

Abstract
Posttranslational protein modifications can play a major role in disease pathogenesis; phosphorylation, sumoylation, and acetylation modulate the toxicity of a variety of proteotoxic proteins. The androgen receptor (AR) is substantially modified, in response to hormone binding, by phosphorylation, sumoylation, and acetylation; these modifications might thus contribute to DHT-dependent polyglutamine (polyQ)-expanded AR proteotoxicity in spinal and bulbar muscular atrophy (SBMA). SIRT1, a nuclear protein and deacetylase of the AR, is neuroprotective in many neurodegenerative disease models. Our studies reveal that SIRT1 also offers protection against polyQ-expanded AR by deacetylating the AR at lysines 630/632/633. This finding suggested that nuclear AR acetylation plays a role in the aberrant metabolism and toxicity of polyQ-expanded AR. Subsequent studies revealed that the polyQ-expanded AR is hyperacetylated and that pharmacologic reduction of acetylation reduces mutant AR aggregation. Moreover, genetic mutation to inhibit polyQ-expanded AR acetylation of lysines 630/632/633 substantially decreased its aggregation and completely abrogated its toxicity in cell lines and motor neurons. Our studies also reveal one means by which the AR acetylation state likely modifies polyQ-expanded AR metabolism and toxicity, through its effect on DHT-dependent AR stabilization. Overall, our findings reveal a neuroprotective function of SIRT1 that operates through its deacetylation of polyQ-expanded AR and highlight the potential of both SIRT1 and AR acetylation as powerful therapeutic targets in SBMA.
Introduction
The sirtuins are a family of NAD-dependent deacetylases that are linked to the regulation of lifespan and protection against proteotoxicity (for review, see Haigis and Sinclair, 2010; Huber and Superti-Furga, 2011). SIRT1, in particular, plays a protective role in several models of neurodegeneration, including Wallerian degeneration (Araki et al., 2004), Alzheimer's disease (Qin et al., 2006; Kim et al., 2007; Min et al., 2010), and ALS (Kim et al., 2007). The mechanistic basis for SIRT1 neuroprotection may involve its role as a deacetylase of several nonhistone proteins, including PGC1α (Rodgers et al., 2005), HSF1 (Westerheide et al., 2009), and Atg5, Atg7, and Atg8 (Lee et al., 2008).
Protein acetylation and other posttranslational modifications play an important role in the dynamic regulation of protein function, trafficking, and turnover and can thus directly modify the toxicity of disease-causing proteins. Acetylation can play a role in neurodegeneration by directly modifying proteotoxic proteins. Degradation of tau is inhibited by its acetylation and enhanced by SIRT1-dependent deacetylation (Min et al., 2010), consistent with the protective effect of sirtuins in models of AD and tauopathies (for review, see Haigis and Sinclair, 2010). Lysine acetylation also regulates the degradation of two polyQ-expanded proteins, huntingtin and ataxin-7, by promoting (Jeong et al., 2009) or inhibiting (Mookerjee et al., 2009), respectively, the targeting of these proteins to the autophagic pathway. The fact that one posttranslational modification can elicit distinct effects on different proteins containing the same genetic mutation highlights both the significance of specific protein context and the role of normal protein metabolism in polyQ expansion disease.
The androgen receptor (AR), in which polyQ expansion causes the neurodegenerative neuromuscular disease spinal and bulbar muscular atrophy (SBMA) (La Spada et al., 1991), is a nuclear receptor that has been well studied for its roles in endocrine dysfunction and prostate cancer (for review, see Gao, 2010). Investigations into its normal function and metabolism have revealed important aspects of disease mechanism in SBMA, including hormone binding (Katsuno et al., 2002; Takeyama et al., 2002; Chevalier-Larsen et al., 2004), nuclear localization (Takeyama et al., 2002; Montie et al., 2009; Nedelsky et al., 2010), AF2-coactivator interactions (Nedelsky et al., 2010), and the AR amino-carboxyl interaction (Orr et al., 2010). The AR undergoes many posttranslational modifications, both hormone-dependent and hormone-independent, throughout its normal metabolism. These modifications, which include phosphorylation, sumoylation, ubiquitylation, and acetylation, impact aspects of AR metabolism, including trafficking, protein interactions, transcriptional activation, and degradation.
In our studies here to determine the role of SIRT1 in SBMA pathogenesis, we found that SIRT1 is strongly neuroprotective in cell models of SBMA and that the function of SIRT1 to deacetylate the AR is a critical component of its neuroprotection. Moreover, we found that polyQ-expanded AR is hyperacetylated and that acetylation modulates its nuclear aggregation and toxicity. One mechanism by which acetylation may impact the aggregation and toxicity of polyQ-expanded AR is through the regulation of AR degradation. Our studies presented here highlight the therapeutic potential of SIRT1 and its direct role of modulating AR acetylation in SBMA.
Materials and Methods
Creation of stable SIRT1 and SIRT1(H363Y) Tet-On AR112Q and AR10Q PC12 cell lines.
Stable transfection of SIRT1-myc-his and SIRT1(H363Y)-myc-his (pcDNA3.1 plasmid backbone) into Tet-On AR112Q or AR10Q PC12 cells was performed using a calcium phosphate protocol. Cotransfection with pBABE-puro plasmid was used to confer resistance for the selection of colonies. Stable transformants were selected with 2 μg/ml puromycin (Invitrogen). Single colonies were isolated and expanded and screened for SIRT1-myc-his protein expression by Western blot analysis using SIRT1 antibody (Millipore).
Creation of AR acetylation-mutant Tet-On PC12 cells.
pTRE-AR(10-CAG) was used as a template for site-directed mutagenesis (Quick Change II XL, Stratagene). Nucleic acids that encode Lys-630 were mutated to encode an acetylation-null arginine within the protein. NruI/BstBI digests were used to clone this mutation into pTRE-NLSX3-AR(111-CAG) (Montie et al., 2009), yielding pTRE-NLSX3-AR-K630A(111-CAG). pTRE-AR(10-CAG) was used as a template for site-directed mutagenesis of nucleic acids encoding Lys-630, Lys-632, and Lys-633 to acetylation-null arginine (R) and acetylation-mimic threonine (T) or glutamine (Q). The acetylation mutations were cloned into the pTRE-NLSX3-AR(111-CAG) plasmid with NruI and BstBI, yielding NLSX3-AR-K630,632,633R(111-CAG), NLSX3-AR-K630,632,633T(111-CAG), and NLSX3-AR-K630,632,633Q(111-CAG). All constructs were sequenced to verify mutation and CAG length.
