

Abstract
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by progressive memory loss and dementia. Accumulating evidence suggests that inflammation is involved in the pathogenesis of AD. Epidemiological studies suggest that use of anti-inflammatory drugs is associated with a lower incidence of AD. However, clinical trials with anti-inflammatory drugs have not been successful. Recent studies have shown that inflammation is resolved by a process that is mediated by a group of lipid mediators, so called specialized pro-resolving lipid mediators (SPMs). Unlike anti-inflammatory strategies, which usually involve inhibition of the synthesis of inflammatory mediators, stimulating the resolution of inflammation is aimed at ending inflammation in a similar fashion as under normal physiological conditions. We have previously shown that pathways of resolution are impaired in AD. Moreover, we found that SPMs can improve neuronal survival and increase microglial phagocytosis of amyloid beta (Aβ) in in vitro studies, indicating that stimulating resolution of inflammation may be a potential therapeutic target in AD. In this review, we summarize recent findings regarding resolution of inflammation in AD. We also discuss possible strategies to stimulate the resolution of inflammation in AD, specifically focusing on signaling pathways, including SPMs, their receptors and enzymes involved in their formation.
Keywords: resolution of inflammation, inflammation, specialized pro-resolving lipid mediators, Alzheimer’s disease
Introduction
Alzheimer’s disease (AD) is a common neurodegenerative disease characterized by progressive memory loss. The pathological characteristics of AD include the deposition of amyloid beta (Aβ), neurofibrillary tangles, and neuronal loss.1–4 Evidence accumulated during the last three decades has shown the existence of inflammation in AD, including activated microglia within and surrounding senile plaques,5,6 and an increase in the concentration of molecules involved in innate immunity and inflammation such as human leukocyte antigen (HLA)-DR7 and interleukin (IL)-1.8 Moreover, results from studies employing imaging techniques that detect microglial activation,9 and from genome-wide association studies, have shown that gene mutations involved in inflammation and innate immunity such as complement receptor (CR)1 and CD33 are associated with an increased incidence of AD.10 In addition, data from in vitro studies show that Aβ can activate microglia and astrocytes,11,12 leading to increased production of proinflammatory cytokines, which can in turn increase the neuronal production of Aβ precursor protein (APP)13–15 and the amyloidogenic processing of APP, resulting in increased levels of Aβ. Furthermore, inflammation is further potentiated by cellular debris and the microenvironment of damaged tissue. Therefore, a self-reinforcing cycle has been proposed to exist in the pathogenesis of AD13 (Figure 2). Further evidence supporting the involvement of inflammation in AD has been provided by epidemiological studies showing that among patients with rheumatoid arthritis with long-term medication of nonsteroidal anti-inflammatory drugs (NSAIDs) the incidence of AD is lower.16 However, clinical trials based on NSAIDs in patients with AD have not been successful,17–19 or only beneficial in a subgroup of patients with higher baseline plasma levels of tumor necrosis factor (TNF)-α and C-reactive protein.20 Several explanations for this have been suggested, such as treatment starting when the pathology was already too extensive, the duration of the intervention having been too short, or that the patient group was too heterogeneous. It may be that the beneficial effects of NSAIDs in AD only emerge in situations where a strong, chronic, peripheral inflammation is present.
Figure 2.

Aβ can activate microglia, leading to increased production of proinflammatory cytokines, which in turn can increase the neuronal production and amyloidogenic processing of APP, resulting in increased levels of Aβ. Furthermore, inflammation is further potentiated by cellular debris and the microenvironment of damaged tissue. Therefore, a self-reinforcing cycle has been proposed to exist in the pathogenesis of AD. SPMs have been shown to be beneficial in different cellular and animal models of AD. Several mechanisms that mediate the protective effects of SPMs have been reported. Firstly, SPMs can modify the microglial phenotype from a proinflammatory to anti-inflammatory phenotype and can increase microglial phagocytosis of Aβ. Secondly, SPMs can inhibit the production of proinflammatory cytokines. Thirdly, SPMs can shift APP processing from amyloidogenic to nonamyloidogenic pathway. Lastly, SPMs can improve neuronal survival.
Aβ, beta amyloid; AD, Alzheimer’s disease; APP, Aβ precursor protein; SPM, specialized pro-resolving lipid mediator.
Inflammation is a defensive response of the body to harmful stimuli, such as infection and injury, with the purpose of eliminating the threat, after which restorative processes take place. Ultimately, the damaged tissue is meant to be repaired and to return to homeostasis. Inflammation is a dynamic process under the strict control of regulatory mechanisms that under normal conditions orchestrate the progression of the response of detection, activation, destructive defense, and lastly of downregulation and restoration.21–24 In addition to inflammatory proteins such as proinflammatory or anti-inflammatory cytokines, lipids represent groups of molecules that are involved in the regulation of inflammation, such as prostaglandins (PGs) that mediate fever and pain. Recent studies have shown that a group of lipid mediators (LMs) termed ‘specialized pro-resolving lipid mediators’ (SPMs) mediate the ending of inflammation. This process is called ‘resolution of inflammation’.23–27 A self-limiting inflammatory response normally occurs, where pro-resolving activities are sufficient to counteract the proinflammatory response. However, in the situation of a chronic inflammatory disease, pro-resolving activities are not sufficient to counteract the proinflammatory signaling, leading to persistent inflammation which may lead to chronic inflammatory disease. Therefore, understanding the regulation of resolution of inflammation under normal and pathological conditions is important. In this review, we summarized current knowledge about SPMs in AD, and discuss the feasibility of possible novel treatment strategies through stimulating resolution in AD.
SPMs and their signaling pathways
Since inflammation is a protective but potentially self-destructive biological response, it should be ended by resolution after the pathogen is eliminated. Inflammation has commonly been held to end by a passive dissipation of the inflammatory mediators. However, accumulating evidence supports the theory that the resolution of inflammation is a highly regulated process.28 Using a liquid chromatography tandem mass spectrometry (LC-MS-MS) technique, a new group of LMs derived from polyunsaturated fatty acid (PUFA) were discovered, the levels of which were found to be increased at the later phase of inflammation.29–31 The chemical structures of these molecules have been elucidated.23 The molecules were named SPMs, and four classes of SPMs have been identified so far. They include lipoxin A4 (LXA4), which is derived from arachidonic acid (AA), the D-series resolvins, protectins, and maresins derived from docosahexenoic acid (DHA), and the E-series resolvins derived from eicosapentenoic acid (EPA).28,32–34
SPMs are synthesized from their PUFA precursors via the activities of lipoxygenases (LOXs) and cyclooxygenases (COXs; Figure 1). LOXs add oxygen to the carbon chain of PUFAs, and depending on the site of incorporation, the enzyme is classified as either 5-LOX or 15-LOX.35 Notably, besides the role in synthesis of SPMs, 5-LOX is also involved in the synthesis of proinflammatory leukotrienes (LTs).35,36 COXs are crucial enzymes involved in the synthesis of PGs, which mediate inflammation and pain. Interestingly, COXs and LOXs can give rise to aspirin-triggered forms of SPMs in response to aspirin. Thus, the production profile of LMs by this enzymatic machinery can change from proinflammatory classes of LMs to pro-resolving classes of LMs, a process termed ‘class-switching’.37 Increased knowledge on the activation and regulatory mechanisms of these enzymes, and how this can prevent or result in class-switching, is necessary to increase our understanding of chronic inflammatory diseases, and how tissue regeneration may be stimulated.
Figure 1.
