

Abstract
Recent advances in next-generation sequencing have identified novel mutations and revealed complex genetic architectures in human hematological malignancies. Moving forward, new methods to quickly generate animal models that recapitulate the complex genetics of human hematological disorders are needed to transform the genetic information to new therapies. Here, we used a ribonucleoprotein-based CRISPR/Cas9 system to model human clonal hematopoiesis of indeterminate potential and acute myeloid leukemia (AML). We edited multiple genes recurrently mutated in hematological disorders, including those encoding epigenetic regulators, transcriptional regulators, and signaling components in murine hematopoietic stem/progenitor cells. Tracking the clonal dynamics by sequencing the indels induced by CRISPR/Cas9 revealed clonal expansion in some recipient mice that progressed to AML initiated by leukemia-initiating cells. Our results establish that the CRISPR/Cas9-mediated multiplex mutagenesis can be used to engineer a variety of murine models of hematological malignancies with complex genetic architectures seen in human disease.
INTRODUCTION
Acute myelogenous leukemia (AML) is a group of heterogeneous diseases characterized by a clonal expansion of immature myeloid blasts, causing hematopoietic failure (Ferrara and Schiffer, 2013). Recent efforts in sequencing the genomes of multiple cancers have revealed that although adult AML specimens have relatively fewer mutations than solid tumors, AML on average had approximately 13 mutations per case, among which five were recurrently found (Cancer Genome Atlas Research et al., 2013). The recurrently found mutations included those involved in DNA methylation (DMNT3A, TET1, TET2, IDH1, IDH2), chromatin modification (EZH2, ASXL1, KDM6A), signaling pathways (FLT3, NRAS, PTPN11, KRAS), hematopoietic development (CEBPA, RUNX1, GATA2), tumor suppressors (TP53, WT1, PHF6), RNA splicing (SRSF2, U2AF1, SF3B1), cohesion complex (SMC1A, SMC3, STAG2, RAD21) and genes with unknown function (NPM1). These classes of mutations exhibit complex genetic interactions with each other, some showing high propensity of co-occurrence and some showing mutual exclusivity (Chen et al., 2013). Efforts to model the complex mutational landscapes of human AML in animal models have relied largely on mouse genetics by crossing few mutant strains or by introducing the mutant allele by gene transfer, which requires large time- and resource-commitments (Guryanova et al., 2016; Meyer et al., 2016; Shih et al., 2015; Yang et al., 2016). New methods to systematically generate multiple AML animal models with complex mutational landscapes should accelerate the discovery of vulnerabilities shared among AMLs with different genotypes and those that are genotype specific.
The advent of high-throughput sequencing not only enlightened the mutational landscapes of hematological malignancies such as AML or myelodysplastic syndromes (MDS), it also uncovered clonal mutations associated with hematological malignancies in people with no hematological symptoms (Genovese et al., 2014; Jaiswal et al., 2014; Xie et al., 2014b). The incidence of this condition, termed clonal hematopoiesis of indeterminate potential (CHIP), increased with age and was associated with significantly higher risk of developing hematological malignancies compared to those without clonal mutations (Jaiswal et al., 2014; Steensma et al., 2015). The mutations found in CHIP included those found in AML and MDS, including DNMT3A, TET2, and ASXL1, suggesting that these mutations allowed the expansion of pre-malignant clones in otherwise healthy persons. In contrast to AML clones, which often carry multiple detrimental mutations, only one mutation was detected in the majority of persons with CHIP, suggesting that cooperating mutations transform the expanding clones in persons with CHIP into AML clones.
The CRISPR/Cas9 system has transformed the way in which gene function and the regulatory mechanisms are studied. The Cas9 endonuclease complexed with a single-guide RNA (sgRNA) generates DNA breaks that are repaired mostly by error-prone mechanisms to create insertions or deletions (indels) (Hsu et al., 2014; Jiang and Doudna, 2017). To model hematological malignancies and CHIP, primary hematopoietic stem/progenitor cells (HSPCs) need to be gene edited. HSPC editing has been achieved in several methods, including lentiviral transduction (Heckl et al., 2014), plasmid DNA transfection (Mandal et al., 2014), and chemically modified RNA (Hendel et al., 2015). Several groups including ours have used a ribonucleoprotein (RNP)-based delivery method to perform gene editing in both murine and human HSPCs (Dever et al., 2016; DeWitt et al., 2016; Gundry et al., 2016).
Here we used and RNP-based CRISPR/Cas9 system to perform multiplex editing of genes involved in hematological malignancies followed by transplantation to develop murine models of CHIP and AML. We monitored clonal expansion of mutated cells by sequencing the indels induced by Cas9, and found multiple mutant clones that expanded in recipient mice over time. Few clones expanded substantially, causing hematopoietic failure or leukemogenesis. In these models, expansion of a leukemia-initiating cell clone preceded AML development. Our study opens up a new opportunity to engineer murine models of pre-malignant and malignant hematological disorders with a wide variety of co-occurring mutations.
MATERIALS AND METHODS
Mice
The mouse alleles used in this study were Ubc-GFP (C57BL/6-Tg(UBC-GFP)30Scha/J, JAX Stock #004353)(Schaefer et al., 2001) on a C57BL/6 background. CD45.1 mice (B6.SJL-Ptprca Pepcb/BoyJ, JAX Stock #002014) were used as transplant recipient mice. Mice of both sexes were used. Mice were housed in AAALAC-accredited, specific-pathogen-free animal care facilities at Baylor College of Medicine (BCM), and all procedures were approved by the BCM Institutional Animal Care and Use Committee. All other experimental procedures were approved by the BCM Institutional Biosafety Committee.
CRISPR sgRNA Design and Production
CRISPRscan algorithm (www.crisprscan.org) was used to identify the proto-spacer sequences for our target genes (Moreno-Mateos et al., 2015). sgRNAs were produced according to a previous protocol (Gundry et al., 2016). Briefly, a forward primer containing a T7 promoter, a reverse primer specific for the 3’ end of the improved scaffold, and a pKLV2-U6gRNA5(BbsI)-PGKpuro2AmCherry-W plasmid (#67977, Addgene) was used in a PCR reaction. PCR products were purified with a RNeasy Mini kit (#217004, Qiagen) and in vitro transcribed with a HiScribe T7 High Yield RNA synthesis kit (E2040S, NEB) according to the manufacturer’s instruction. The sequences of the most efficient sgRNAs used in this study are shown in Table S1.