Stable transfection of Tet-On PC12 cells (Clontech) with NLSX3-AR constructs and a plasmid conferring hygromycin resistance (pTK-hygromycin) was performed using a calcium phosphate protocol. Stable transformants were selected with 200 μg/ml hygromycin. Single colonies were isolated and expanded and screened for doxycycline-inducible AR protein expression by slot blot and Western analysis using an antibody against AR (AR-H280; Santa Cruz Biotechnology). AR expression in the various mutant cell lines was adjusted using doxycycline to achieve protein levels equivalent to that in NLSX3-AR110Q-expressing PC12 cells. Genomic DNA was extracted from each clone to verify mutation and CAG length via sequence analysis. Cells were maintained in normal growth media [DMEM with 10% heat-inactivated horse serum, 5% heat-inactivated fetal bovine serum, 2 mm l-glutamine, 100 U/ml penicillin/streptomycin, 200 μg/ml hygromycin (Invitrogen), and 100 μg/ml G418 (Mediatech)] at 37°C, 10% CO2.
Creation of adeno-associated viruses.
pcDNA3.1-SIRT1-myc-his and pcDNA3.1-SIRT1(H363Y)-myc-his were digested with BamHI and PmeI to remove SIRT1-myc-his and SIRT1(H363Y)-myc-his cDNA and the ends were filled in with Klenow. The cis-adeno-associated viral (AAV) plasmid (serotype 1), with CMV enhancer and chicken β-actin promoter, was digested with EcoRV and EcoRI and the ends were blunted with Klenow. Ligations were performed to produce cis-AAV-SIRT1-myc-his and cis-AAV-SIRT1(H363Y)-myc-his. To create adeno-associated viruses expressing AR, pTRE plasmids containing NLSX3-AR(111-CAG), NLSX3-AR-K630,632,633R(111-CAG) (R3), or NLSX3-AR-K630,632,633Q(111-CAG) (Q3) cDNAs were digested with NheI and BamHI to remove the AR-encoding fragment and the ends filled in with Klenow. The AR-encoding fragment was ligated into the cis-AAV plasmid that was digested as described above. Mutations and CAG repeat lengths were confirmed by sequence analysis.
All AAV cis-plasmids described above were co-transfected into HEK393T cells with trans (H21) and adenovirus helper (pFδ6) plasmids using a calcium phosphate protocol. After 5 d in culture, cells were lysed by repeated freeze/thaw in Opti-MEM (Invitrogen) to release AAV from cells, and lysates were centrifuged and filtered to remove cellular debris. The appropriate volume of cell lysate to achieve the highest, equivalent infection of motor neurons within dissociated spinal cord cultures was determined by serial dilution infections of wild-type (nontransgenic) cultures and subsequent immunostaining for AR (AR-318), c-myc (Santa Cruz Biotechnology), and neurofilament-heavy chain (SMI32).
siRNA reduction of SIRT1.
Stable Tet-On PC12 cells were transfected with SIRT1 siRNA by electroporation, using an Amaxa nucleoporator (Mirus), following the specified protocol for PC12 cells. Fifty picomoles of either nontargeting siRNA (ON-TARGETplus Nontargeting siRNA #1; Dharmacon) or SIRT1 siRNA (AAGATGAAGTTGACCTCCTCA) (Rodgers et al., 2005) (Qiagen) were electroporated into 4 million AR112Q-PC12 cells. Cells were plated and treated 24 h later with doxycycline and 10 nm DHT for an additional 48 h. Cells were harvested and evaluated by Western blot for SIRT1 protein and by immunofluorescence for AR aggregation. All experiments were performed in triplicate at least three times. Student's t test was used to determine statistical significance.
Treatment of inducible PC12 cell lines for AR aggregation analysis.
Stable Tet-On PC12 cell lines were treated with doxycycline to express equivalent levels of NLSX3-AR110Q (K3), NLSX3-AR(K630A)110Q (K630A), NLSX3-AR(K630,632,633R)110Q (R3), NLSX3-AR(K630,632,633T)110Q (T3), and NLSX3-AR(K630,632,633Q)110Q (Q3) for 48 h with EtOH or 10 nm DHT in charcoal-stripped serum-containing cell culture media. Immunofluorescence was performed to detect AR (AR-H280) and cells were visualized with a Leica microscope equipped with epifluorescence. Three hundred cells from random fields were counted from three replicate wells by an observer blinded to the experimental condition, and the percentage of cells with nuclear inclusions of AR was calculated and graphed. Cells treated in parallel were harvested for biochemical analysis. All experiments were performed in triplicate at least three times. Statistical significance was determined with ANOVA and Tukey post hoc analysis (SigmaStat).
PC12 cell toxicity assay.
Stable Tet-On PC12 cell lines were treated with doxycycline to express equivalent levels of AR, in the presence and absence of 10 nm DHT, for 12 d. At the end of the treatment regimen, cells were harvested and stained with Trypan blue to reveal dead cells. Two hundred cells were counted by an observer blinded to the experimental condition and the percentage of Trypan blue-positive (dead) cells was determined. All experiments were performed in triplicate at least three times. Statistical significance was determined by ANOVA and Tukey post hoc analysis (SigmaStat).
AR stabilization by DHT.
Stable Tet-On PC12 cell lines were treated with doxycycline to express equivalent levels of AR for 48 h in charcoal-stripped serum-containing media. Cells were then washed three times with PBS to remove doxycycline and inhibit new AR protein translation. At this time triplicate samples from each cell line were harvested to serve as the DHT-untreated sample. The remaining wells of cells received fresh media containing 10 nm DHT for an additional 36 h, at which time these samples were harvested and AR levels determined by Western analysis. Student's t test was used to determine statistical significance.