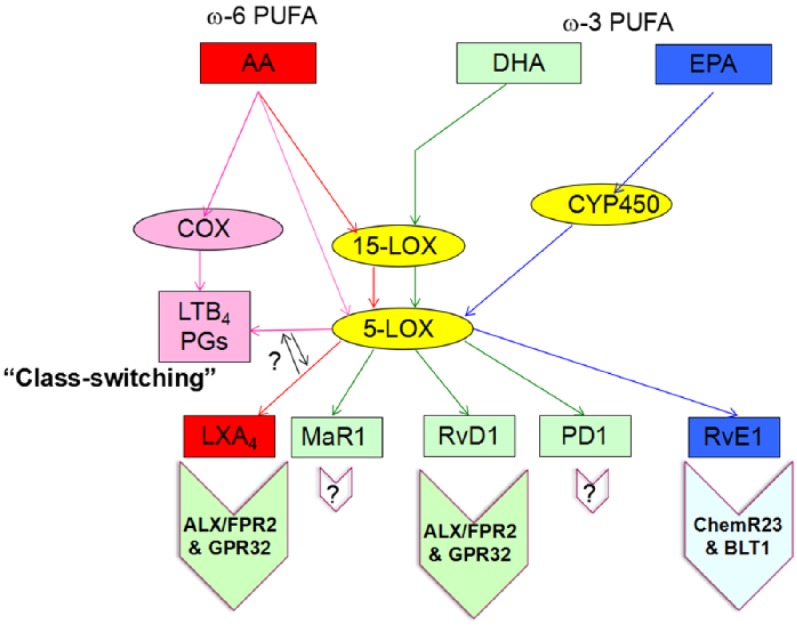
Synthesis and signaling of specialized pro-resolving lipid mediators. LXA4 is derived from AA by sequential processing and the activity of 15- and 5-LOXs. On the other hand, AA can also give rise to proinflammatory LMs such as LTB4 and prostaglandins by the action of COXs. Under certain circumstances, the production of proinflammatory LMs can be switched to the pro-resolving LMs, a process called ‘class-switching’. The D-series resolvins PD1 and MaR1 are derived from DHA by the enzymatic processing of 15- and 5-LOXs. The E-series resolvins are derived from EPA by the action of CYP450. LXA4 and RvD1 have been found to bind to ALX/FPR2 and GPR32, and RvE1 binds to ChemR23 and BLT1. However, the receptors for PD1 and for MaR1 have not been identified.
ω resiFA, omega-3 polyunsaturated fatty acid; AA, arachidonic acid; ALX/FPR2, lipoxin A4/formyl peptide receptor 2; BLT1, leukotriene B4 receptor 1; ChemR23, chemerin receptor 23; COX, cyclooxygenase; CYP450, cytochrome P450; DHA, docosahexenoic acid; EPA, eicosapentenoic acid; GPR32, G protein receptor 32; LM, lipid mediator; LOX, lipoxygenase; LTB4, leukotriene B4; LXA4, lipoxin A4; MaR1, maresin 1; omeUFA, omega-6 polyunsaturated fatty acid; PD1, protectin D1; RvD1, resolvin D1; RvE1, resolvin E1; SPM, specialized pro-resolving lipid mediator.
Receptors for LXA4, RvD1, and RvE1 have been identified, while the receptors for other SPMs are unknown. The receptors identified so far belong to the 7-transmembrane G protein-coupled receptor family. LXA4 and RvD1 have been found to bind to lipoxin A4 (LXA4)/formyl peptide receptor 2 (ALX/FPR2) and G protein receptor (GPR) 32,38,39 and RvE1 binds to chemerin receptor 23 (ChemR23) and the leukotriene B4 receptor 1 (BLT1).40,41 RvE1 binds to BLT1 as a partial agonist, and counteracts proinflammatory signals transduced by BLT1 to mediate the resolution of inflammation.41 These receptors bind other ligands upon which they can transduce a proinflammatory signal. ALX/FPR2 has been identified as a receptor for Aβ,42 which activates microglia, and thereby transducing proinflammatory signals. ChemR23 binds chemerin,43 a chemotactic peptide,43 associated with increased inflammation.44 Furthermore, the nuclear receptor peroxisome proliferator-activated receptor (PPAR)-γ has been reported to mediate protective effects of SPMs.45 GPR120 binds long chain fatty acids (FAs) including DHA and EPA,46 and is also a candidate receptor for SPMs.
The biological functions of SPMs have been investigated in different systems. In patients with severe asthma, a chronic inflammatory airway allergic disease, the levels of LXA4 and neuroprotectin (NP) D1were found to be reduced,47,48 and lower plasma levels were described in patients with localized aggressive periodontitis compared with healthy individuals.49 Utilizing SPMs as a means of treating inflammatory diseases has been investigated in several models of inflammatory diseases. Thus, treatments with SPMs have yielded protective effects in models for asthma,50–52 colitis,53,54 and peritonitis.41,55 Furthermore, there is evidence of beneficial effects of SPMs in cellular and animal models involving the nervous system (i.e. for cerebral ischemia,56–61 pain,62–66 Parkinson’s disease,67 and AD).68,69
Resolution of inflammation is impaired in AD
A deficiency in resolution has been described in chronic inflammatory diseases, including AD. SPMs are small molecular lipids derived from PUFA, and lipid dysregulation is a feature of several conditions in which chronic inflammation is present, including obesity, vascular disease, and diabetes. Interestingly, these diseases share many risk factors and features with AD, suggesting a complex picture of overlapping and interacting etiologies. A dysregulation of lipids in AD is suggested by studies showing decreased levels of DHA in the human AD hippocampus compared with those of control participants,70 and by the dysfunctional conversion of dietary FAs to omega-3 FAs in the liver in AD.71 Furthermore, Bazan and collaborators have previously reported reduced levels of both DHA and its derivative neuroprotectin (NP) D1 in the hippocampus of patients with AD,70 and more recently we showed reduced LXA4 levels in both the hippocampus and cerebrospinal fluid (CSF) of patients with AD.72 Interestingly, CSF levels of LXA4 and RvD1 correlated with cognitive function,72 indicating that these SPMs may play a role in preserving memory functions. More recently, studies on the entorhinal cortex showed lower levels of maresin 1 (MaR1) in AD.73 Furthermore, RvD5 was detected in the human brain for the first time, and at lower levels in AD patients as compared with age-matched controls.73 In contrast, the levels of the proinflammatory PGD2 were higher in AD. Altogether these data indicate a disturbance in the resolution of inflammation in AD, and counteracting this by stimulation of pro-resolving activities may be of therapeutic value in AD.
Supplementation with omega-3 FAs in AD patients
Several epidemiological studies suggest that an increased intake of omega-3 FAs is associated with a reduced risk of dementia.74–77 Furthermore, omega-3 FAs have been showed beneficial in AD-related disease models.78 However, clinical trials in which patients with AD are treated with omega-3 FAs have not been clearly successful,79–81 although in patients with the mildest cognitive disturbance and in elderly people with minor cognitive disturbance82 there was improvement. A possible explanation may be that there are factors inhibiting the beneficial effects of omega-3 FAs in patients with late-stage AD. A lack of conversion from omega-3 FAs to SPMs could be one such factor, in the case that SPMs represent the effective molecular components that mediate the beneficial effects of omega-3 FAs. Further studies are required to investigate whether indeed this conversion is inhibited in the AD brain.
Restoring resolution of inflammation
Administration of SPMs in animal and cellular models of AD
In view of a deficiency in resolution in AD, it is important to understand the biological function of SPMs in the brain. In studies on transgenic animal models of AD, treatment with aspirin-triggered LXA4 was shown to ameliorate Aβ and tau pathology, and to improve memory function.68,69 Moreover, our group has shown that SPMs can increase microglial phagocytosis of Aβ and improve neuron survival in in vitro models.73 Stimulating resolution of inflammation represents a novel strategy that differs from treatments with anti-inflammatory drugs, which inhibit a pathway or block the synthesis of proinflammatory mediators. Instead, stimulating pro-resolving activities represents a way to end inflammation in a similar fashion as under ‘normal’ physiological conditions. In the following sections, several possible means of stimulating the resolution of inflammation are discussed, direct treatment with SPMs, namely treatment with precursors for SPMs, activation of receptors of SPMs, modulation of biosynthetic enzyme activity, and stimulating class-switching.
With a normal diet, omega-3 FAs can be converted from other essential FAs, and it is hypothesized that this conversion is disturbed in AD, which, together with a decreased capacity to convert omega-3 FAs to SPMs, would lead to decreased levels of SPMs. It has been shown that the formation of DHA in the liver is disturbed in AD.71 SPMs have also been reported protective in AD in different animal and cellular models68–70,72,73,83,84 (Table 1 and Figure 2), indicating that treatment with SPMs may represent a therapeutic strategy. The mechanism underlying the protective effects of SPMs have also been intensively studied, in the following sections we will discuss the protective roles of SPMs and the underlying mechanisms of them.