Murine HSPCs cells isolation
Bone marrow cells were either flushed from long bones (tibias and femurs) or isolated by crushing the long bones (tibias and femurs), pelvic bones and vertebrae with mortar and pestle in Hank’s buffered salt solution (HBSS) without calcium and magnesium, supplemented with 2% heat-inactivated bovine serum (Gibco). Cells were triturated and filtered through a nylon screen (100μm, Sefar America) or 40μm cell strainer (ThermoFisher Scientific) to obtain a single-cell suspension. The cells were stained with biotin conjugated c-kit (CD117) antibody, anti-biotin microbeads (Miltenyi Biotec) and then positively separated using autoMACS (Miltenyi Biotec). c-Kit-selected cells were stained with PE-conjugated Gr1 (RB6–8C5), CD11b (M1/70), B220 (RA3–6B2), Ter119 (RER-119) and CD3 (145–2C11), APC-conjugated Sca-1 (D7) (all from eBioscience), Stravtividin-APC-Cy7 (Biolegend), and LSK cells were sorted on a BD FACSAria II. To identify myeloid progenitor cells, bone marrow cells were incubated with PE-conjugated lineage markers, eFluor660-conjugated CD34 (RAM34), PE-Cy7-conjugated CD16/32 (93), APC-eFluor780-conjugated c-kit (2B8) and PerCP-Cy5.5-conjugated Sca-1(D7) antidbodies.
To analyze cells in peripheral blood, red blood cells were lysed with an ACK solution, and then stained with PE-Cy7-conjugated Gr-1 (RB6–8C5), APC-eFluor780-conjugated CD11b (M1/70), PerCP-Cy5.5-conjugated B220 (RA3–6B2) and PE-conjugated CD3 (145–2C11).
Electroporation
c-kit+ and LSK cells were cultured in X-Vivo 15 media (Lonza) supplemented with 2% FBS, murine SCF (50 ng/ml), mTPO (50 ng/ml), mIL-3 (10 ng/ml), and mIL-6 (10 ng/ml) (all from Peprotech) for 3 or 16–24 hours before electroporation. Before electroporation, sgRNAs were heated to 95°C for 2 min and immediately chilled on ice for 2 min, followed by incubation with 1 μg Cas9 protein (PNA Bio, 1 μg/μL in Buffer T) at room temperature for 15 minutes to obtain the Cas9-sgRNA RNP complex. 1 × 105 HSPCs were re-suspended in 10 μL of Buffer T, mixed with Cas9-sgRNA RNP, and then electroporated by using the Neon transfection system (ThermoFisher Scientific). Condition of 1700V, 20ms, 1 pulse was used in all experiments.
T7 endonuclease assay and TIDE analysis
To determine Cas9 cleavage efficiency with the T7 endonuclease assay, the PCR products spanning target cleavage site were PCR amplified, diluted 1:4 in 1× Buffer 2 (NEB) and hybridized slowly in a thermal cycler. The hybridized fragments were digested with T7 endonuclease I (NEB), and separated by polyacrylamide gel electrophoresis. Band intensities were analyzed using the Image J software.
PCR amplicons spanning Cas9 cleavage sites were Sanger sequenced and the TIDE program was used as previously described(Brinkman et al., 2014).
Mouse bone marrow transplantation
After 2–3 hours of electroporation, 10,000 LSK cells were collected and retro-orbitally injected together with 2 × 105 competitor cells into lethally irradiated mice (500cGy twice, with at least 3 hours interval). Peripheral blood was collected monthly to determine the donor-type contribution in myeloid, B-cells and T-cells. Secondary transplantation was performed by transplanting 5 × 106 primary bone marrow cells into sub-lethally irradiated mice (650cGy). For the limiting dilution assay, serial doses of bulk leukemia cells (5×102, 5×103, 5×104 and 5×105) were transplanted into sub-lethally irradiated mice (650cGy). For L-GMP transplantation, 5×104 GMP together with 2×105 competitor cells were transplanted into sub-lethally irradiated mice (650cGy).
Immunoblotting
Donor-derived GFP+ cells from whole bone marrow of secondary transplants were sorted for total protein extraction. Normal bone marrow cells were collected and used as a control for protein expression level. The total protein samples were lysed with 1 × 106 cells in 100 μL of 1× lysis buffer (20 mM HEPES, 150 mM sodium chloride, 1% Triton-X-100, 10% glycerol, 1 mM EDTA, 100 mM sodium fluoride, 1 mM phenylmethylsulfonyl fluoride and 17.5 mM glycerophosphate) supplemented with protease and phosphatase inhibitors cocktail. Antibodies used are mouse anti-β-actin (1:1000; Clone, AC-15; ThermoFisher Scientific), rabbit anti-DNMT3A (1:1000; Clone, D23G1; Cell Signaling Technology), rabbit anti-Runx1 (1:1000; Clone, ab35962; Abcam), rabbit anti-EZH2 (1:1000; Clone, ab186006; Abcam), rabbit anti-Ep300 (1:1000; Clone, ab59240; Abcam), rabbit anti-p44/42 MAPK (1:1000; Clone, 137F5; Cell Signaling Technology), and rabbit anti-Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (1:1000; Clone, D13.14.4E; Cell Signaling Technology).
Colony forming assays
Colony assays were performed in cytokine-supplemented methylcellulose (M3434, StemCell Technologies) by plating 10,000 cells per well. Colony numbers were counted after seven days. For serial replating assays, cells were washed with PBS and 10,000 cells were plated.
To determine gene editing at a clonal level, single HSPCs after electroporation or whole bone marrow cells from primary leukemia mice were sorted into 96-well plates with methylcellulose (M3234, StemCell Technologies) supplemented with mIL-3 (10 ng/ml), mIL-6 (10 ng/ml), mSCF (20 ng/ml) and mG-CSF (10 ng/ml). Genomic DNA was extracted from colonies after 7 days and then genotyped as described above.
High-throughput sequencing
Donor-derived GFP+ cells were sorted from peripheral blood and lysed in buffer L (10 mM Tris-HCl pH8, 1 mM EDTA and 25 mM NaCl) supplemented with Proteinase K (200 μg/ml, Millipore). Samples were incubated at 55°C for 30 minutes, followed by 95°C for 3 minutes to inactivate Proteinase K. The genomic regions flanking the Cas9 target site for each gene were amplified by two-rounds of PCR (amplicon size 200–300bp) to attach Illumina adapters to the amplicon. All PCR products were pooled and purified with Sera-Mag Magnetic SpeedBeads (ThermoFisher Scientific). 150 bp pair-end sequencing runs were performed with a MiSeq or a NextSeq500 instrument (Illumina). Reads were adaptor trimmed and aligned using BLAT to reference amplicon sequences. Indels were surveyed using Integrative Genomics Viewer. To determine the mutations in clonally derived AML or in single AML-derived colonies, we performed high-throughput sequencing as described above. Since Nf1 had a large deletion that deleted the primer binding sites, we designed primers to amplify a larger amplicon (approximately 1 kb). PCR with these primer pairs revealed two bands, one corresponding to alleles with small indels and the other to the mutant allele with 306bp deletion that were confirmed by Sanger sequencing.