Immunofluorescence.
PC12 cells and dissociated spinal cord cultures were fixed with 4% paraformaldehyde for 15 min, washed in PBS, permeabilized with 0.3% Triton X-100 for 15 min, blocked in 2% goat serum (Jackson ImmunoResearch) for 20 min, and incubated for 60 min in primary antibody diluted in 1.5% goat serum. Cells were washed in PBS and incubated for 30 min with secondary antibodies (FITC-, Texas Red-, or Alexa Fluor 350-conjugated; Jackson ImmunoResearch and Invitrogen), washed in PBS, incubated for 10 min with Hoechst (2 μg/ml), washed in PBS, and mounted with Vectashield (Vector Laboratories). Fluorescence was visualized with a Leica (Leica Microsystems) microscope; images were captured with a Leica camera and compiled with IP Lab software (BD Biosciences). Antibodies used include AR-H280 (1:100; Santa Cruz Biotechnology), AR-318 (1:50; Vector Laboratories), SMI32 (1:1000; Sternberger Monoclonal), anti-His-tag (1:200; Millipore), anti-His-tag (1:50; Cell Signaling Technology), and anti-c-Myc (1:100; Santa Cruz Biotechnology).
Western blot analysis.
Protein extracts were prepared by lysing cells with Triton-DOC lysis buffer (1% sodium deoxycholate and 0.5% Triton X-100 in PBS with protease inhibitors) or lysis buffer for acetyl-lysine immunoprecipitation (20 mm Tris, pH 7.5, 150 nm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm sodium orthovanadate, 1 μg/ml Leupeptin, 1 mm PMSF, and 5 mm sodium butyrate). All lysates were sonicated three times for 10 s using a Branson cup sonifier and a DC protein assay (Bio-Rad) was performed to determine protein concentration. Lysates were electrophoresed by SDS-PAGE and transferred to 0.45 μm PVDF (Immobilon-P). Western hybridization was performed using the following antibodies diluted in 5% nonfat milk: AR-H280 (1:1000; Santa Cruz Biotechnology), GAPDH (1:100,000; Fitzgerald), tubulin (1:2000; Sigma), SIRT1 (1:2000; Millipore), acetyl-lysine (1:1000; Cell Signaling Technology), and HMGB1 (1:500; Novus). Detection was performed with ECL [Pierce (Thermo Fisher)].
Nuclear/Cytoplasmic fractionation of PC12 cells.
Stable Tet-On PC12 cell lines were treated with doxycycline to express equivalent levels of AR10Q or AR112Q in the presence of 10 nm DHT for 48 h. Ten thousand cells from each condition were lysed in NP-40 buffer (10 mm Tris, pH 7.4, 10 mm NaCl, 3 mm MgCl2, 0.5% NP-40, 5 mm sodium butyrate and protease inhibitors) and fractionated by centrifugation. Twenty percent of the nuclear and cytoplasmic fractions were removed and electrophoresed by SDS-PAGE, and Western analysis was performed (AR-H280, Santa Cruz Biotechnology) to determine AR localization. The integrity of the fractions was determined using antibodies specific to nuclear (HMGB1, Novus) and cytoplasmic (GAPDH, Fitzgerald) fractions. The remainder of the fractions underwent immunoprecipitation of acetyl-lysine as outlined below.
Immunoprecipitation of acetyl-lysine.
Cells were washed with PBS and lysed with acetyl-lysine buffer or NP40 buffer (described above). Lysates were sonicated three times for 10 s using a Branson cup sonifier. A DC protein assay (Bio-Rad) was performed to determine protein concentration. Two-hundred and fifty or five-hundred micrograms of protein (or in the case of nuclear/cytoplasmic fractions, 80% of the fraction) from each cell line was precleared with 20 μl of protein G Dynabeads (Invitrogen) in a total volume of 250 μl and rotated for 45 min at room temperature. Dynabeads were removed from lysates (with Dynal magnet) and 2.5 μl of anti-acetyl-lysine antibody (Cell Signaling Technology) was added to the samples and incubated at 4°C overnight. Dynabeads (20 μl) were added to the lysates and rotated at room temperature for 45 min. Dynabeads were then removed from the lysates and washed four times with lysis buffer, then proteins were eluted by boiling in 2× Laemmli buffer for 5 min. Samples were evaluated by SDS-PAGE and Western blot for AR (AR-H280) as outlined above. Following evaluation of AR detection, membranes were stripped and reprobed with acetyl-lysine antibody to determine the efficiency of the immunoprecipitation. To determine relative levels of acetylated AR, densitometric values of AR following acetyl-lysine IP were divided by the AR value from the total lysate for each sample. Such analysis was performed on triplicate samples and a Student's t test was performed to determine statistical significance.
Dissociated spinal cord cultures.
Dissociated spinal cord cultures were established according to Roy et al. (1998) and as described by Montie et al. (2009). Transgenic mouse genotyping was performed from a tail biopsy as described by Chevalier-Larsen et al. (2004); male and female embryos were not distinguished. Transgenic AR112Q and nontransgenic spinal cords were pooled separately, plated for culture, and incubated for 3 weeks in media conditioned by embryonic 13.5 d nontransgenic brain glial cells [MEM, 35 mm NaHCO3, 0.5% dextrose, 1% N3] supplemented with 10 nm 2.5 S NGF. After 3 weeks, AR112Q cultures were treated with AAV-EGFP, AAV-SIRT1, or AAV-SIRT1(H363Y). Non-transgenic cultures were treated with AAV-NLSX3-AR111CAG, AAV-NLSX3-AR111CAG (R3), AAV-NLSX3-AR111CAG (Q3), AAV-SIRT1, or AAV-EGFP for 5 d. At this time, replicate wells were treated with 10 μm DHT for 6 h and subsequently analyzed by immunofluorescence to determine the percentage infectivity of each viral construct. The remaining cultures were treated with EtOH or 10 μm DHT for an additional 7 d. Three wells of cells from each treatment group were immunostained as described above. Motor neurons were identified by analysis of neurofilament-heavy chain (SMI32 antibody) and morphology. Motor neurons from 10 random fields from each of three separate wells were counted using 20,000× magnification by an observer blinded to the experimental condition. Statistical significance was determined by ANOVA and post hoc Tukey test (SigmaStat). All procedures involving mice were performed following the guidelines of the Institutional Animal Care and Use Committee of Thomas Jefferson University.