Table 1.
Specialized pro-resolving lipid mediators in Alzheimer’s disease and their effects in animal and cellular models.
| SPMs | Disease model | Action | Methods/mechanisms | Reference |
|---|---|---|---|---|
| Observational study | ||||
| PD1 | human post-mortem brain | reduced levels of PD1 in hippocampus of AD patients | LC-MS-MS | Lukiw and colleagues70 |
| LXA4, RvD1 | human post-mortem brain, CSF samples from AD patients | reduced levels of LXA4 in hippocampus and CSF of AD patients | EIA, LC-MS-MS | Wang and colleagues72 |
| MaR1, RvD5 | human post-mortem brain | reduced levels of MaR1 and RvD5 in entorhinal cortex of AD patients | LC-MS-MS | Zhu and colleagues73 |
| LXA4 | 3×Tg AD mice model | reduced levels of LXA4 levels with age and levels of LXA4 were significantly more impacted in 3×Tg AD mice | LC-MS-MS | Dunn and colleagues69 |
| RvE1, LXA4 | 5×FAD mice model | reduced levels of RvE1 and LXA4 in the hippocampus of 5×FAD mice | EIA | Kantarci and colleagues76 |
| PD1 | 3×Tg AD mice model | reduced levels of PD1 in hippocampus of AD patients | LC-MS-MS | Zhao and colleagues45 |
| Treatment study | ||||
| ATL | Tg2576 APP transgenic mice model | reduced Aβ pathology, improved cognition | reduced NF-κB activation, inhibited proinflammatory cytokines and chemokines production, modulated microglia phenotype | Medeiros and colleagues68 |
| ATL | 3×Tg AD mice model | reduced Aβ and tau pathology, improved cognition | inhibited GSK-3β and P38 MAPK activity, inhibited microglia and astrocyte reactivity | Dunn and colleagues69 |
| LXA4 | intracerebroventricular injection of Aβ in mice | inhibited IL-1β and TNFα production in the cortex and hippocampus of mice | blocked IκBα degradation and NF-κB p65 subunit translocation into the nucleus stimulated by Aβ | Wu and colleagues77 |
| PD1 | human neuronal cells | promoted neuronal cell survival | induced the anti-apoptotic and neuroprotective genes expression | Lukiw and colleagues70 |
| PD1 | human neuron-glia coculture | suppressed Aβ induced apoptosis | downregulated COX-2 and B-94, upregulated ADAM10 while downregulated BACE1 | Zhao and colleagues45 |
| RvD1 | PBMC | increase the phagocytosis of Aβ | downregulated the proinflammatory cytokines and chemokines production | Mizwicki and colleagues74 |
| RvE1, LXA4 | 5×FAD mice model | restored the levels of SPMs, decreased Aβ pathology | reversed the inflammatory process, and decreased the neuroinflammation associated with Aβ pathology | Kantarci and colleagues76 |
| LXA4, RvD1, PD1, MaR1 | human microglia and neuronal cells | improved neuronal survival and increased microglia phagocytosis of Aβ | modulated microglia phenotype | Zhu and colleagues73 |
Aβ, beta amyloid; AD, Alzheimer’s disease; ADAM10, A disintegrin and metalloproteinase domain-containing protein 10; ATL, aspirin-triggered lipoxin; B-94, a TNF-B inducible proinflammatory element; BACE1, β secretase 1; ChemR23, chemerin receptor 23; COX-2, cyclooxygenase 2; CSF, cerebrospinal fluid; EIA, enzyme immune assay; I cyc nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha; IL, interleukin; LC-MS-MS, liquid chromatography tandem mass spectrometry; LXA4, lipoxin A4; MaR1, maresin 1; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B-cells; p38 MAPK, p38 mitogen-activated protein kinase; PBMC, peripheral blood mononuclear cell; PD1, protectin D1; SPM, specialized pro-resolving lipid mediator; RvD1, resolvin D1; RvD5, resolvin D5; Tg, transgenic; TNF-α tumor necrosis factor alpha.
LXA4
The level of LXA4 has been shown to be reduced in post-mortem AD brains and in various AD animal models inluding 3×Tg mice and 5×FAD mice.72,73,85 Furthermore, LXA4 has been shown to be protective in AD-related animal and cellular studies.68,69,85 Treatment of aspirin-triggered LXA4 (ATL) has been shown improved cognition in Tg2576 transgenic AD mice through mechanisms related to reduced nuclear factor kappa B (NF-κB) activation and levels of proinflammatory cytokines and chemokines, as well as increased levels of anti-inflammatory cytokines IL-10 and transforming growth factor-β. In addition, ATL was shown to modify microglia phenotypes from the proinflammatory M1 to the anti-inflammatory M2.68,69 In line with these findings, another study using 3×Tg AD mice showed that ATL treatment improved the memory impairment of 3×Tg AD mice, reduced Aβ deposition, and decreased tau phosphorylation. Moreover, the decrease in p-tau was associated with the inhibition of the tau kinases GSK-3β and p38 mitogen-activated protein kinase (MAPK). In addition, ATL also reduced microglial and astrocyte activation.69 Most recently, another study utilizing 5×FAD mice model showed that LXA4 treatment decreased Aβ pathology as well as the inflammation induced by Aβ.85 In Aβ-stimulated BV2 cells, LXA4 has been shown to decrease the levels of proinflammatory cytokines IL-1β and TNFα, inhibiting the degradation of IκBα, and the translocation of the NF-κB p65 subunit into the nucleus induced by Aβ.86 We have previously found that LXA4 promotes neuronal survival in staurosporine (STS)-induced neuronal death.73 In summary, the levels of LXA4 are decreased in AD and treatment with LXA4 is protective in AD by reducing Aβ deposition and tau phosphorylation, inhibiting inflammatory cytokine production, modifying microglial phenotypes and inhibiting NF-κB signaling.
Resolvins
The levels of RvE1 were found to be significantly lower in the hippocampus of 5×FAD mice than in control mice and administration of RvE1 restored the levels of RvE1 in these mice. Furthermore, RvE1 has been shown to decrease Aβ pathology and to restore the homeostasis for inflammatory cytokines and chemokines such as GM-CSF, interferon (IFN)-γ, IL-1β, MCP-1, MIP-1α, and MIP-1β.85 Another study utilizing macrophages derived from AD patients showed that RvD1 increased the phagocytosis of Aβ and inhibited fibrillar Aβ-induced apoptosis via MEK1/2, PKA, and PI3K signaling.83 In the same study, the authors also showed that RvD1 downregulated soluble Aβ (sAβ) induced secretions of proinflammatory cytokines and chemokines.83 In accord with these reports, our group has observed that RvD1 improved neuronal survival induced by STS in differentiated SH-SY5Y cells. We also showed RvD1 downregulated Aβ42-induced up-regulation of CD11b in human microglia cells.73 In summary, the levels of resolvins are decreased in AD and treatment with resolvins are protective in AD by reducing antibody pathology, decreasing inflammatory mediators producing modifying microglia phenotype, and promoting microglia phagocytosis via mechanisms associated with MEK, PKA and PI3K signaling.
PD1
The levels of PD1 have been shown to be decreased in human post-mortem AD brains and in 3×Tg AD mice.70 Moreover, it has been demonstrated that PD1 promoted neuronal cell survival via the induction of anti-apoptotic genes encoding Bcl-2, Bcl-xl, and Bfl-1(A1) and repressed Aβ42-triggered activation of proinflammatory genes in a human neuron-glia (HNG) coculture model.70 In addition, the authors also showed that PD1 downregulated Aβ42-triggered expression of the proinflammatory enzyme cyclooxygenase-2 (COX-2) and of B-94 (a TNF-α-inducible proinflammatory element) and apoptosis in HNG cells. In human neural cells transfected with βAPPsw (Swedish double mutation APP695sw, K595N-M596L), PD1 was shown to suppress Aβ42 production by down-regulating β-secretase (BACE1) while activating α-secretase (ADAM10), thus shifting the APP cleavage from the amyloidogenic pathway into nonamyloidogenic pathway.45 The PD1-mediated downregulation of BACE1 and Aβ42 peptide release is PPARγ-dependent in 3×Tg AD model.45 In summary, the levels of PD1 are decreased in AD and treatment with PD1 is protective by inducing the anti-apoptotic gene and inhibiting inflammatory gene expression, modify APP processing, and promoting PPAR-γ signaling. More studies are needed to further clarify the mechanism of PD1 on AD.