Statistical analysis
All data are shown as mean±SEM in this study. GraphPad Prism software was used to perform statistical analysis. Unpaired, two-sided student’s t-test was used to compare data from two groups. Log rank test was used to analyze the survival data.
Data availability
High-throughput sequencing data of the amplicon sequencing is deposited to the SRA (SRP132807).
RESULTS
Optimization of murine HSPC gene editing for hematopoietic reconstitution
We previously have showed efficient genome editing of murine c-kit+ and human CD34+ HSPCs by a RNP-based CRISPR/Cas9 delivery method (Gundry et al., 2016). To determine whether a more defined population of immature HSPCs could be edited using our approach, we optimized the condition to edit murine lineage-c-kit+Sca-1+ (LSK) cells that are enriched for hematopoietic stem cells (HSCs). We used LSK cells isolated from Ubc-GFP mice and electroporated the cells with Cas9 protein together with sgGFP. The editing condition we used previously for c-kit+ cells (Gundry et al., 2016) resulted in approximately 70% editing of LSK cells (Fig. 1A). We determined that extending the culture of LSK cells to 16–24 hours improved the editing efficiency of LSK cells, resulting in more than 95% ablation of GFP (Fig. 1A). The gene editing procedure did not appreciably affect the reconstitution potential of LSK cells, since LSK cells edited with sgRNA targeting Rosa26 (sgRosa26) exhibited robust engraftment and did not show any differences in the reconstitution levels of B-, T-, and myeloid cells, compared to the mice reconstituted with cells electroporated with no sgRNA (Fig. S1A). These conditions were used in the subsequent experiments.
Figure 1. Clonal expansion modeled with CRISPR/Cas9-mediated gene editing.
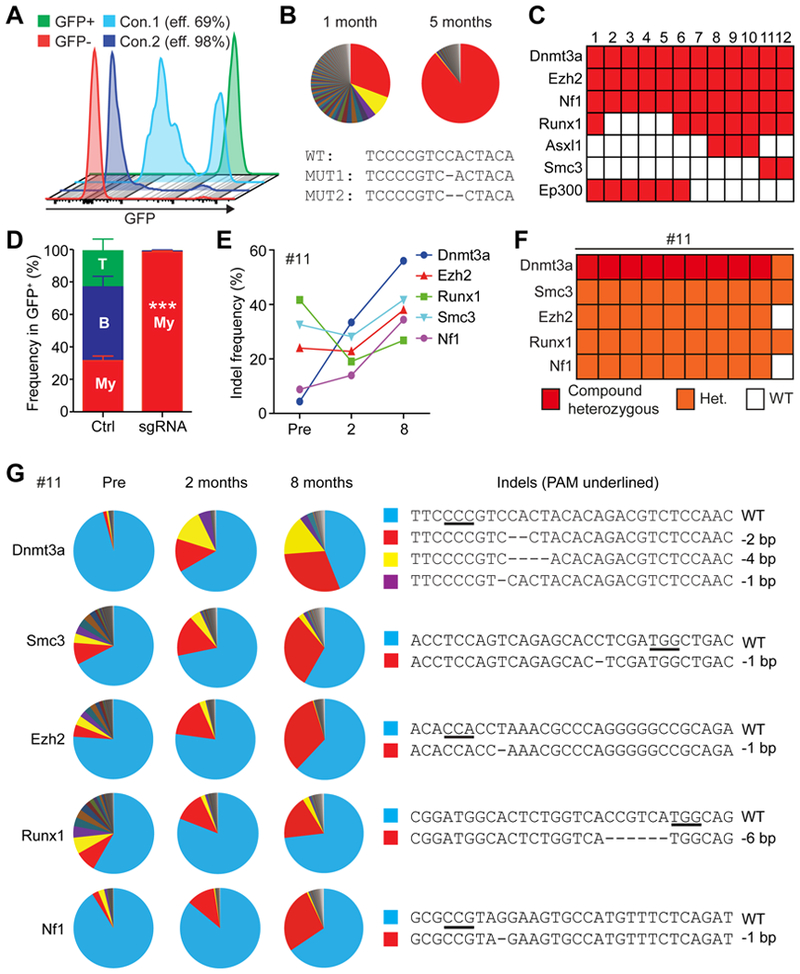
(A) Optimization of the gene editing protocol for immature LSK cells. With the optimized condition (Condition 2), more than 95% of the LSK cells can be edited. (B) Changes in clonal compositions at 1 and 5 months after transplantation as determined by amplicon sequencing of Dnmt3a (One representative figure out of total 3 mice). Each sector represents the fraction of reads with different indels. An allele represented by a red sector (MUT1) expanded at 5 months after transplantation, while MUT2 (yellow sector) reduced its frequency. The sequences around the Cas9 cleavage site (with PAM sequences underlined) are shown below. (C) A chart describing the combination of genes targeted in mice that developed clonal hematopoiesis or AML. The rows represent edited genes indicated by the red boxes and columns represent recipient mice. (D) Relative frequencies of myeloid cells (My), B-cells, and T-cells at 8 month after transplantation of un-edited control cells or LSK cells after multiplex editing (mouse no.6–12, n=7). (E) Indel frequencies in each gene of mouse no. 11 at the indicated time points, as determined by TIDE analysis. (F) Genotyping of clones derived from the peripheral blood of mouse no. 11. Nine out of 10 colonies had mutations in all five genes, including compound heterozygous mutations in Dnmt3a. (G) Amplicon sequencing result of the five genes targeted in mouse no. 11. Each sector represents the fraction of allele with a unique indel, shown on right. All data represent mean±SEM; ***, p<0.001 by Student’s t-test.
CRISPR/Cas9-mediated gene editing of murine HSPCs to model CHIP
To generate mouse models of human hematological disorders, we sought to create loss-of-function mutations in six genes that are found recurrently mutated in CHIP and/or AML (Dnmt3a, Ezh2, Nf1, Runx1, Asxl1, and Smc3) (Cancer Genome Atlas Research et al., 2013; Genovese et al., 2014; Jaiswal et al., 2014) (Fig. S1B). We also included Ep300, since Ep300 deletion accelerated the progress of MDS and AML, suggesting that Ep300 may function as a tumor suppressor in myeloid malignancies (Cheng et al., 2017). We designed four sgRNA per gene and electroporated them individually into c-kit+ HSPCs. We performed T7 endonuclease assays on genomic DNA isolated from cells two days after electroporation to identify the most efficient sgRNA for each gene (Fig. S1B). The selected sgRNAs were used in all subsequent experiments.