Results
SIRT1 abrogates polyQ-expanded AR aggregation and toxicity
SIRT1 is neuroprotective in numerous neurodegenerative diseases (for review, see Haigis and Sinclair, 2010). We wished to investigate the ability of SIRT1 to modulate aggregation and toxicity in cell models of SBMA. PC12 cells that inducibly express polyglutamine (polyQ)-expanded AR (AR112Q) form polyQ length- and DHT-dependent nuclear inclusions of proteolyzed polyQ-expanded AR (Walcott and Merry, 2002), similar to those found in patient tissue (Li et al., 1998), and die in response to DHT (Montie et al., 2009). Stable overexpression of SIRT1 in this cell model resulted in a substantial decrease in the number of cells with nuclear inclusions (Fig. 1A), reduced the amount of SDS-insoluble AR112Q observed by Western analysis (Fig. 1B), and rescued cells from DHT-dependent death (Fig. 1C). This ability of SIRT1 to reduce AR112Q aggregation and to inhibit toxicity was dependent upon its deacetylase domain, as expression of SIRT1 bearing a mutation previously shown to eliminate its deacetylase activity [SIRT1(H363Y)] (Tanny et al., 1999) failed to reduce AR112Q aggregation and toxicity (Fig. 1). Moreover, expression of deacetylase-deficient SIRT1(H363Y) substantially increased the number of cells with nuclear inclusions (Fig. 1A) and enhanced cell death (Fig. 1C). Overexpression of SIRT1 or of SIRT1(H363Y) had no effect on the nuclear localization of expanded AR following 30 min, 1, 2, or 48 h of DHT treatment, assessed by immunostaining of AR protein (data not shown).
Figure 1.

SIRT1 abrogates mutant AR aggregation and toxicity in a manner dependent upon its deacetylase activity. PC12 cells expressing AR112Q were stably transfected with either SIRT1 or deacetylase-deficient SIRT1(H363Y). A, Stable overexpression of SIRT1 in AR112Q-expressing PC12 cells resulted in a significant reduction in the number of cells containing nuclear inclusions, compared with control, following treatment with 10 nm DHT for 48 h, while deacetylase-deficient SIRT1(H363Y) increased nuclear inclusions. ANOVA with post hoc Tukey test was performed to determine statistical significance. *p < 0.01 B, Western analysis of AR in cells treated in parallel with those in A reveals a substantial decrease in SDS-insoluble AR112Q upon overexpression of SIRT1, and an increase in high molecular weight species upon overexpression of SIRT1(H363Y). C, Cells were treated for 12 d with either 10 nm DHT or EtOH. Two hundred cells were counted from triplicate wells for each treatment and the percentage of Trypan blue-positive cells was graphed. SIRT1 rescued AR112Q-expressing PC12 cells from DHT-dependent death, while SIRT1(H363Y) did not. Also note that SIRT1(H363Y) enhanced DHT-dependent death. ANOVA with post hoc Tukey test was performed to determine statistical significance. *p < 0.05.
Given the effect of SIRT1 overexpression on polyQ-expanded AR aggregation and toxicity, we wished to determine the role of endogenous SIRT1 in these events. We used siRNA to knock down SIRT1 (Rodgers et al., 2005) in the PC12 cell model of SBMA, described above. We achieved a 77% knockdown of SIRT1 protein using a single siRNA (Fig. 2A). This reduction in SIRT1 expression resulted in a significant increase in polyQ-expanded AR aggregation (Fig. 2B), revealing a role for endogenous SIRT1 in the prevention of polyQ-expanded aggregation.
Figure 2.
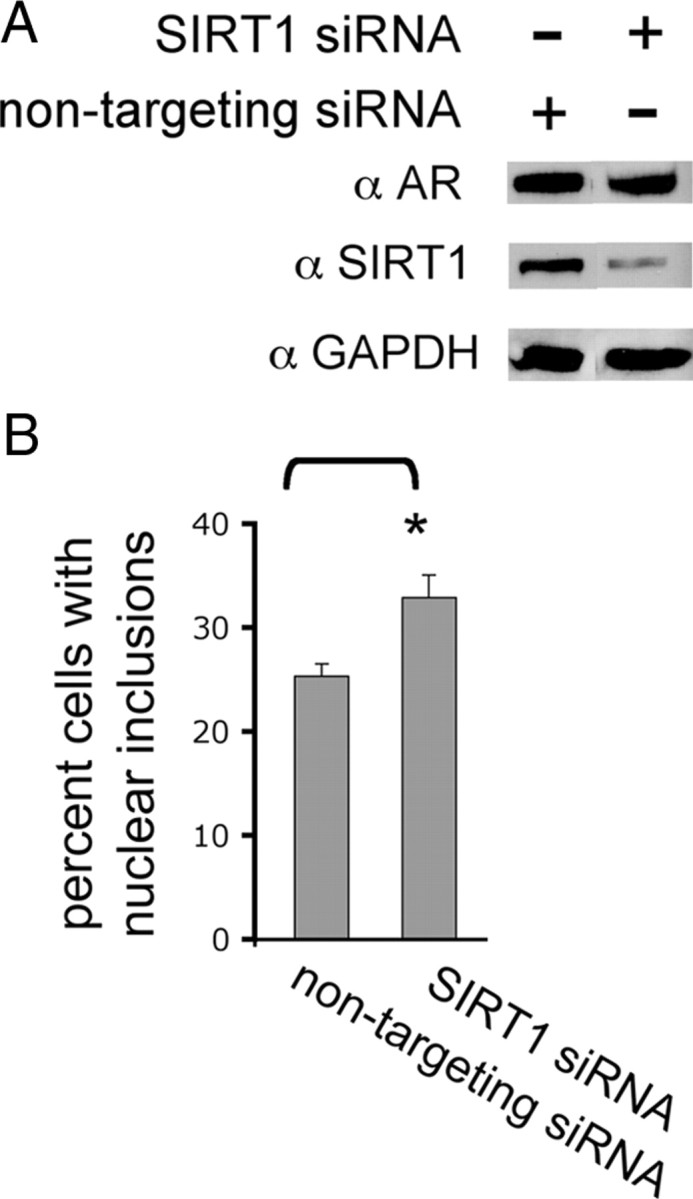
Reduction of SIRT1 in cells expressing polyglutamine-expanded AR increases nuclear inclusions. PC12 cells were treated with siRNA against SIRT1 or nontargeting siRNA. Twenty-four hours later cells were treated with doxycycline to induce AR112Q expression and 10 nm DHT. A, After 48 h, cells were harvested and expression of SIRT1 and AR (AR-H280) was evaluated by Western analysis. Seventy-seven percent knockdown of SIRT1 was achieved. GAPDH was used as a loading control. B, Cells treated as in A were fixed and immunostained for AR (AR-H280), and 300 cells from each of three wells per treatment were counted and the number of cells with AR nuclear inclusions was determined. Reduction of SIRT1 significantly increased the number of cells with nuclear inclusions of AR112Q. Student's t test was performed to determine statistical significance. *p < 0.01.
