulate oxidative stress responses and bioenergetic profiles of human retinal epithelial cells cultured in normal or high glucose conditions. Pharmacol Res. 2015;99:296–307. [DOI] [PubMed] [Google Scholar]
- 50.Zick M, Rabl R, Reichert AS. Cristae formation-linking ultrastructure and function of mitochondria. Biochim Biophys Acta. 2009;1793:5–19. [DOI] [PubMed] [Google Scholar]
- 51.Belevich I, Verkhovsky MI, Wikstrom M. Proton-coupled electron transfer drives the proton pump of cytochrome c oxidase. Nature. 2006;440:829–32. [DOI] [PubMed] [Google Scholar]
- 52.Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15:786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vacek TP, Vacek JC, Tyagi SC. Mitochondrial mitophagic mechanisms of myocardial matrix metabolism and remodelling. Archives of physiology and biochemistry. 2012;118:31–42. • Mitochondrial damage and matrix metalloproteinases and metalloproteinases and [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mohammad G, Kowluru RA. Matrix metalloproteinase-2 in the development of diabetic retinopathy and mitochondrial dysfunction. Lab Invest. 2010;90:1365–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kowluru RA, Mishra M. Regulation of Matrix Metalloproteinase in the Pathogenesis of Diabetic Retinopathy. Prog Mol Biol Transl Sci. 2017;148:67–85. [DOI] [PubMed] [Google Scholar]
- 56.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bebenek K, Kunkel TA. Functions of DNA polymerases. Adv Protein Chem. 2004;69:137–65. [DOI] [PubMed] [Google Scholar]
- 58.Scarpulla RC. Nucleus-encoded regulators of mitochondrial function: integration of respiratory chain expression, nutrient sensing and metabolic stress. Biochim Biophys Acta. 2012;1819:1088–97. •• Mitochondria-nuclear cross talk [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Santos JM, Tewari S, Goldberg AFX, et al. Mitochondria biogenesis and the development of diabetic retinopathy. Free Radic Biol Med. 2011;51:1849–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kazak L, Reyes A, Holt IJ. Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol. 2012;13:659–71. [DOI] [PubMed] [Google Scholar]
- 61.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–38. [DOI] [PubMed] [Google Scholar]
- 62.Graziewicz MA, Longley MJ, Copeland WC. DNA polymerase gamma in mitochondrial DNA replication and repair. Chem Rev. 2006;106:383–405. [DOI] [PubMed] [Google Scholar]
- 63.Tewari S, Santos JM, Kowluru RA. Damaged mitochondrial DNA replication system and the development of diabetic retinopathy. Antioxid Redox Signal. 2012;17:492–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Ann Rev Gene. 2012;46:265–87. [DOI] [PubMed] [Google Scholar]
- 65.Chen H, Vermulst M, Wang YE, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhong Q, Kowluru RA. Diabetic retinopathy and damage to mitochondrial structure and transport machinery. Inves Ophthalmol Vis Sci. 2011;52:8739–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Devi TS, Somayajulu M, Kowluru RA, et al. TXNIP regulates mitophagy in retinal Muller cells under high-glucose conditions: implications for diabetic retinopathy. Cell Death Dis. 2017;8:e2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Matsuda N, Sato S, Shiba K, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–32. [DOI] [PubMed] [Google Scholar]
- 70.Kowluru RA, Abbas SN. Diabetes-induced mitochondrial dysfunction in the retina. Invest Ophthalmol Vis Sci. 2003;44:5327–34. [DOI] [PubMed] [Google Scholar]
- 71.Barber AJ, Gardner TW, Abcouwer SF. The significance of vascular and neural apoptosis to the pathology of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2011;52:1156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Timmins JM, Ozcan L, Seimon TA, et al. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Inves. 2009;119:2925–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cai J, Yang J, Jones DP. Mitochondrial control of apoptosis: the role of cytochrome c. Biochim Biophys Acta. 1998;1366:139–49. [DOI] [PubMed] [Google Scholar]
- 74.Kern TS, Tang J, Mizutani M, et al. Response of capillary cell death to aminoguanidine predicts the development of retinopathy: Comparison of diabetes and galactosemia. Invest Ophthalmol Vis Sci. 2000;41:3972–78. [PubMed] [Google Scholar]