MaR1
Our group has previously shown that the levels MaR1 were lower in the entorhinal cortex (ENT) of AD patients as compared with age-matched controls. In vitro studies showed MaR1exerted neuroprotective activity, and that MaR1down-regulated Aβ42-induced up-regulation of CD40 in human microglia.73 Moreover, MaR1 increased microglial phagocytosis of Aβ42.73 However, animal studies regarding the effects of MaR1 on AD animal model are lacking. However, the effects of MaR1 on other inflammatory models have been reported. MaR1 has been shown to inhibit neutrophil infiltration in murine peritonitis as well as enhancing human macrophage uptake of apoptotic neutrophils.87 Interestingly, MaR1 also accelerated surgical regeneration in Planaria sp., increasing the rate of head reappearance.64 In experimental colitis, MaR1 has been showed to be protective by inhibiting the NF-κB pathway and decreasing the production of proinflammatory mediators, as well as by shifting the macrophage phenotype from M1 to M2.54 In IL-10−/− mice with spontaneous colitis, MaR1 ameliorates iron-deficient anemia by the inhibition of hepcidin expression though the IL-6/STAT3 pathway.88 In the future, the effects of MaR1 on AD animal and cellular models and its mechanisms need further investigation.
Modulation of enzymes involved in the formation of SPMs
5-LOX
In the brain, 5-LOX is expressed by all cell types,89 and higher levels have been shown in the AD brain and that of an AD mouse model.90 The role of 5-LOX in AD is not completely understood. Crossing 5-LOX knockout (KO) mice with a mouse model for AD, based on transgenic expression of mutated human APP, resulted in reduced Aβ pathology and improved cognitive performance.91 Interestingly, 5-LOX inhibitor minocycline has been shown to inhibit β-secretase (BACE) 1, and reduce Aβ pathology and inflammation in APP transgenic mice.92 5-LOX is activated by phosphorylation at three currently known sites, Ser663, Ser271, and Ser523. Ser271 is phosphorylated by P38 MAPK, and extracellular-signal-regulated kinases phosphorylate 5-LOX at Ser663.36 Interestingly, phosphorylation of 5-LOX at Ser523 has been reported to shift the production of LMs towards LXA4, while the production of LTB4 is decreased, and Ser523 is therefore considered to be an anti-inflammatory phosphorylation site involved in class-switching.93,94 Above all, 5-LOX functions as a double edged sword in AD. 5-LOX modulators capable of inhibiting its proinflammatory activity or increasing phosphorylation at Ser523, and promoting class-switching without increasing the production of proinflammatory LMs, are of potential interest.
15-LOX-1 and 15-LOX-2
There are two isoforms of 15-LOX found in the brain: 15-LOX-1 and 15-LOX-2.72,95 15-LOX is implicated in many diseases, including AD.96 It has been reported that in the AD brain, in pathologically affected areas such as the frontal and temporal regions, the levels of 15-LOX-1 are higher, and that the concentration of its metabolic product 15-hydroxyeicosatetraenoic acid (15-HETE) is markedly elevated in the CSF of patients with AD compared with controls.95,97 The levels of 15-LOX-2 were found to be elevated in the AD brain as well.72 The role of 15-LOX in AD is unclear, since 15-LOX-1 can directly oxidize lipids in the cell membrane,98 generating oxidative stress that can be detrimental to neurons. However, 15-LOX is also involved in the synthesis of SPMs and is thus related to anti-inflammatory and pro-resolving activities. Inhibition of 15-LOX-1 in APP-over-expressing neuroblastoma cells significantly reduced the levels of soluble APP and BACE.99 Moreover, crossing APP transgenic mice with 15-LOX KO mice resulted in a progeny with reduced Aβ pathology.100 To summarize, 15-LOX-1 may play a dual role in AD, but further studies are needed to elucidate the biology of these enzymes and how it is regulated.
COX-1 and COX-2
The genes encoding COX-1 and COX-2 are located on chromosome 9 and 1, respectively. In an immunohistochemical study on post-mortem brain tissue from the frontal and temporal cortex COX-1 immunoreactivity was found both in neurons and microglial cells, while COX-2 immunoreactivity was found only in neurons.101 The number of COX-2-positive neurons was higher in AD brains compared with control brains.101 Later studies showed increased levels of COX-2 in the early stages of AD, whereas decreased levels were observed at a late stage of the disease.102,103 Aspirin is a well-known NSAID, widely used for the treatment of inflammation, and low doses are prescribed for the prevention of cardiovascular events. The mechanism of aspirin’s action is one of irreversible acetylation of COX-1 and COX-2, thereby affecting their catalytic activity and inhibiting the synthesis of PGs and thromboxanes (TXs).104 Interestingly, aspirin was found to stimulate the formation of a novel series of aspirin-triggered lipoxins (ATLs) during the co-incubation of human umbilical vein endothelial cells with neutrophils.105 Later studies showed that ATLs inhibited neutrophil infiltration in a mouse ear inflammation model, showing anti-inflammatory and pro-resolving properties.106 Further studies on the mechanisms of ATL synthesis showed that acetylated COX-2 is still active and can convert AA to 15-RHETE and give rise to ATL.107 ATL has also been shown to improve memory function and ameliorate AD pathology in the 3xTg mouse model.68 It is hoped that the development of more specific modulators of COXs could help to promote the production of SPMs without causing the side effects, such as gastrointestinal (GI) irritation,108 associated with aspirin.
Activation of receptors for SPMs
We have previously shown alterations in the levels of receptors for SPMs in the AD brain.72 Both neurons and glial cells express ChemR23 in the human brain, with higher levels found in AD compared with control participants.72 Similarly, ALX/FPR2 was detected in both neurons and glia in the human brain, with higher levels in AD.72 Furthermore, the levels of PPAR-γ were higher in the hippocampus of patients with AD.72 The increased levels of SPM receptors in the AD brain may be interpreted as a compensatory mechanism for the reduced levels of SPMs.72 Interestingly, at least some of the anti-inflammatory effects of SPMs seem to be due to the competition of SPMs with proinflammatory ligands such as LTB4, which act as partial agonists or antagonists.41 The PPAR-γ agonist rosiglitazone has been shown to increase the production of LXA4,57 however, PPAR-γ has been shown to bind other ligands and have other biological functions, such as a glucose-reducing capacity. Thus, rosiglitazone has been used for the treatment of diabetes. Also, other receptors of SPMs can mediate transduction of a proinflammatory signal, for example, ALX/FPR2 has been found to bind Aβ,42 which is known to activate microglia. However, whether this binding results in proinflammatory signals remains to be shown. ChemR23 binds chemerin,43 a chemotactic peptide43 associated with increased inflammation.44 To summarize, owing to the multi-ligand binding capacity of the receptors for SPMs, the mechanism underpinning the transduction of pro-resolving signaling must be investigated if we are to target this pathway. Partial agonists of these receptors capable of stimulating the resolution of inflammation without activating proinflammatory signals would be ideal for this purpose.
Comparison between anti-inflammatory and pro-resolving strategies in treating AD
As we have discussed before, different from an anti-inflammatory strategy, which is based on inhibiting the production of proinflammatory mediators, a pro-resolving strategy is one where positive activation of the resolution of inflammation is more likely to naturally occur in physiological conditions. Anti-inflammatory drugs have been tested in the AD field, although some of these drugs showed positive effects in animal models or in phase I/II clinical trials, not all of them showed efficacy in large double blind randomized large phase III clinical trials. Many reasons have been proposed for this, such as differences between animal models and human beings or because the treatment was too late in the pathological phase of AD; indeed, inflammation has been shown decades before the clinical symptoms onset. Although the pro-resolving strategy has not yet been tested in clinical trials, in principle, it has many advantages towards an anti-inflammatory strategy. In this section, we will summarize the anti-inflammatory drugs and discuss the advantages of the pro-resolving strategy over the anti-inflammatory strategy.