Next, we targeted LSK cells with a single sgRNA against Dnmt3a (sgDnmt3a), which is one of the most frequently mutated genes in AML and CHIP (Cancer Genome Atlas Research et al., 2013; Genovese et al., 2014; Jaiswal et al., 2014). We observed robust engraftment of edited LSK cells, and while we observed no differences in total white blood cell numbers between mice transplanted with Dnmt3a-edited or control cells (Fig. S1C–D), Dnmt3a-edited myeloid cells expanded over the course of five months after transplantation (Fig. S1E). To determine the ablation efficiency of Dnmt3a by Cas9, we sorted donor-derived cells from peripheral blood and performed TIDE analysis (Brinkman et al., 2014). The indel frequency of Dnmt3a was significantly higher at 5 months compared to 1-month post transplantation, suggesting that Dnmt3a mutant clones expanded over time (Fig. S1F). We further performed high-throughput sequencing to characterize and quantify the indels. This revealed that while the donor-derived cells at one month are comprised of a large number of small clones and one larger clone, the composition became dominated by one dominant clone with one base deletion at position 2605 (creating p.His869Trpfs*6) (Fig. 1B). These findings demonstrate that our RNP-based CRISPR/Cas9 system achieves efficient cleavage at target loci, and creates a murine model of CHIP by mutagenizing genes directly in murine HSPCs.
Modeling human myeloid malignancies with multiplex editing
To explore whether multiplex editing of HSPCs can be used to develop mouse models of hematological disorders, we first examined the dose-response cleavage activity for multiplex editing. Overall, the cleavage efficacy of sgRNAs increased sharply from 125ng to 250ng complexed with 1μg of Cas9 protein, without any significant increases with larger amount of the Cas9-sgRNA complex (Fig. S2A–B). Thus, we used 200–250ng per sgRNA (total 1μg of sgRNA) with 1μg of Cas9 protein for downstream experiments, and combined four to five sgRNAs together for multiplex editing. We electroporated LSK cells isolated from Ubc-GFP mice with Cas9 protein together with different combinations of sgRNAs (Fig. 1C) and transplanted them into lethally irradiated mice. After transplantation, we monitored donor cell contribution to the hematopoietic system analyzing monthly the frequency of donor-derived GFP+ cells in peripheral blood. We also performed amplicon sequencing with samples collected at pre-transplantation and GFP+ cells in peripheral blood at different time points after transplantation to determine the mutation frequencies of the targeted genes.
Several mice (no. 6–12) exhibited increased myeloid chimerism over time, such that by 8 months post transplantation more than 95 % of the donor-derived cells were in the myeloid lineage, which was not observed in the recipients of control cells (Fig. 1D). Amplicon sequencing of the donor derived cells from these mice revealed changes in clonal dynamics reminiscent of CHIP. Mouse no. 11 (transplanted with LSK cells targeted for Dnmt3a, Smc3, Ezh2, Runx1, and Nf1) exhibited increased overall indel frequencies in Dnmt3a and Nf1 upon transplantation, as determined by TIDE (Fig. 1E). Examination of the specific mutations in the five genes by sequencing revealed that Dnmt3a and Nf1 exhibited increased overall mutant allele frequencies upon transplantation, consistent with the TIDE analysis (Fig. 1G). The overall mutant allele frequencies of Smc3 and Ezh2 did not change upon transplantation, while the mutation frequency of Runx1 was reduced. Nonetheless, these three genes showed expansion of one dominant allele, suggesting that clonal selection occurred in vivo (Fig. 1G). To determine whether these mutations co-occur in the same clones, we collected peripheral blood mononuclear cells and grew colonies, then genotyped the colonies. Most (9 out of 10) of the colonies showed the same genotype; compound heterozygous for Dnmt3a, and heterozygous for Smc3, Ezh2, Runx1, and Nf1 (Fig. 1F). This suggests that a clone with these mutations expanded, and also indicate that our method is capable of mutating at least 6 alleles in a clone. The mutant alleles identified in individual colonies corresponded to the dominant alleles detected by high-throughput sequencing (Fig. 1F, Dnmt3a −2bp/−4bp, Smc3 −1bp, Ezh2 −1bp, Runx1 −6bp, Nf1 −1bp). The compound heterozygous alleles of Dnmt3a were composed of the two dominant mutant alleles detected by high-throughput sequencing (red and yellow sectors of Fig. 1G). Thus, multiplex gene editing of LSK cells generates clones with complex mutational profiles that exhibit clonal expansion.
Other mice (mouse no. 2, 3, 4, and 5) also exhibited myeloid-biased reconstitution at one month after transplantation (Fig. 2A and Fig. S2C–E), but developed severe anemia and died 1 to 4 month after transplantation (Fig. 2B–C). Serial transplantation with whole bone marrow cells from these mice did not cause any diseases (data not shown), suggesting that the edited cells had not fully transformed to acute leukemia by the time mice died of anemia. Amplicon sequencing revealed that LSK cells transplanted into mouse no. 2 had high rates of Ep300 and Ezh2 mutation before transplantation, whereas Dnmt3a and Nf1 initially had mutation rates of 12–20% (Fig. 2D; Pre). At one month after transplantation, the mutation allele frequencies of two mutant Ep300 variants increased (red and yellow sectors), while one Ezh2 variant increased (red sector) (Fig. 2D). A Dnmt3a mutant allele (1 bp deletion causing p.His869Trpfs*6, red sector) increased substantially upon transplantation (to 82%), and maintained the allele frequency until the mouse died of anemia before 4 months of transplantation. Nf1 only had small clones with low overall mutation rate, suggesting that Nf1 mutation did not contribute to the expansion of the large clone(s) harboring Dnmt3a with Ep300 and/or Ezh2 mutations. Mouse no. 3 also exhibited increased mutant allele frequencies upon transplantation, creating large clones with Dnmt3a, Ep300, Ezh2, and Nf1 mutations (Fig. 2E). Of note, some of the expanding clones carried detrimental mutations in Ep300 (such as the −1 bp mutation in no. 2 [p.Asn359Metfs*4] or the +1 bp mutation in no. 3 [p.Asn359Argfs*51]). This is consistent with a recent study demonstrating a tumor suppressor role of Ep300 in myeloid malignancies (Cheng et al., 2017). Additionally, although EP300 mutations in CHIP have not been reported in large studies, a case of clonal hematopoiesis with EP300 mutation was recently reported (Gondek et al., 2016). Together, these results suggest that Ep300 mutation may have cooperated with other mutations (such as Dnmt3a or Ezh2) in our cohort of mice to cause clonal expansion.
Figure 2. Clonal myeloid diseases cause fatal anemia.