- 75.Del Olmo-Aguado S, Nunez-Alvarez C, Osborne NN. Blue Light Action on Mitochondria Leads to Cell Death by Necroptosis. Neurochemical resear 2016;41:2324–35. [DOI] [PubMed] [Google Scholar]
- 76.Shosha E, Xu Z, Yokota H, et al. Arginase 2 promotes neurovascular degeneration during ischemia/reperfusion injury. Cell Death Dis. 2016;7:e2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gemenetzi M, Lotery AJ. The role of epigenetics in age-related macular degeneration. Eye (Lond). 2014;28:1407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang P, Huang B, Xu X, et al. Ten-eleven translocation (Tet) and thymine DNA glycosylase (TDG), components of the demethylation pathway, are direct targets of miRNA-29a. Biochem Biophys Res Comm. 2013;437;368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dykxhoorn DM, Novina CD, Sharp PA. Killing the messenger: short RNAs that silence gene expression. Nat Rev Mol Cell Biol. 2003;4:457–67. [DOI] [PubMed] [Google Scholar]
- 80.Agardh E, Lundstig A, Perfilyev A, et al. Genome-wide analysis of DNA methylation in subjects with type 1 diabetes identifies epigenetic modifications associated with proliferative diabetic retinopathy. BMC Med. 2015;13:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mishra M, Kowluru RA. Epigenetic modification of mitochondrial DNA in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2015;56:5133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tewari S, Zhong Q, Santos JM, et al. Mitochondria DNA replication and DNA methylation in the metabolic memory associated with continued progression of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2012;53:4881–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kowluru RA, Shan Y, Mishra M. Dynamic DNA methylation of matrix metalloproteinase-9 in the development of diabetic retinopathy. Lab Invest. 2016;96:1040–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhong Q, Kowluru RA. Regulation of matrix metalloproteinase-9 by epigenetic modifications and the development of diabetic retinopathy. Diabetes. 2013;62:2559–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Duraisamy AJ, Mishra M, Kowluru R. Crosstalk between histone and DNA methylation in regulation of retinal matrix metalloproteinase-9 in diabetes. Invest Opthalmol Vis Sci. 2017;In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhong Q, Kowluru RA. Epigenetic changes in mitochondrial superoxide dismutase in the retina and the development of diabetic retinopathy. Diabetes. 2011;60:1304–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Devi TS, Lee I, Huttemann M, et al. TXNIP links innate host defense mechanisms to oxidative stress and inflammation in retinal Muller glia under chronic hyperglycemia: implications for diabetic retinopathy. Exp Diabetes Res. 2012;2012:438238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kowluru RA, Mishra M. Epigenetic regulation of redox signaling in diabetic retinopathy: Role of Nrf2. Free Radic Biol Med. 2017;103:155–164. • Epigenetics and diabetic retinopathy [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mishra M, Zhong Q, Kowluru RA. Epigenetic modifications of Nrf2-mediated glutamate-cysteine ligase: implications for the development of diabetic retinopathy and the metabolic memory phenomenon associated with its continued progression. Free Radic Biol Med. 2014;75C:129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mastropasqua R, Toto L, Cipollone F, et al. Role of microRNAs in the modulation of diabetic retinopathy [Review]. Prog Retin Eye Res. 2014;43C:92–107. [DOI] [PubMed] [Google Scholar]
- 91.Geiger J, Dalgaard LT. Interplay of mitochondrial metabolism and microRNAs. Cell Mol Life Sci. 2017;74:631–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cai S, Bressler NM. Aflibercept, bevacizumab or ranibizumab for diabetic macular oedema: recent clinically relevant findings from DRCR.net Protocol T. Curr Opin Ophthalmol. 201;28:636–43. [DOI] [PubMed] [Google Scholar]
- 93.Kanwar M, Chan PS, Kern TS, et al. Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci. 2007;48:3805–11. [DOI] [PubMed] [Google Scholar]