Different stages of inflammation in AD
Neuroinflammation, discovered in AD more than 30 years ago, has now become a major field of AD research today. Inhibiting neuroinflammation in AD may be the key to successful treatment of AD. Epidemiological studies showed an association between the intake of NSAIDs and a lower prevalence of AD.109 A similar association was found for the intake of n-3 FAs.110 Inspired by the epidemiological studies, several studies have been carried out to investigate the possibility to treat and prevent AD by NSAIDs.111 However, these studies have rendered varying results. Heterogeneous study populations may mask small improvements in cognition, and it may be that there are forms of pathology in AD that inhibit the beneficial effects of NSAIDs.
The question whether inflammation is a cause or the consequence of the pathology in AD is controversial, and little is known regarding the role of resolution in AD. To pinpoint the role of inflammation and resolution of inflammation in AD, we need to know how they change during the pathological course of the disease in comparison with normal aging. Aging is associated with a gradual increase in inflammation as shown by increased microglial activation112,113 and increased plasma levels of proinflammatory cytokines such as IL-6 and TNF-α.114 Resolution of inflammation in normal human aging has not been investigated. However, in a senescence-accelerated mouse model (SAMP8), resolution of inflammation was suggested to be insufficient to cope with the increased levels of inflammation associated with aging.115 AD is associated with increased inflammation compared with age-matched controls.116 However, in general, longitudinal studies that monitor inflammation in the pathological course of AD are lacking. Mild cognitive impairment (MCI) is considered as the prodromal stages of AD, and therefore comparing inflammation-related changes in MCI with AD and with healthy controls can give some hints about changes of inflammation during the progression AD. It seems that the changes of inflammatory cytokines during the disease are very dynamic. At the individual cytokine level, there are data showing an increase, a temporary increase, unchanged or decreased levels117 during the pathological course of AD, indicating the complexity of the inflammatory process in AD. The differentially altered inflammatory cytokines during the progression of AD may play distinct roles at different stages of the disease. The levels of LXA4 in CSF were found to be lower in AD compared with both MCI and control participants. It may be speculated that the pro-resolution signal steadily decreases as AD pathology progresses117
Advantages of the pro-resolving strategy over the anti-inflammatory strategy
Clinical trials regarding anti-inflammatory treatment have given disappointing results. Even though no clinical trials have been tried regarding pro-resolving therapy in AD, it may have advantages over the anti-inflammatory therapy. First of all, the anti-inflammatory therapy with NSAIDs is based on inhibiting the inflammation pathway, which may have its own physiological role, and therefore, may have side effects. On the contrary, the pro-resolving strategy is based on activating the pro-resolution pathway as it naturally occurs. It is plausible that the pro-resolving method may have fewer side effects. Secondly, as we have discussed, the failure of the anti-inflammatory therapy may partly be due to the treatment coming too late in the AD pathological phase, when the pathological changes already exist and are difficult to revise. The pro-resolving strategy includes two aspects: anti-inflammation and repairing. Since SPMs have been shown as potentially promoting the regeneration in animal models,64 it can be hypothesized that SPMs can stimulate neuronal regeneration, therefore, giving hope to patients who were at the later phase of AD. However, clinical trials of SPMs on AD are needed to further evaluate the role of SPMs in AD.
Questions need to be considered before SPMs can be used on AD patients
Before their use on patients with AD can be assessed, several questions must be addressed. Safety is one of the most important aspects to consider. Omega-3 FAs have been shown to contribute to an increased risk of bleeding and hemorrhagic stroke, owing to their effects on platelet aggregation.118 Omega-3 FA supplementation has also been associated with suppressed immune responses to infections.119–121 No side effects of SPMs have been reported so far; however, since they are the downstream products of omega-3 FAs, this must be investigated. Considering that SPMs are relatively new subjects of research, more studies focused on safety are needed to rule out potential harmful effects. The administration route must also be considered. There is no direct evidence showing that SPMs can pass the blood–brain barrier (BBB). However, since SPMs are small lipophilic molecules, it is conceivable that they can cross the BBB, similarly to their precursors, DHA and EPA. The choice of SPMs for use in treatments is another important point of consideration. Our in vitro studies have shown different efficacy with respect to neuroprotection, anti-inflammation, and phagocytosis.73 In addition, there are SPM analogs for which the biological activities are largely unknown. Whether one SPM is more effective than another, or whether a combination is stronger, requires further investigation.
Conclusions and future perspectives
To summarize, stimulation of resolution of inflammation is beneficial by increasing removal of Aβ via phagocytosis, counter-regulating Aβ induced proinflammatory microglial activation, and preventing neuronal cell death. However, factors affecting resolution such as AD-related pathology need to be taken into consideration when translating the results from cellular studies to the clinical situation. Promoting resolution of inflammation represents a novel strategy for treatment of inflammatory disorders. However, numerous studies are required to enable us to fully understand the mechanisms of resolution before it can be successfully translated to the clinic. First, resolution of inflammation includes two factors: anti-inflammation and repair. The repair mechanisms have received less attention. In the case of diseases involving chronic inflammation, such as AD, in which the tissue has already been damaged, it is important to determine whether stimulating pro-resolving activities can initiate repair and neuronal regeneration. Second, promoting endogenous production of SPMs, potentially by stimulating class-switching mechanisms, is of great interest since the stability of SPMs in the brain is not known. Modulators of COXs and LOXs, as well as PPAR-γ agonists, are also of potential interest. Furthermore, SPMs are produced under physiological conditions, but it remains to be determined whether they have functions separate from their role in resolving inflammation. Lastly, SPMs are end products of PUFAs in the metabolic pathways of COXs and LOXs, and there are many intermediates in these pathways. The functions of these molecules are largely unknown, and merit further investigation.
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing. Mingqin Zhu and Xiuzhe Wang contributed equally to this work.
Footnotes
Funding: The authors are grateful for the support from the grants from the National Science Foundation of China (No. 31600820, NO. 81501089); The Health and Family Planning Commission of Jilin Province (No. 2016Q036); The Science and technology planning project of Jilin Province (No. 20180520110JH); The Swedish Research Council (22743, 22744); Swedish Brain Power; The Chinese Scholarship Council, P.R. China; The Knut and Alice Wallenberg Foundation; Karolinska Institutet research funds; Stiftelsen för Gamla Tjänarinnor; The Swedish Alzheimer Foundation; Gun och Bertil Stohnes Stiftelse; and Barmore Fund (MUSC).
Conflict of interest statement: The authors declare that there is no conflict of interest.
Contributor Information
Mingqin Zhu, Department of Neurology and Neuroscience Center, First Hospital of Jilin University, Xinmin Street No 71, Changchun 130000, China.
Xiuzhe Wang, Department of Neurology, Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, Shanghai, China.
Li Sun, Department of Neurology and Neuroscience Center, First Hospital of Jilin University, Changchun, China.
Marianne Schultzberg, Department of Neurobiology, Care Sciences & Society, Section of Neurodegeneration, Karolinska Institutet, Center for Alzheimer Research, Sweden.
Erik Hjorth, Department of Neurobiology, Care Sciences & Society, Section of Neurodegeneration, Karolinska Institutet, Center for Alzheimer Research, Sweden.