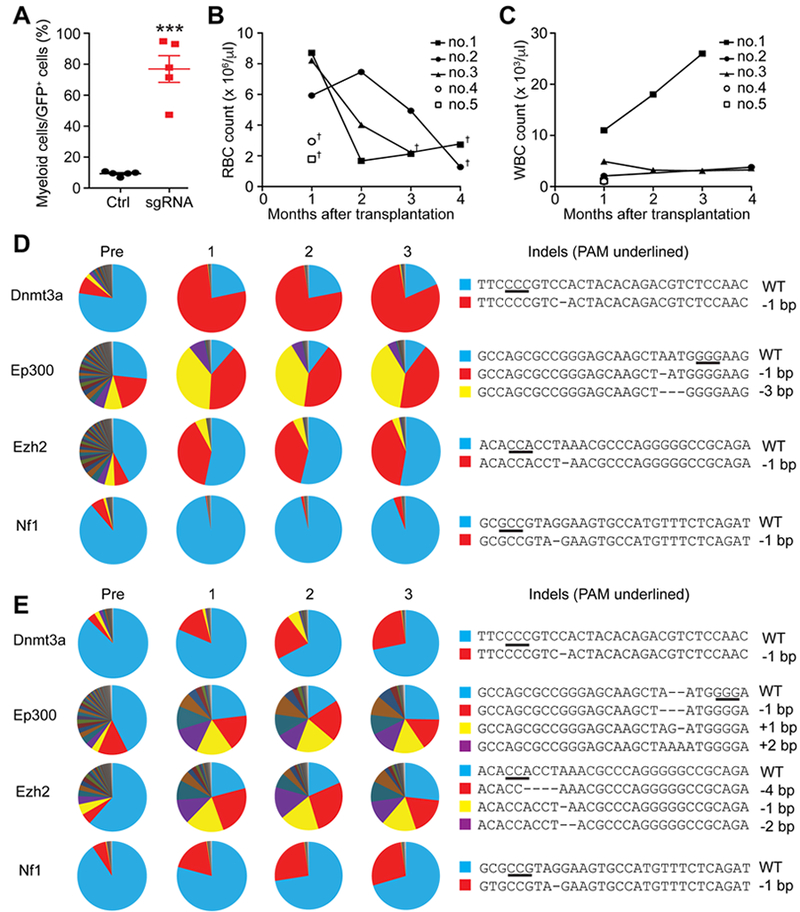
(A) Quantification of the relative frequencies of myeloid cells within donor-derived cells of mouse no. 1, 2, 3, 4, and 5 one month after transplantation. Red blood cell counts (B) and white blood cell counts (C) of the indicated mice. These mice exhibited anemia and died at 1 to 4 months after transplantation, as indicated by the dagger symbols. Normal C57BL/6 mice typically have around 107 RBC/μl. (D-E) Clonal dynamics as determined by amplicon sequencing of donor-derived cells at pre-transplantation, 1-, 2-, and 3-months post transplantation of mouse no. 2 (D) and no. 3 (E). The sequences of wild-type and the dominant clones are shown in right, with the PAM sequences underlined. All data represent mean±SEM; ***, p<0.001 by Student’s t-test.
Modeling AML with multiplex gene editing
Multiplex gene editing of LSK cells also caused AML. Mouse no. 1 transplanted with LSK cells targeted for Dnmt3a, Ezh2, Runx1, Nf1, and Ep300 showed high white blood cell counts over time and succumbed around three months after transplantation (Fig. 2B–C, Table S2). Peripheral blood examination revealed an expansion of myeloid cells and reduction of B- and T-cells within the GFP+ cell population (Fig. 3A–B). 95% of the GFP+ donor-derived cells in the peripheral blood were Mac-1+Gr-1+ myeloid cells at the time of euthanasia. This mouse exhibited splenomegaly and had more than 20% of myeloid blasts in the bone marrow, indicating that leukemic transformation occurred by the multiplex genome editing of HSPCs (Fig. 3C, and Fig. S3A–B).
Figure 3. Development of AML clones after multiplex gene editing of HSPCs.
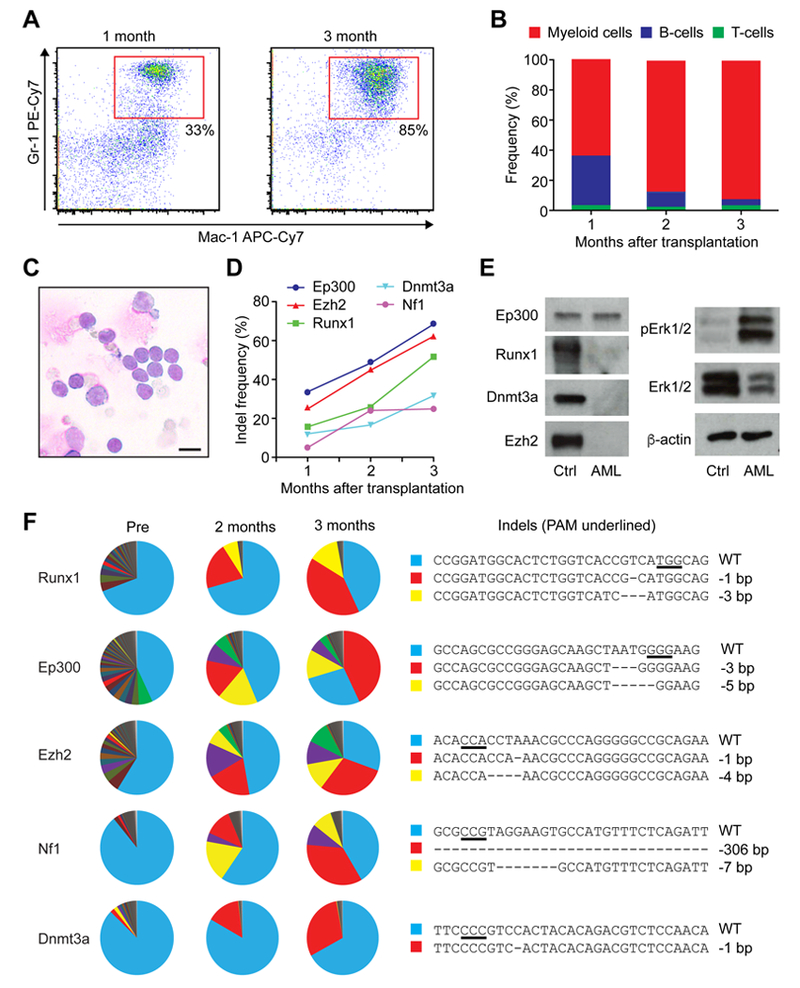
(A) Flow cytometry plots showing the expansion of Mac-1+Gr-1+ myeloid cells in mouse no. 1 at 1- and 3-months post transplantation. (B) Frequencies of myeloid, B-, and T-cells within the GFP+ donor-derived cells at 1-, 2-, and 3-months post transplantation. (C) Wright-giemsa staining of bone marrow cells showing myeloid blasts (n=3 biological replicates). Scale bar; 10μm. (D) Increased indel frequency within each gene over time, as determined by TIDE analysis. (E) Representative western blots showing protein level of Dnmt3a, Ezh2, Runx1, Ep300 and activated Erk1/2(pErk1/2) in donor-derived cells from secondary recipient mice (n=3 biological replicates). (F) Changes in clonal compositions at pre-transplantation, 2-, and 3-months post transplantation as determined by amplicon sequencing. Each sector represents unique clones with unique indels. The right panels show the sequences of the wild-type and the dominant mutants. The PAM sequences are underlined.