SIRT1 protects motor neurons expressing polyQ-expanded AR from DHT-dependent death
Having demonstrated a beneficial role of SIRT1 in abrogating the aberrant aggregation and toxicity of polyQ-expanded AR in PC12 cells, we wished to determine whether SIRT1 would also have a therapeutic benefit in the disease-relevant cell type, motor neurons. Thus, we initiated mixed spinal cord cultures containing motor neurons from transgenic mice expressing polyQ-expanded AR, as previously described (Mojsilovic-Petrovic et al., 2009; Montie et al., 2009; Orr et al., 2010). Motor neurons expressing AR112Q in these cultures die in response to DHT (Montie et al., 2009). Treatment of AR112Q-expressing cultures with AAV-EGFP also resulted in DHT-dependent death (Fig. 3). However, overexpression of SIRT1 abrogated DHT-dependent death of motor neurons in a manner dependent upon its deacetylase activity, as deacetylase-deficient SIRT1(H363Y) did not rescue expanded AR-expressing motor neurons from DHT-dependent death (Fig. 3). These data further substantiate a neuroprotective role of SIRT1 in SBMA and highlight its potential as a promising candidate for therapeutic development.
Figure 3.
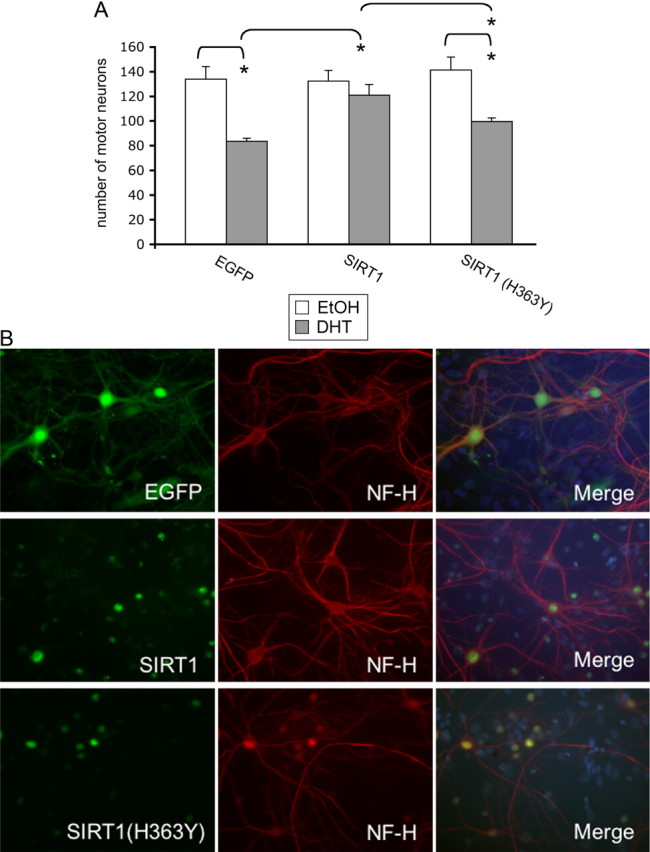
SIRT1 protects motor neurons expressing polyglutamine-expanded AR from DHT-dependent death. Dissociated spinal cord cultures were initiated from transgenic mouse embryos expressing AR112Q. SIRT1, deacetylase deficient SIRT1 (SIRT1(H363Y); both tagged with myc-his) or EGFP were overexpressed using AAV infection. Five days after infection, cultures were treated with DHT or EtOH for an additional 7 d and motor neuron number was determined. SIRT1 expression was assayed using an anti-myc antibody. A, Motor neurons from 10 random fields of cells were counted under 20,000× magnification from triplicate wells for each treatment and the average number of motor neurons was determined. SIRT1 protected AR112Q-expressing motor neurons from DHT-dependent death, while SIRT1(H363Y) did not. B, Images represent motor neurons treated with DHT as described in A. NF-H, Unphosphorylated neurofilament-heavy chain. Percentage infectivity of motor neurons with AAV: 92% EGFP (control), 88% SIRT1, and 94% SIRT1(H363Y). ANOVA with post hoc Tukey test was performed to determine statistical significance. *p < 0.01.
SIRT1 reduces AR acetylation at lysines 630, 632, and 633
We demonstrated that the deacetylase activity of SIRT1 is essential for its role in ameliorating polyQ-expanded AR aggregation and toxicity (Figs. 1, 3). Of the many known SIRT1 substrates, one of note is the AR itself (Fu et al., 2006). SIRT1 has been shown to specifically deacetylate lysines 630, 632, and 633 within an AR with normal polyglutamine length (Fu et al., 2006). Thus, we evaluated the effect of SIRT1 overexpression on the acetylation of polyQ-expanded AR. We found that SIRT1 overexpression decreased AR112Q acetylation in a manner dependent upon its deacetylase activity (Fig. 4A,B).
Figure 4.