References
- 1. Hardy J. A hundred years of Alzheimer’s disease research. Neuron 2006; 52: 3–13. [DOI] [PubMed] [Google Scholar]
- 2. Lue LF, Rydel R, Brigham EF, et al. Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia 2001; 35: 72–79. [DOI] [PubMed] [Google Scholar]
- 3. Reitz C. Alzheimer’s disease and the amyloid cascade hypothesis: a critical review. Int J Alzheimers Dis 2012; 2012: 369808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zilka N, Kazmerova Z, Jadhav S, et al. Who fans the flames of Alzheimer’s disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J Neuroinflammation 2012; 9: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McGeer PL, Itagaki S, Tago H, et al. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett 1987; 79: 195–200. [DOI] [PubMed] [Google Scholar]
- 6. McGeer PL, Itagaki S, Boyes BE, et al. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988; 38: 1285–1291. [DOI] [PubMed] [Google Scholar]
- 7. Eikelenboom P, Stam FC. Immunoglobulins and complement factors in senile plaques. An immunoperoxidase study. Acta Neuropathol 1982; 57: 239–242. [DOI] [PubMed] [Google Scholar]
- 8. Griffin WS, Stanley LC, Ling C, et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in down syndrome and Alzheimer disease. Proc Natl Acad Sci USA 1989; 86: 7611–7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cagnin A, Brooks DJ, Kennedy AM, et al. In-vivo measurement of activated microglia in dementia. Lancet 2001; 358: 461–467. [DOI] [PubMed] [Google Scholar]
- 10. Tosto G, Reitz C. Genome-wide association studies in Alzheimer’s disease: a review. Curr Neurol Neurosci Rep 2013; 13: 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meda L, Cassatella MA, Szendrei GI, et al. Activation of microglial cells by β-amyloid protein and interferon-γ. Nature 1995; 374: 647–650. [DOI] [PubMed] [Google Scholar]
- 12. Hu J, Akama KT, Krafft GA, et al. Amyloid-β peptide activates cultured astrocytes: morphological alterations, cytokine induction and nitric oxide release. Brain Res 1998; 785: 195–206. [DOI] [PubMed] [Google Scholar]
- 13. Del Bo R, Angeretti N, Lucca E, et al. Reciprocal control of inflammatory cytokines, IL-1 and IL-6, and Aβ production in cultures. Neurosci Lett 1995; 188: 70–74. [DOI] [PubMed] [Google Scholar]
- 14. Blasko I, Veerhuis R, Stampfer-Kountchev M, et al. Costimulatory effects of interferon-γ and interleukin-1β or tumor necrosis factor α on the synthesis of Aβ1–40 and Aβ1–42 by human astrocytes. Neurobiol Dis 2000; 7: 682–689. [DOI] [PubMed] [Google Scholar]
- 15. Dash PK, Moore AN. Enhanced processing of APP induced by IL-1β can be reduced by indomethacin and nordihydroguaiaretic acid. Biochem Biophys Res Commun 1995; 208: 542–548. [DOI] [PubMed] [Google Scholar]
- 16. McGeer PL, McGeer E, Rogers J, et al. Anti-inflammatory drugs and Alzheimer disease. Lancet 1990; 335: 1037. [DOI] [PubMed] [Google Scholar]
- 17. Group AR, Lyketsos CG, Breitner JC, et al. Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology 2007; 68: 1800–1808. [DOI] [PubMed] [Google Scholar]
- 18. Reines SA, Block GA, Morris JC, et al. Rofecoxib: no effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology 2004; 62: 66–71. [DOI] [PubMed] [Google Scholar]
- 19. Scharf S, Mander A, Ugoni A, et al. A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer’s disease. Neurology 1999; 53: 197–201. [DOI] [PubMed] [Google Scholar]
- 20. O’Bryant Sea. A proinflammatory endophenotype predicts treatment response in a multicenter trial of NSAIDs in AD. Alzheimers Dement 2014; 10: P273–P274. [Google Scholar]
- 21. Scott A, Khan KM, Cook JL, et al. What is “inflammation”? Are we ready to move beyond Celsus? Br J Sports Med 2004; 38: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schultzberg M, Lindberg C, Aronsson AF, et al. Inflammation in the nervous system - physiological and pathophysiological aspects. Physiol Behav 2007; 92: 121–128. [DOI] [PubMed] [Google Scholar]
- 23. Serhan CN. Discovery of specialized pro-resolving mediators marks the dawn of resolution physiology and pharmacology. Mol Aspects Med 2017; 58: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Serhan CN. Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. FASEB J 2017; 31: 1273–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chiurchiu V, Leuti A, Dalli J, et al. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci Transl Med 2016; 8: 353ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Colas RA, Dalli J, Chiang N, et al. Identification and actions of the maresin 1 metabolome in infectious inflammation. J Immunol 2016; 197: 4444–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Q, Zhu B, Li Y. Resolution of cancer-promoting inflammation: a new approach for anticancer therapy. Front Immunol 2017; 8: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014; 510: 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hedqvist P, Raud J, Palmertz U, et al. Lipoxin A4 inhibits leukotriene B4-induced inflammation in the hamster cheek pouch. Acta Physiol Scand 1989; 137: 571–572. [DOI] [PubMed] [Google Scholar]
- 30. Chiang N, Takano T, Clish CB, et al. Aspirin-triggered 15-epi-lipoxin A4 (ATL) generation by human leukocytes and murine peritonitis exudates: development of a specific 15-epi-LXA4 ELISA. J Pharmacol Exp Ther 1998; 287: 779–790. [PubMed] [Google Scholar]
- 31. Chiang N, Gronert K, Clish CB, et al. Leukotriene B4 receptor transgenic mice reveal novel protective roles for lipoxins and aspirin-triggered lipoxins in reperfusion. J Clin Invest 1999; 104: 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Serhan CN. Novel chemical mediators in the resolution of inflammation: resolvins and protectins. Anesthesiol Clin 2006; 24: 341–364. [DOI] [PubMed] [Google Scholar]
- 33. Serhan CN, Petasis NA. Resolvins and protectins in inflammation resolution. Chem Rev 2011; 111: 5922–5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Asatryan A, Bazan NG. Molecular mechanisms of signaling via the docosanoid neuroprotectin D1 for cellular homeostasis and neuroprotection. J Biol Chem 2017; 292: 12390–12397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Radmark O, Werz O, Steinhilber D, et al. 5-Lipoxygenase, a key enzyme for leukotriene biosynthesis in health and disease. Biochim Biophys Acta 2015; 1851: 331–339. [DOI] [PubMed] [Google Scholar]
- 36. Radmark O, Werz O, Steinhilber D, et al. 5-Lipoxygenase: regulation of expression and enzyme activity. Trends Biochem Sci 2007; 32: 332–341. [DOI] [PubMed] [Google Scholar]
- 37. Levy BD, Clish CB, Schmidt B, et al. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol 2001; 2: 612–619. [DOI] [PubMed] [Google Scholar]
- 38. Maddox JF, Hachicha M, Takano T, et al. Lipoxin A4 stable analogs are potent mimetics that stimulate human monocytes and THP-1 cells via a G-protein-linked lipoxin A4 receptor. J Biol Chem 1997; 272: 6972–6978. [DOI] [PubMed] [Google Scholar]
- 39. Krishnamoorthy S, Recchiuti A, Chiang N, et al. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc Natl Acad Sci USA 2010; 107: 1660–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Arita M, Bianchini F, Aliberti J, et al. Stereochemical assignment, anti-inflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med 2005; 201: 713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Arita M, Ohira T, Sun YP, et al. Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J Immunol 2007; 178: 3912–3917. [DOI] [PubMed] [Google Scholar]
- 42. Le Y, Gong W, Tiffany HL, et al. Aβ42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1. J Neurosci 2001; 21: RC123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bondue B, Wittamer V, Parmentier M. Chemerin and its receptors in leukocyte trafficking, inflammation and metabolism. Cytokine Growth Factor Rev 2011; 22: 331–338. [DOI] [PubMed] [Google Scholar]