To determine the clonal dynamics of the mutant cells over time, donor-derived GFP+ cells from peripheral blood were sorted and sgRNA target sites were analyzed by TIDE and by amplicon sequencing. TIDE analysis showed increased indel rates at each Cas9 target site over-time (Fig. 3D), suggesting that clones with mutations expanded in vivo. Amplicon sequencing detected many indels in five genes at each time point, with numerous small clones detected in the pre-transplantation sample (Fig. 3F). Substantial clonal selection was observed after transplantation, with expanded clones emerging for each targeted gene at 2 months and further expanded at 3 months post transplantation (Fig. 3F, red sectors). To determine whether these five genes are co-mutated in the same clone, we sorted lineage-c-kit+ myeloid progenitor cells from the bone marrow and genotyped individual colonies. We found that most colonies (14 out of 19) had identical mutations in all five genes, while some colonies had different mutations and some were un-edited, indicating that some degree of clonal heterogeneity existed within the myeloid progenitor cell population in the primary recipient (Fig. S4B). Runx1, Ezh2, and Dnmt3a in the major clone all had −1bp deletion causing frame shifts (Fig. 3F and Fig. S4A). Nf1 had a large −306bp deletion, which deleted a whole exon encoding the GTPase-activating protein related domain (GRD) that converts the active GTP-bound RAS to its inactive GDP-bound form (Fig. 3F and Fig. S4A). The dominant clone in this AML mouse had an in-frame −3bp deletion in Ep300 that removed asparagine 359 (p.Asn359del), leaving the role of Ep300 in this AML clone unclear (Fig. 3F and Fig. S4A). The mutations present in the major clone identified by genotyping the colonies were identical to the major indels (red sectors in Fig. 3F) identified by high-throughput sequencing of the bulk of AML cells.
To examine the consequences of the mutations on protein expression levels, we performed immunoblotting of freshly isolated AML cells (Fig. 3E). We did not detect any changes in the protein level or the size of Ep300, consistent with the in-frame 3bp deletion detected in Ep300. Protein levels of Runx1, Dnmt3a and Ezh2 were all significantly reduced, due to the truncation of the antigens recognized by the antibodies. Consistent with the role of Nf1 as a negative regulator of the Ras-MAPK signaling pathway, we found that Erk1/2 is activated in AML cells. Thus, multiplex editing of murine HSPCs can be used to recapitulate the complex genetic lesions of human myeloid leukemia and further allows the assessment of clonal dynamics during leukemogenesis.
Leukemogenesis by multiplex editing is derived from LICs
To examine whether the AML we generated is transplantable, we performed a secondary transplantation assay. Secondary recipient mice receiving primary AML cells succumbed to the disease 22–35 days post transplantation with increased white blood cell numbers and decreased red blood cells and platelets (Fig. 4A–B). Recipient mice showed splenomegaly and expanded myeloid cells in the spleen (Fig. 4C–E). Infiltration of myeloid cells was found in the spleen and the liver (Fig. S3C). Colony forming assays showed the AML cells had extensive colony-forming ability compared to normal bone marrow cells (Fig. S4E). Limiting dilution transplantation revealed that the frequency of leukemia-initiating cells (LICs) was approximately one in 72,000 primary AML cells (Fig. 4G). LICs have been identified in the immunotypically-defined GMP fraction, termed leukemic-GMPs (L-GMPs), in multiple murine AML models (Cozzio et al., 2003; Krivtsov et al., 2006). Flow cytometry revealed that the AML generated by multiplex gene editing had expanded GMPs, while the frequencies of CMPs and MEPs were significantly reduced (Fig. 4H–I). To examine whether the expanded GMP population had the LIC activity, we performed purified GMP transplantation. All recipient mice transplanted with purified GMP succumbed to AML within 40 days of transplantation (Fig. S4F), indicating that GMPs have the LIC activity. Therefore, multiplex CRISPR/Cas9 editing induces AML with an immunophenotypically-defined LIC population.
Figure 4. Generation of LICs by CRISPR/Cas9-mediated gene editing.
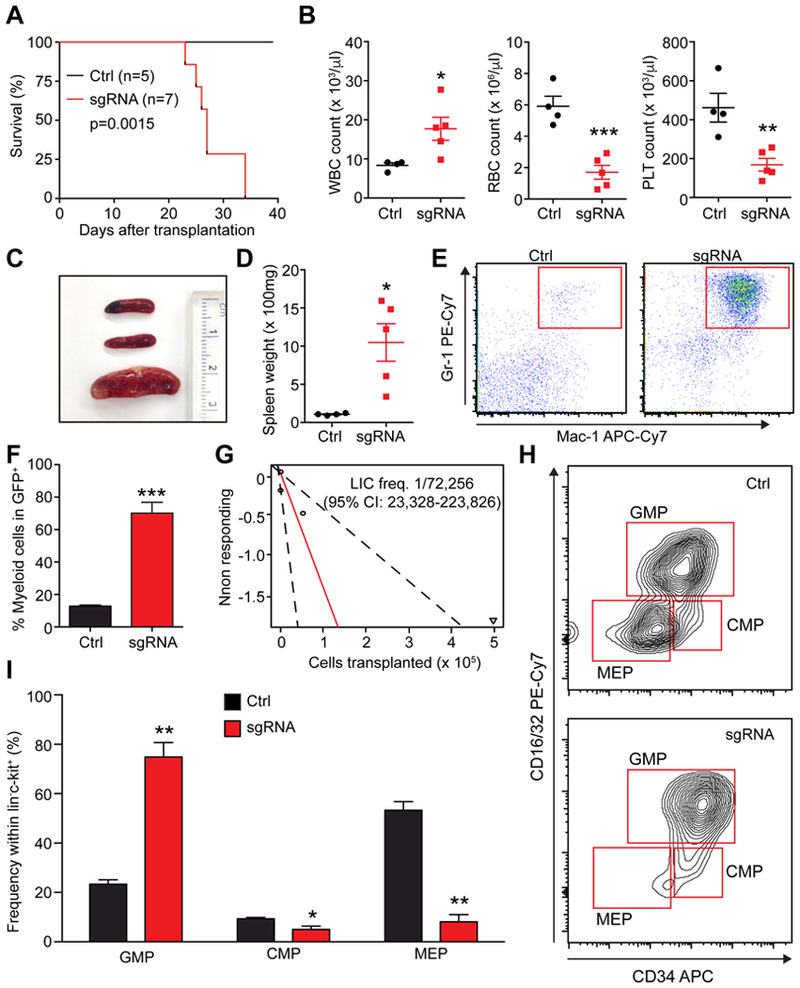
(A) Survival curves of mice receiving 5 × 106 whole bone marrow cells that were collected from primary AML mouse (sgRNAs) and control mice (Ctrl) (p=0.0015, log-rank test) (n=5 for ctrl and n=7 for leukemia mice). (B) Comparison of white blood cell (WBC), red blood cell (RBC), and platelet (PLT) numbers in secondary recipients (n=4–5). (C) A representative image of the spleens from control recipients (top two) and AML recipients (bottom). (D) Quantification of the spleen weight of control and AML recipient mice (n=4–5). (E) Representative flow cytometry plots showing expansion of myeloid cells in the spleens of AML recipient mice compared to control recipient mice (n=5). (F) Quantification of the frequencies of myeloid cells relative to the entire donor-derived GFP+ fraction in secondary recipient mice (n=5). (G) Limiting dilution assay demonstrated that the LIC frequency in whole bone marrow of primary AML mouse was about 1 in 72,000 (n=5 recipient mice per cell dose). (H) Representative flow cytometry plots showing expansion of GMPs, while CMPs and MEPs were depleted in AML mice. (I) Quantification of the results shown in (H) (n=5). All data represent mean±SEM; *, p<0.05; **, p<0.01; ***, p<0.001 by Student’s t-test.