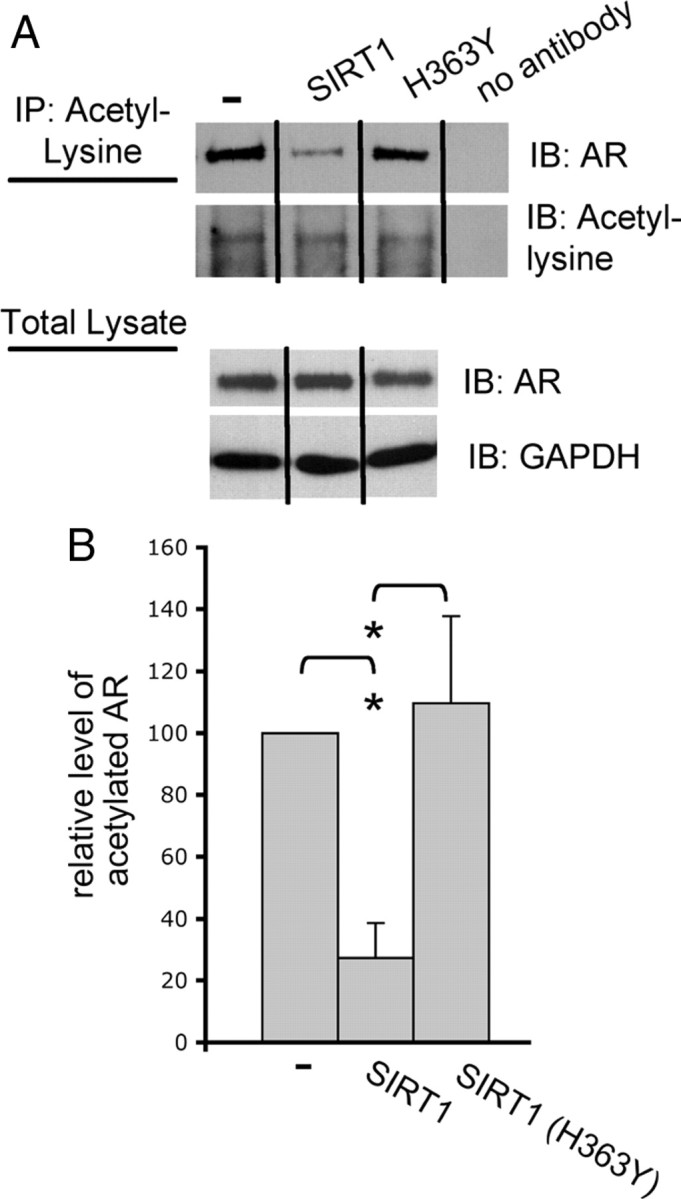
SIRT1 reduces acetylation of polyQ-expanded AR. A, Cells were treated with 10 nm DHT or EtOH for 48 h. Immunoprecipitation (IP) of acetyl-lysine and immunoblotting (IB) for AR (AR-H280) revealed a substantial reduction in AR112Q acetylation when SIRT1, but not SIRT1 (H363Y), was overexpressed. Western analysis revealed equivalent levels of AR between cell lines. B, Relative densitometric values of AR following acetyl-lysine IP and Western analysis of AR in AR112Q-expressing cells and in AR112Q-expressing cells that coexpress either SIRT1 or SIRT1 (H363Y). The level of acetylated AR in AR112Q cells was set as 100. A significant reduction in AR112Q acetylation was observed in the presence of SIRT1. Student's t test was performed to determine statistical significance. *p < 0.05.
We next sought to determine whether the effect of SIRT1 on polyQ-expanded AR acetylation is due to its ability to deacetylate lysines 630, 632, and 633. The three lysines within the AR that are acetylated in response to hormone are clustered in the KLKK motif located in the hinge region at positions 630/632/633 (Fu et al., 2000). This sequence is part of a bipartite nuclear localization signal (Simental et al., 1991; Zhou et al., 1994) and mutation of these lysines within normal Q-length AR reduces hormone-dependent nuclear AR transit (Zhou et al., 1994; Thomas et al., 2004; Shiota et al., 2010). To circumvent the complication of reduced nuclear AR localization, we introduced a heterologous nuclear localization signal [cloned in triplicate (NLSX3)], fused to the AR N terminus, as previously described (Montie et al., 2009). We used Tet-On PC12 cells expressing AR110Q with either intact lysines 630, 632, and 633 (K3) or acetylation-null mutation at amino acids 630, 632, and 633 (R3), resulting in the inability of this AR protein to be acetylated at lysines 630, 632, 633, as previously shown (Fu et al., 2006). We also created two cell lines with an additional modification by stably overexpressing SIRT1 in AR110Q-K3- and AR110Q-R3-expressing PC12 cells. Evaluation of AR acetylation following DHT treatment revealed a substantial decrease in acetylated AR110Q-K3 upon SIRT1 overexpression, but no effect of SIRT1 overexpression on the already low levels of AR acetylation observed for AR110Q(R3) (Fig. 5). These results indicate that lysines 630/632/633, previously shown to be deacetylated by SIRT1 (Fu et al., 2006), are critical for SIRT1 to decrease the acetylation of polyglutamine-expanded AR.
Figure 5.

Lysines 630, 632, and 633 within the AR are required for SIRT1 to decrease polyglutamine-expanded AR acetylation. PC12 cells expressing polyQ-expanded AR110Q with an exogenous nuclear localization signal in triplicate (NLSX3) were used for these studies. PC12 cells expressing AR110Q or AR110Q with lysines 630, 632, and 633 mutated to acetylation-null arginine (AR110Q-R3) in the presence and absence of stable overexpression of SIRT1 were treated with 10 nm DHT for 48 h. Acetyl-lysine was immunoprecipitated from protein lysates and AR levels determined by Western analysis (AR-H280). Overexpression of SIRT1 reduced the levels of acetylated AR110Q, while it had no effect on levels of acetylated AR110Q-R3. Note that levels of acetylated AR111Q-R3 were reduced compared with those of AR110Q. Equivalent levels of acetyl-lysine were immunoprecipitated from each sample (data not shown). Western analysis of total protein lysates revealed equivalent levels of AR between cell lines.