- 44. Fulop P, Seres I, Lorincz H, et al. Association of chemerin with oxidative stress, inflammation and classical adipokines in non-diabetic obese patients. J Cell Mol Med 2014; 18: 1313–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhao Y, Calon F, Julien C, et al. Docosahexaenoic acid-derived neuroprotectin D1 induces neuronal survival via secretase- and PPARγ-mediated mechanisms in Alzheimer’s disease models. PLoS One 2011; 6: e15816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oh DY, Talukdar S, Bae EJ, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010; 142: 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miyata J, Fukunaga K, Iwamoto R, et al. Dysregulated synthesis of protectin D1 in eosinophils from patients with severe asthma. J Allergy Clin Immunol 2013; 131: 353–360 e351–352. [DOI] [PubMed] [Google Scholar]
- 48. Planaguma A, Kazani S, Marigowda G, et al. Airway lipoxin A4 generation and lipoxin A4 receptor expression are decreased in severe asthma. Am J Respir Crit Care Med 2008; 178: 574–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fredman G, Oh SF, Ayilavarapu S, et al. Impaired phagocytosis in localized aggressive periodontitis: rescue by Resolvin E1. PLoS One 2011; 6: e24422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Aoki H, Hisada T, Ishizuka T, et al. Resolvin E1 dampens airway inflammation and hyperresponsiveness in a murine model of asthma. Biochem Biophys Res Commun 2008; 367: 509–515. [DOI] [PubMed] [Google Scholar]
- 51. Levy BD, Lukacs NW, Berlin AA, et al. Lipoxin A4 stable analogs reduce allergic airway responses via mechanisms distinct from CysLT1 receptor antagonism. FASEB J 2007; 21: 3877–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Flesher RP, Herbert C, Kumar RK. Resolvin E1 promotes resolution of inflammation in a mouse model of an acute exacerbation of allergic asthma. Clin Sci (Lond) 2014; 126: 805–814. [DOI] [PubMed] [Google Scholar]
- 53. Fiorucci S, Wallace JL, Mencarelli A, et al. A β-oxidation-resistant lipoxin A4 analog treats hapten-induced colitis by attenuating inflammation and immune dysfunction. Proc Natl Acad Sci USA 2004; 101: 15736–15741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Marcon R, Bento AF, Dutra RC, et al. Maresin 1, a proresolving lipid mediator derived from omega-3 polyunsaturated Fatty acids, exerts protective actions in murine models of colitis. J Immunol 2013; 191: 4288–4298. [DOI] [PubMed] [Google Scholar]
- 55. Yamada T, Tani Y, Nakanishi H, et al. Eosinophils promote resolution of acute peritonitis by producing proresolving mediators in mice. FASEB J 2011; 25: 561–568. [DOI] [PubMed] [Google Scholar]
- 56. Bazan NG, Eady TN, Khoutorova L, et al. Novel aspirin-triggered neuroprotectin D1 attenuates cerebral ischemic injury after experimental stroke. Exp Neurol 2012; 236: 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sobrado M, Pereira MP, Ballesteros I, et al. Synthesis of lipoxin A4 by 5-lipoxygenase mediates PPARγ-dependent, neuroprotective effects of rosiglitazone in experimental stroke. J Neurosci 2009; 29: 3875–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wu Y, Wang YP, Guo P, et al. A lipoxin A4 analog ameliorates blood-brain barrier dysfunction and reduces MMP-9 expression in a rat model of focal cerebral ischemia-reperfusion injury. J Mol Neurosci 2012; 46: 483–491. [DOI] [PubMed] [Google Scholar]
- 59. Ye XH, Wu Y, Guo PP, et al. Lipoxin A4 analogue protects brain and reduces inflammation in a rat model of focal cerebral ischemia reperfusion. Brain Res 2010; 1323: 174–183. [DOI] [PubMed] [Google Scholar]
- 60. Wu L, Miao S, Zou LB, et al. Lipoxin A4 inhibits 5-lipoxygenase translocation and leukotrienes biosynthesis to exert a neuroprotective effect in cerebral ischemia/reperfusion injury. J Mol Neurosci 2012; 48: 185–200. [DOI] [PubMed] [Google Scholar]
- 61. Belayev L, Mukherjee PK, Balaszczuk V, et al. Neuroprotectin D1 upregulates Iduna expression and provides protection in cellular uncompensated oxidative stress and in experimental ischemic stroke. Cell Death Differ 2017; 24: 1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Huang L, Wang CF, Serhan CN, et al. Enduring prevention and transient reduction of postoperative pain by intrathecal resolvin D1. Pain 2011; 152: 557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hu S, Mao-Ying QL, Wang J, et al. Lipoxins and aspirin-triggered lipoxin alleviate bone cancer pain in association with suppressing expression of spinal proinflammatory cytokines. J Neuroinflammation 2012; 9: 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Serhan CN, Dalli J, Karamnov S, et al. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J 2012; 26: 1755–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xu ZZ, Liu XJ, Berta T, et al. NpD/PD1 protects against neuropathic pain in mice after nerve trauma. Ann Neurol 2013; 74: 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Park CK, Lu N, Xu ZZ, et al. Resolving TRPV1- and TNF-α-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. J Neurosci 2011; 31: 15072–15085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Calandria JM, Sharp MW, Bazan NG. The docosanoid neuroprotectin D1 induces TH-positive neuronal survival in a cellular model of Parkinson’s disease. Cell Mol Neurobiol 2015; 35: 1127–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Medeiros R, Kitazawa M, Passos GF, et al. Aspirin-triggered lipoxin A4 stimulates alternative activation of microglia and reduces Alzheimer disease-like pathology in mice. Am J Pathol 2013; 182: 1780–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dunn HC, Ager RR, Baglietto-Vargas D, et al. Restoration of lipoxin A4 signaling reduces Alzheimer’s disease-like pathology in the 3xTg-AD mouse model. J Alzheimers Dis 2015; 43: 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lukiw WJ, Cui JG, Marcheselli VL, et al. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest 2005; 115: 2774–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kang J, Rivest S. Lipid metabolism and neuroinflammation in Alzheimer’s disease: a role for liver X receptors. Endocr Rev 2012; 33: 715–746. [DOI] [PubMed] [Google Scholar]
- 72. Wang X, Zhu M, Hjorth E, et al. Resolution of inflammation is altered in Alzheimer’s disease. Alzheimers Dement 2015; 11: 40–50 e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhu M, Wang X, Hjorth E, et al. Pro-resolving lipid mediators improve neuronal survival and increase abeta42 phagocytosis. Mol Neurobiol 2016; 53: 2733–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kalmijn S, Launer LJ, Ott A, et al. Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Ann Neurol 1997; 42: 776–782. [DOI] [PubMed] [Google Scholar]
- 75. Engelhart MJ, Geerlings MI, Ruitenberg A, et al. Diet and risk of dementia: does fat matter?: the Rotterdam Study. Neurology 2002; 59: 1915–1921. [DOI] [PubMed] [Google Scholar]
- 76. Barberger-Gateau P, Letenneur L, Deschamps V, et al. Fish, meat, and risk of dementia: cohort study. BMJ 2002; 325: 932–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fiala M, Kooij G, Wagner K, et al. Modulation of innate immunity of patients with Alzheimer’s disease by omega-3 fatty acids. FASEB J 2017; 31: 3229–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Famenini S, Rigali EA, Olivera-Perez HM, et al. Increased intermediate M1-M2 macrophage polarization and improved cognition in mild cognitive impairment patients on omega-3 supplementation. FASEB J 2017; 31: 148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Freund-Levi Y, Eriksdotter-Jonhagen M, Cederholm T, et al. Omega-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD study: a randomized double-blind trial. Arch Neurol 2006; 63: 1402–1408. [DOI] [PubMed] [Google Scholar]
- 80. Chiu CC, Su KP, Cheng TC, et al. The effects of omega-3 fatty acids monotherapy in Alzheimer’s disease and mild cognitive impairment: a preliminary randomized double-blind placebo-controlled study. Prog Neuropsychopharmacol Biol Psychiatry 2008; 32: 1538–1544. [DOI] [PubMed] [Google Scholar]
- 81. Rogers PJ, Appleton KM, Kessler D, et al. No effect of n-3 long-chain polyunsaturated fatty acid (EPA and DHA) supplementation on depressed mood and cognitive function: a randomised controlled trial. Br J Nutr 2008; 99: 421–431. [DOI] [PubMed] [Google Scholar]