Recipients of the limiting dilution transplantation allowed us to determine the mutation profiles of LIC clones. GFP+ bone marrow cells from the recipients were isolated and were sequenced. All samples had an identical mutation profile (Dnmt3a −1bp, Ezh2 −1bp, Runx1 −1bp, Nf1 −306bp, Ep300 −3bp) that corresponded to the major clone in the primary recipient mice (Fig. S4C). We also sorted single L-GMPs and genotyped the colonies derived from L-GMPs. These clonal L-GMPs had the same combinatorial mutations as found in the major clone (Fig. S4D). These results indicate that one major clone develops over time, out-competing other mutant clones in the primary recipient and ultimately develops into LICs.
DISCUSSION
Here we described an approach to model human CHIP and AML by CRISPR/Cas9-mediated multiplex mutagenesis of murine HSPCs. With an optimized RNP-based delivery system, we achieved more than 90% gene ablation at target loci. By targeting multiple genes recurrently mutated in AML combined with high-throughput amplicon sequencing, we observed expansion of mutant clones, resembling CHIP, some of which continued to expand to cause hematopoietic failure or AML.
Mutating Dnmt3a singly or in combination with other recurrently mutated genes led to expansion of Dnmt3a mutant clones in recipient mice, resembling CHIP in humans. Some of these recipient mice showed high mutant allele frequencies, and by the time they had mutant allele frequencies of more than 70%, the mice succumbed to hematopoietic failure or AML. Several features of these mice, including myeloid expansion and anemia, resembled that of MDS. Thus, our method may be useful in generating new mouse models of MDS. However, since these cells were not transplantable, more work is needed to characterize these mice. In human CHIP, MDS, and AML, a large fraction of the DNMT3A mutations are missense mutations affecting codon R882 in the C-terminal methyltransferase domain (Cancer Genome Atlas Research et al., 2013; Jaiswal et al., 2014; Ley et al., 2010; Walter et al., 2011). The C-terminus of DNMT3A also promotes tetramerization of DNMT3A, and DNMT3A tetramer possesses higher processivity than the dimer or the monomer (Holz-Schietinger et al., 2011; Holz-Schietinger et al., 2012; Jia et al., 2007). DNMT3A R882H mutation acts as a dominant negative mutation, by preventing the formation of the highly processive DNMT3A tetramer (Holz-Schietinger et al., 2012; Russler-Germain et al., 2014). Although we did not attempt to introduce the R878 (mouse equivalent of human R882) mutation by homology-directed repair mechanisms, we targeted the C-terminal domain of murine Dnmt3a involved in tetramerization. In multiple experiments with independent gene editing experiments, we repeatedly observed expansion of clones with Dnmt3a p.His869Trpfs*6 mutation, causing CHIP and AML. Whether Dnmt3a p.His869Trpfs*6 has a particularly strong oncogenic property, or whether it is the most likely mutation induced by Cas9 (at −3 bp of the PAM sequence), remains unknown. Since multiple mutations in the tetramer interface inhibit DNMT3A tetramerization (Holz-Schietinger et al., 2011; Holz-Schietinger et al., 2012), we speculate that p.His869Trpfs*6 affects the catalytic activity and/or the tetramerization of Dnmt3a. Future study with saturating mutagenesis of Dnmt3a in HSPCs followed by transplantation should shed light on the oncogenic properties of Dnmt3a mutants.
Some of the recipients that exhibited high mutant allele frequency succumbed to hematopoietic failure, and some developed AML. The AML described here in detail carried mutations in Dnmt3a, Ezh2, Runx1, Nf1, and Ep300, all of which except for Ep300 caused deleterious changes in the sequences, confirmed by protein expression levels and activation of the MAPK pathway. Deletion of Dnmt3a, Ezh2, and Runx1 blocks differentiation, while deletion of Nf1 activates the Ras-MAPK pathway (Behrens et al., 2017; Challen et al., 2011; Heckl et al., 2014; Tanaka et al., 2012; Xie et al., 2014a). The combinatorial effects of these mutations likely generated a LIC population defined as L-GMP, initially characterized in MLL-rearranged murine AML (Cozzio et al., 2003; Krivtsov et al., 2006; Somervaille and Cleary, 2006). Further investigation is needed to determine the contribution of each mutation in myeloid transformation, since we cannot formally exclude the possibility that some mutations are bystander mutations that do not contribute to transformation. Generation of immunophenotypically-defined clonal LIC population carrying mutations in genes recurrently mutated in human AML should make it possible to not only determine the effects of each mutations in leukemogenesis, but also to conduct comparative studies to identify genotype-specific and common vulnerabilities of LICs.
One limitation with the current method is that it remains challenging to create specific point mutations that are frequently found in hematological malignancies, such as gain-of-function mutations in NRAS, KRAS, or KIT, or neomorphic mutations found IDH1 and IDH2 (Cancer Genome Atlas Research et al., 2013). Further optimization of the CRISPR/Cas9-editing technology should enable precise modeling of human hematological malignancies by creating specific point mutations in a combinatorial manner.