The ability of SIRT1 to protect motor neurons expressing polyQ-expanded AR from death is dependent upon intact AR lysines 630, 632, 633
Given the role of AR lysines 630, 632, and 633 in the reduction of AR acetylation by SIRT1, we questioned whether the neuroprotective effect of SIRT1 in SBMA cell models depends on its ability to deacetylate these three lysines. We used dissociated spinal cord cultures to determine whether SIRT1 would protect motor neurons expressing polyQ-expanded AR with a mutation at lysines 630, 632, and 633 that both mimics acetylation and prevents their deacetylation (K630Q/K632Q/K633Q; Q3) (Fu et al., 2003). SIRT1 overexpression in motor neurons expressing polyQ-expanded AR with intact lysines (AR111Q-K3) rescued them from DHT-dependent death (Fig. 6). However, SIRT1 failed to protect motor neurons expressing AR111Q-Q3 (acetylation-mimic) from DHT-dependent AR toxicity (Fig. 6). These data indicate that lysines 630, 632, and 633 are necessary for the neuroprotective effect of SIRT1 on polyQ-expanded AR toxicity in motor neurons.
Figure 6.

The neuroprotective role of SIRT1 in motor neurons expressing polyQ-expanded AR is dependent upon AR lysines 630, 632, and 633. Dissociated spinal cord cultures were initiated from nontransgenic embryonic mice. AAVs were made to express EGFP, SIRT1-myc-his, AR111Q, or acetylation-mimic AR111Q-Q3 [both forms of AR contain an exogenous nuclear localization signal in triplicate (NLSX3) at the N terminus]. After 5 d, the percentage infectivity of motor neurons with AR, EGFP, and SIRT1 was determined by immunofluorescence. The rest of the cultures were then treated with either EtOH or DHT for an additional 7 d. Immunofluorescence analysis was performed to reveal motor neurons (with antibody SMI32) and AR (AR-318) and Hoechst 33258 was used to stain nuclei. Motor neurons were counted from 10 random fields of cells under 20,000× magnification from triplicate wells for each treatment. The number of viable motor neurons expressing AR is represented in the graph. DHT treatment significantly reduced the number of motor neurons expressing AR111Q/EGFP, as well as those expressing AR111Q-Q3/EGFP. SIRT1 overexpression in motor neurons expressing AR111Q (lysine intact) rescued these motor neurons from DHT-dependent death, while SIRT1 did not protect motor neurons expressing AR111Q-Q3 (acetylation-mimic). ANOVA with post hoc Tukey test was performed to determine statistical significance. *p < 0.001.
Polyglutamine-expanded AR is hyperacetylated
Our finding that SIRT1 decreases the acetylation of lysine 630, 632, and 633 within polyQ-expanded AR and that these three lysines are critical for the neuroprotective effect of SIRT1 in motor neurons expressing polyQ-expanded AR led us to question whether polyQ-expanded AR is aberrantly acetylated. We compared the acetylation status of polyQ-expanded AR to that of normal Q-length AR by using Tet-on PC12 cells that express normal Q-length (AR10Q) or expanded Q-length AR (AR112Q) (Fig. 7). We found that AR112Q, in the presence of hormone, is acetylated to a greater extent than AR10Q (2.4-fold greater, p < 0.05). Moreover, consistent with the importance of nuclear localization of the expanded AR for disease manifestation, we observed, using fractionated cell extracts, the exclusive localization of hyperacetylated AR112Q within the nucleus (Fig. 7B).
Figure 7.
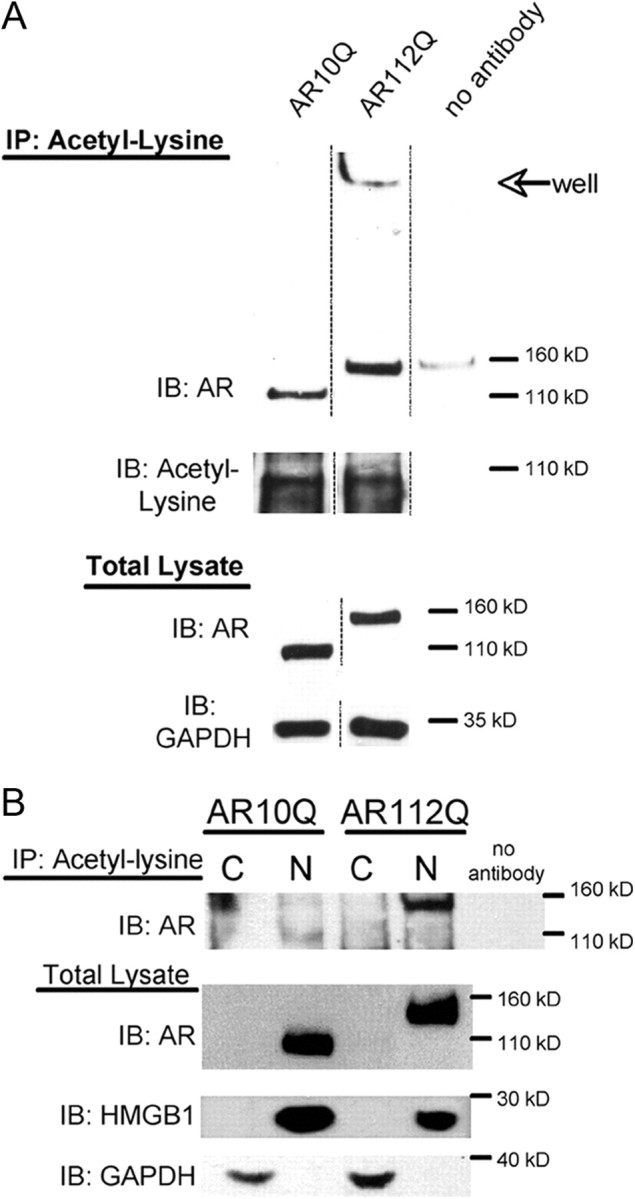
Nuclear polyglutamine-expanded AR is hyperacetylated compared with normal glutamine-length AR. AR10Q and AR112Q-expressing PC12 cells were treated with 10 nm DHT for 48 h. A, Acetylation of AR was evaluated by immunoprecipitation (IP) of acetyl-lysine from cell lysates and subsequent Western analysis. A representative Western blot shows that a larger proportion of AR112Q is acetylated compared with AR10Q, and that acetylated AR112Q is observed in an SDS-insoluble form in the well and stacking layer of the gel. Reprobing of the membrane revealed equivalent acetyl-lysine immunoprecipitated from AR10Q and AR112Q lysates. Western analysis of total lysate reveals equivalent AR levels. GAPDH was used as a loading control for total lysates. B, Nuclear and cytoplasmic fractions of cell lysates were used for immunoprecipitation (IP) of acetyl-lysine, and the immunoprecipitates were electrophoresed by SDS-PAGE and immunoblotted (IB) for AR (ARH280). Nuclear, but not cytoplasmic, AR10Q and AR112Q were immunoprecipitated with acetyl-lysine antibody. The nuclear/cytoplasmic fractions were verified by running a portion of the fractions on an SDS-page gel and probing the membrane for AR, nuclear (HMGB1), and cytoplasmic (GAPDH) markers. Note that the signal in the upper-left portion of the ARH280-probed membrane represents nonspecific background signal. There is only a faint signal of AR10Q in the nuclear fraction at 110 kDa. C, Cytoplasmic fraction; N, nuclear fraction.