- 94.Mishra M, Duraisamy AJ, Kowluru RA. Sirt1-a guardian of the development of diabetic retinopathy. Diabetes. 2018;In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kowluru RA, Tang J, Kern TS. Abnormalities of retinal metabolism in diabetes and experimental galactosemia. VII. Effect of long-term administration of antioxidants on the development of retinopathy. Diabetes. 2001;50:1938–42. [DOI] [PubMed] [Google Scholar]
- 96.Hammes HP, Du X, Edelstein D, et al. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med. 2003;9:294–99. [DOI] [PubMed] [Google Scholar]
- 97.Kowluru RA, Santos JM, Zhong Q. Sirt1, a negative regulator of matrix metallo-einase-9 in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2014;55:5653–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Moshetova LK, Vorob’eva IV, Alekseev IB, et al. Results of the use of antioxidant and angioprotective agents in type 2 diabetes patients with diabetic retinopathy and age-related macular degeneration. Vestn Oftalmol. 2015;131:33–44. [DOI] [PubMed] [Google Scholar]
- 99.Chous AP, Richer SP, Gerson JD, et al. The Diabetes Visual Function Supplement Study (DiVFuSS). Br J Ophthalmol. 2016;100:227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hu BJ, Hu YN, Lin S, et al. Application of Lutein and Zeaxanthin in nonproliferative diabetic retinopathy. Int J Ophthalmol. 2011;4:303–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rodriguez-Carrizalez AD, Castellanos-Gonzalez JA, Martinez-Romero EC, et al. The effect of ubiquinone and combined antioxidant therapy on oxidative stress markers in non-proliferative diabetic retinopathy: A phase IIa, randomized, doubleblind, and placebo-controlled study. Redox Rep. 2016;21:155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Domanico D, Fragiotta S, Cutini A, et al. Circulating levels of reactive oxygen species in patients with nonproliferative diabetic retinopathy and the influence of antioxidant supplementation: 6-month follow-up. Ind J Ophthalmol. 2015;63:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Adlam VJ, Harrison JC, Porteous CM, et al. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–95. [DOI] [PubMed] [Google Scholar]
- 104.Mayer-Davis EJ, Bell RA, Reboussin BA, et al. Antioxidant nutrient intake and diabetic retinopathy: the San Luis Vally diabetes study. Ophthalmology. 1998;105:2264–70. [DOI] [PubMed] [Google Scholar]
- 105.Antonetti DA, Klein R, Gardner TW. Diabetic retinopathy. N Eng J Med. 2012;366:1227–39. [DOI] [PubMed] [Google Scholar]
- 106.Frantz M-C, Wipf P. Mitochondria as a target in treatment. Env Mol Mutag. 2010;51:462–475. • Mitochondria as therapeutic target [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Aiello LP. Diabetic retinopathy and other ocular findings in the diabetes control and complications trial/epidemiology of diabetes interventions and complications study. Diabetes Care. 2014;37:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bokkon I Recognition of functional roles of free radicals. Curr Neuroph. 2012;10:287–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chakrabarti AK, Feeney K, Abueg C, et al. Rationale and design of the EMBRACE STEMI study: a phase 2a, randomized, double-blind, placebo-controlled trial to evaluate the safety, tolerability and efficacy of intravenous Bendavia on reperfusion injury in patients treated with standard therapy including primary percutaneous coronary intervention and stenting for ST-segment elevation myocardial infarction. Am Heart J. 2013;165:509–14 [DOI] [PubMed] [Google Scholar]
- 110.Vandenbroucke RE, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov. 2014;13:904–27. [DOI] [PubMed] [Google Scholar]
- 111.Robak T New nucleoside analogs for patients with hematological malignancies. Expert Opin Invest Drug. 2011;20:343–59. [DOI] [PubMed] [Google Scholar]
- 112.Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20:2429–40. [DOI] [PubMed] [Google Scholar]
- 113.Chistiakov DA, Orekhov AN, Bobryshev YV. Treatment of cardiovascular pathology with epigenetically active agents: Focus on natural and synthetic inhibitors of DNA methylation and histone deacetylation. Int J Cardiol. 2017;227:66–82. [DOI] [PubMed] [Google Scholar]
- 114.Zwergel C, Valente S, Mai A. DNA Methyltransferases inhibitors from natural sources. Curr Top Med Chem. 2016;16:680–96. [DOI] [PubMed] [Google Scholar]
- 115.Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17:630–41. • Epigenetic inhibitors and chronic diseases [DOI] [PubMed] [Google Scholar]