A previous study generated mouse AML models by multiplex gene editing using a lentiviral sgRNA delivery method (Heckl et al., 2014). Additionally, a recent study described the generation of CHIP and myeloid disease models by multiplex gene editing of human CD34+ HSPCs using transient plasmid transfection (Tothova et al., 2017). In this model, myeloid malignancies showing expansion of immature myeloid cells or histiocytes emerged when multiplex gene editing was combined with the introduction of oncogenic FLT3-ITD or NPM1 mutants. Our study demonstrates that CRISPR/Cas9 editing of murine LSK cells with a DNA- and integration-free approach can be used to generate full-blown AML that carried clonally derived LICs with the ability to serially transfer AML. Generation of genotype-specific LICs with these methods will provide a valuable platform to explore the interaction between genetic lesions to therapeutic responses.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (CA193235 and DK107413). Flow-cytometry was partially supported by the NIH (NCRR grant S10RR024574, NIAID AI036211 and NCI P30CA125123) for the BCM Cytometry and Cell Sorting Core. H&E stain was performed by BCM Pathology Core partially supported by the NIH (P30CA125123). High-throughput Sequencing was performed in BCM Genomic and RNA Profiling Core partially supported by the NIH (P30CA125123).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
REFERENCES
- Behrens K, Maul K, Tekin N, Kriebitzsch N, Indenbirken D, Prassolov V, Muller U, Serve H, Cammenga J, and Stocking C (2017). RUNX1 cooperates with FLT3-ITD to induce leukemia. The Journal of experimental medicine 214, 737–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N., Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr., Laird PW, et al. (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368, 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, et al. (2011). Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 44, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SJ, Shen Y, and Chen Z (2013). A panoramic view of acute myeloid leukemia. Nat Genet 45, 586–587. [DOI] [PubMed] [Google Scholar]
- Cheng G, Liu F, Asai T, Lai F, Man N, Xu H, Chen S, Greenblatt S, Hamard PJ, Ando K, et al. (2017). Loss of p300 accelerates MDS-associated leukemogenesis. Leukemia 31, 1382–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML, and Weissman IL (2003). Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev 17, 3029–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE, Pavel-Dinu M, Saxena N, Wilkens AB, Mantri S, et al. (2016). CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature 539, 384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt MA, Magis W, Bray NL, Wang T, Berman JR, Urbinati F, Heo SJ, Mitros T, Munoz DP, Boffelli D, et al. (2016). Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science translational medicine 8, 360ra134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara F, and Schiffer CA (2013). Acute myeloid leukaemia in adults. Lancet 381, 484–495. [DOI] [PubMed] [Google Scholar]
- Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 371, 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondek LP, Zheng G, Ghiaur G, DeZern AE, Matsui W, Yegnasubramanian S, Lin MT, Levis M, Eshleman JR, Varadhan R, et al. (2016). Donor cell leukemia arising from clonal hematopoiesis after bone marrow transplantation. Leukemia 30, 1916–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundry MC, Brunetti L, Lin A, Mayle AE, Kitano A, Wagner D, Hsu JI, Hoegenauer KA, Rooney CM, Goodell MA, et al. (2016). Highly Efficient Genome Editing of Murine and Human Hematopoietic Progenitor Cells by CRISPR/Cas9. Cell reports 17, 1453–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guryanova OA, Shank K, Spitzer B, Luciani L, Koche RP, Garrett-Bakelman FE, Ganzel C, Durham BH, Mohanty A, Hoermann G, et al. (2016). DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat Med 22, 1488–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckl D, Kowalczyk MS, Yudovich D, Belizaire R, Puram RV, McConkey ME, Thielke A, Aster JC, Regev A, and Ebert BL (2014). Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat Biotechnol 32, 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendel A, Bak RO, Clark JT, Kennedy AB, Ryan DE, Roy S, Steinfeld I, Lunstad BD, Kaiser RJ, Wilkens AB, et al. (2015). Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol 33, 985–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz-Schietinger C, Matje DM, Harrison MF, and Reich NO (2011). Oligomerization of DNMT3A controls the mechanism of de novo DNA methylation. J Biol Chem 286, 41479–41488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz-Schietinger C, Matje DM, and Reich NO (2012). Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J Biol Chem 287, 30941–30951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, and Zhang F (2014). Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371, 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia D, Jurkowska RZ, Zhang X, Jeltsch A, and Cheng X (2007). Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 449, 248–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, and Doudna JA (2017). CRISPR-Cas9 Structures and Mechanisms. Annual review of biophysics 46, 505–529. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. (2006). Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442, 818–822. [DOI] [PubMed] [Google Scholar]
- Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, et al. (2010). DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363, 2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK, Ferreira LM, Collins R, Meissner TB, Boutwell CL, Friesen M, Vrbanac V, Garrison BS, Stortchevoi A, Bryder D, et al. (2014). Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell 15, 643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer SE, Qin T, Muench DE, Masuda K, Venkatasubramanian M, Orr E, Suarez L, Gore SD, Delwel R, Paietta E, et al. (2016). DNMT3A Haploinsufficiency Transforms FLT3ITD Myeloproliferative Disease into a Rapid, Spontaneous, and Fully Penetrant Acute Myeloid Leukemia. Cancer discovery 6, 501–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Mateos MA, Vejnar CE, Beaudoin JD, Fernandez JP, Mis EK, Khokha MK, and Giraldez AJ (2015). CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods 12, 982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton R, Meyer MR, Erdmann-Gilmore P, Townsend RR, Wilson RK, et al. (2014). The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell 25, 442–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer BC, Schaefer ML, Kappler JW, Marrack P, and Kedl RM (2001). Observation of antigen-dependent CD8+ T-cell/ dendritic cell interactions in vivo. Cellular immunology 214, 110–122. [DOI] [PubMed] [Google Scholar]
- Shih AH, Jiang Y, Meydan C, Shank K, Pandey S, Barreyro L, Antony-Debre I, Viale A, Socci N, Sun Y, et al. (2015). Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell 27, 502–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somervaille TC, and Cleary ML (2006). Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer cell 10, 257–268. [DOI] [PubMed] [Google Scholar]
- Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, and Ebert BL (2015). Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 126, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Miyagi S, Sashida G, Chiba T, Yuan J, Mochizuki-Kashio M, Suzuki Y, Sugano S, Nakaseko C, Yokote K, et al. (2012). Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood 120, 1107–1117. [DOI] [PubMed] [Google Scholar]
- Tothova Z, Krill-Burger JM, Popova KD, Landers CC, Sievers QL, Yudovich D, Belizaire R, Aster JC, Morgan EA, Tsherniak A, et al. (2017). Multiplex CRISPR/Cas9-Based Genome Editing in Human Hematopoietic Stem Cells Models Clonal Hematopoiesis and Myeloid Neoplasia. Cell Stem Cell 21, 547–555 e548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, Fulton R, Schmidt H, Kalicki-Veizer J, O’Laughlin M, et al. (2011). Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 25, 1153–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, and Orkin SH (2014a). Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell 14, 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, et al. (2014b). Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 20, 1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Rodriguez B, Mayle A, Park HJ, Lin X, Luo M, Jeong M, Curry CV, Kim SB, Ruau D, et al. (2016). DNMT3A Loss Drives Enhancer Hypomethylation in FLT3- ITD-Associated Leukemias. Cancer Cell 30, 363–365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
High-throughput sequencing data of the amplicon sequencing is deposited to the SRA (SRP132807).