- 82. Yurko-Mauro K, McCarthy D, Rom D, et al. Beneficial effects of docosahexaenoic acid on cognition in age-related cognitive decline. Alzheimers Dement 2010; 6: 456–464. [DOI] [PubMed] [Google Scholar]
- 83. Mizwicki MT, Liu G, Fiala M, et al. 1α,25-dihydroxyvitamin D3 and resolvin D1 retune the balance between amyloid-β phagocytosis and inflammation in Alzheimer’s disease patients. J Alzheimers Dis 2013; 34: 155–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fiala M, Terrando N, Dalli J. Specialized pro-resolving mediators from omega-3 fatty acids improve amyloid-beta phagocytosis and regulate inflammation in patients with minor cognitive impairment. J Alzheimers Dis 2015; 48: 293–301. [DOI] [PubMed] [Google Scholar]
- 85. Kantarci A, Aytan N, Palaska I, et al. Combined administration of resolvin E1 and lipoxin A4 resolves inflammation in a murine model of Alzheimer’s disease. Exp Neurol 2018; 300: 111–120. [DOI] [PubMed] [Google Scholar]
- 86. Wu J, Wang A, Min Z, et al. Lipoxin A4 inhibits the production of proinflammatory cytokines induced by beta-amyloid in vitro and in vivo. Biochem Biophys Res Commun 2011; 408: 382–387. [DOI] [PubMed] [Google Scholar]
- 87. Serhan CN, Yang R, Martinod K, et al. Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med 2009; 206: 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wang H, Shi P, Huang C, et al. Maresin 1 ameliorates iron-deficient anemia in IL-10(-/-) mice with spontaneous colitis by the inhibition of hepcidin expression though the IL-6/STAT3 pathway. Am J Transl Res 2016; 8: 2758–2766. [PMC free article] [PubMed] [Google Scholar]
- 89. Zhang L, Zhang WP, Hu H, et al. Expression patterns of 5-lipoxygenase in human brain with traumatic injury and astrocytoma. Neuropathol 2006; 26: 99–106. [DOI] [PubMed] [Google Scholar]
- 90. Ikonomovic MD, Abrahamson EE, Uz T, et al. Increased 5-lipoxygenase immunoreactivity in the hippocampus of patients with Alzheimer’s disease. J Histochem Cytochem 2008; 56: 1065–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Firuzi O, Zhuo J, Chinnici CM, et al. 5-Lipoxygenase gene disruption reduces Aβ pathology in a mouse model of Alzheimer’s disease. FASEB J 2008; 22: 1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ferretti MT, Allard S, Partridge V, et al. Minocycline corrects early, pre-plaque neuroinflammation and inhibits BACE-1 in a transgenic model of Alzheimer’s disease-like amyloid pathology. J Neuroinflammation 2012; 9: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ye Y, Lin Y, Perez-Polo JR, et al. Phosphorylation of 5-lipoxygenase at ser523 by protein kinase A determines whether pioglitazone and atorvastatin induce proinflammatory leukotriene B4 or anti-inflammatory 15-epi-lipoxin A4 production. J Immunol 2008; 181: 3515–3523. [DOI] [PubMed] [Google Scholar]
- 94. Luo M, Jones SM, Phare SM, et al. Protein kinase A inhibits leukotriene synthesis by phosphorylation of 5-lipoxygenase on serine 523. J Biol Chem 2004; 279: 41512–41520. [DOI] [PubMed] [Google Scholar]
- 95. Yao Y, Clark CM, Trojanowski JQ, et al. Elevation of 12/15 lipoxygenase products in AD and mild cognitive impairment. Ann Neurol 2005; 58: 623–626. [DOI] [PubMed] [Google Scholar]
- 96. Joshi YB, Giannopoulos PF, Pratico D. The 12/15-lipoxygenase as an emerging therapeutic target for Alzheimer’s disease. Trends Pharmacol Sci 2015; 36: 181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Pratico D, Zhukareva V, Yao Y, et al. 12/15-lipoxygenase is increased in Alzheimer’s disease: possible involvement in brain oxidative stress. Am J Pathol 2004; 164: 1655–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ivanov I, Heydeck D, Hofheinz K, et al. Molecular enzymology of lipoxygenases. Arch Biochem Biophys 2010; 503: 161–174. [DOI] [PubMed] [Google Scholar]
- 99. Succol F, Pratico D. A role for 12/15 lipoxygenase in the amyloid-β precursor protein metabolism. J Neurochem 2007; 103: 380–387. [DOI] [PubMed] [Google Scholar]
- 100. Yang H, Zhuo JM, Chu J, et al. Amelioration of the Alzheimer’s disease phenotype by absence of 12/15-lipoxygenase. Biol Psychiatry 2010; 68: 922–929. [DOI] [PubMed] [Google Scholar]
- 101. Hoozemans JJ, Rozemuller AJ, Janssen I, et al. Cyclooxygenase expression in microglia and neurons in Alzheimer’s disease and control brain. Acta Neuropathol 2001; 101: 2–8. [DOI] [PubMed] [Google Scholar]
- 102. Yermakova AV, O’Banion MK. Downregulation of neuronal cyclooxygenase-2 expression in end stage Alzheimer’s disease. Neurobiol Aging 2001; 22: 823–836. [DOI] [PubMed] [Google Scholar]
- 103. Hoozemans JJ, Rozemuller JM, van Haastert ES, et al. Cyclooxygenase-1 and -2 in the different stages of Alzheimer’s disease pathology. Curr Pharm Des 2008; 14: 1419–1427. [DOI] [PubMed] [Google Scholar]
- 104. Botting RM. Inhibitors of cyclooxygenases: mechanisms, selectivity and uses. J Physiol Pharmacol 2006; 57 Suppl 5: 113–124. [PubMed] [Google Scholar]
- 105. Claria J, Serhan CN. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte inter-actions. Proc Natl Acad Sci U S A 1995; 92: 9475–9479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Takano T, Fiore S, Maddox JF, et al. Aspirin-triggered 15-epi-lipoxin A4 (LXA4) and LXA4 stable analogues are potent inhibitors of acute inflammation: evidence for anti-inflammatory receptors. J Exp Med 1997; 185: 1693–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Serhan CN. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot Essent Fatty Acids 2005; 73: 141–162. [DOI] [PubMed] [Google Scholar]
- 108. Traversa G, Walker AM, Ippolito FM, et al. Gastroduodenal toxicity of different nonsteroidal antiinflammatory drugs. Epidemiology 1995; 6: 49–54. [DOI] [PubMed] [Google Scholar]
- 109. Beard CM, Kokman E, Kurland LT. Rheumatoid arthritis and susceptibility to Alzheimer’s disease. Lancet 1991; 337: 1426. [DOI] [PubMed] [Google Scholar]
- 110. Cunnane SC, Plourde M, Pifferi F, et al. Fish, docosahexaenoic acid and Alzheimer’s disease. Prog Lipid Res 2009; 48: 239–256. [DOI] [PubMed] [Google Scholar]
- 111. Imbimbo BP, Solfrizzi V, Panza F. Are NSAIDs useful to treat Alzheimer’s disease or mild cognitive impairment? Front Aging Neurosci 2010; 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Perry VH, Matyszak MK, Fearn S. Altered antigen expression of microglia in the aged rodent CNS. Glia 1993; 7: 60–67. [DOI] [PubMed] [Google Scholar]
- 113. Sheffield LG, Berman NE. Microglial expression of MHC class II increases in normal aging of nonhuman primates. Neurobiol Aging 1998; 19: 47–55. [DOI] [PubMed] [Google Scholar]
- 114. Wei J, Xu H, Davies JL, et al. Increase of plasma IL-6 concentration with age in healthy subjects. Life Sci 1992; 51: 1953–1956. [DOI] [PubMed] [Google Scholar]
- 115. Wang X, Puerta E, Cedazo-Minguez A, et al. Insufficient resolution response in the hippocampus of a senescence-accelerated mouse model–SAMP8. J Mol Neurosci 2015; 55: 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Solito E, Sastre M. Microglia function in Alzheimer’s disease. Front Pharmacol 2012; 3: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Brosseron F, Krauthausen M, Kummer M, et al. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: a comparative overview. Mol Neurobiol 2014; 50: 534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Bright JM, Sullivan PS, Melton SL, et al. The effects of n-3 fatty acid supplementation on bleeding time, plasma fatty acid composition, and in vitro platelet aggregation in cats. J Vet Intern Med 1994; 8: 247–252. [DOI] [PubMed] [Google Scholar]
- 119. Kelley DS, Taylor PC, Nelson GJ, et al. Dietary docosahexaenoic acid and immunocompetence in young healthy men. Lipids 1998; 33: 559–566. [DOI] [PubMed] [Google Scholar]
- 120. Cooper AL, Gibbons L, Horan MA, et al. Effect of dietary fish oil supplementation on fever and cytokine production in human volunteers. Clin Nutr 1993; 12: 321–328. [DOI] [PubMed] [Google Scholar]
- 121. Lee TH, Hoover RL, Williams JD, et al. Effect of dietary enrichment with eicosapentaenoic and docosahexaenoic acids on in vitro neutrophil and monocyte leukotriene generation and neutrophil function. N Engl J Med 1985; 312: 1217–1224. [DOI] [PubMed] [Google Scholar]