Pharmacologic reduction of acetylation decreases nuclear inclusions
To investigate whether the enhanced acetylation of AR112Q plays a role in its aberrant aggregation, we used pharmacologic means to reduce AR112Q acetylation and evaluated the effect on AR aggregation. We observed a significant reduction in the number of cells with nuclear inclusions when AR112Q acetylation was reduced with anacardic acid (36% reduction, p < 0.01), an inhibitor of the histone acetyltransferases CBP/p300 and PCAF (Fig. 8).
Figure 8.
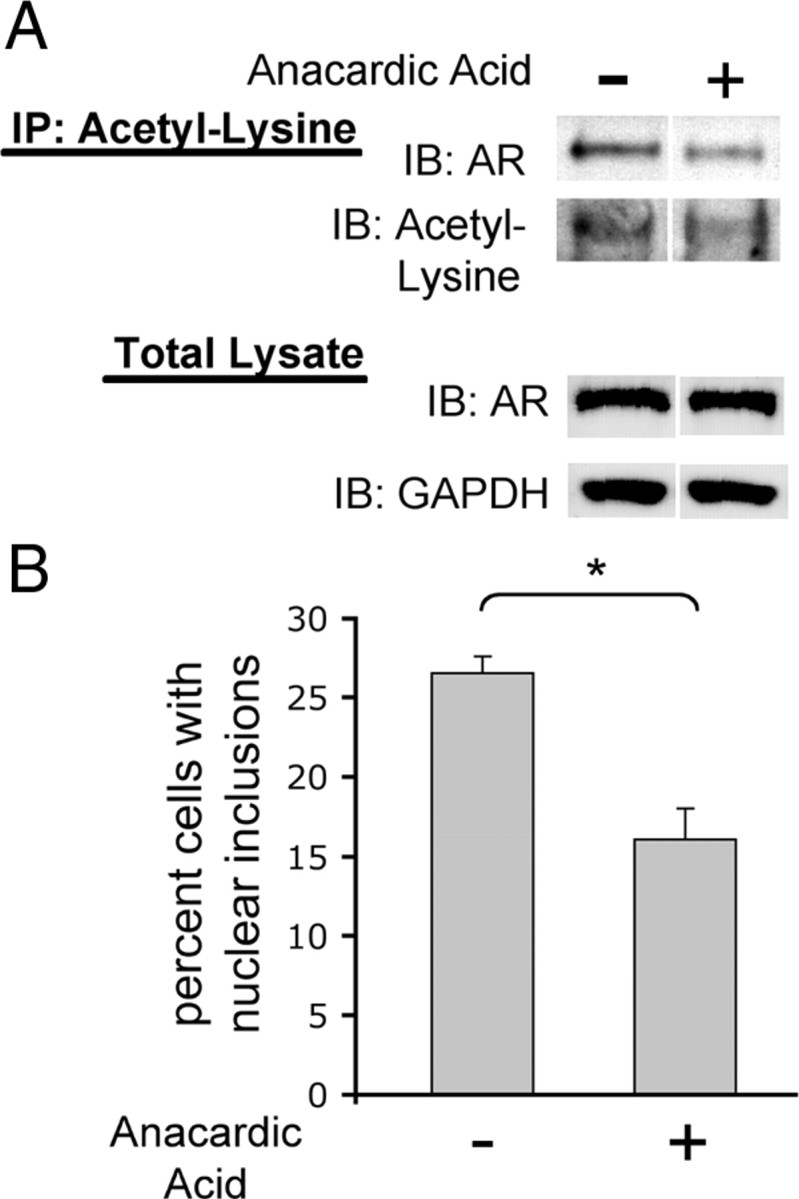
Pharmacologic reduction of polyglutamine-expanded AR reduces nuclear inclusions. AR112Q-expressing PC12 cells were treated in triplicate for 48 h with 10 nm DHT in the presence and absence of 30 μm anacardic acid (inhibitor of CBP/p300 and PCAF). A, Immunoprecipitation of acetyl-lysine and subsequent Western analysis for AR revealed that AA treatment reduced acetylation of AR112Q. Shown here is a representative Western blot. B, AR112Q-expressing cells treated in parallel were fixed and immunostained for AR. Five-hundred cells were counted from each of three wells and the percentage of cells with nuclear inclusions graphed. Anacardic acid reduced the number of cells with nuclear inclusions. Student's t test was performed to determine statistical significance. *p < 0.01.
Genetic inhibition of polyQ-expanded AR acetylation ameliorates its aggregation and toxicity
We next used a genetic approach to directly evaluate the role of polyQ-expanded AR acetylation in its aggregation and toxicity and generated multiple Tet-inducible PC12 cell lines expressing various acetylation-mutant forms of AR. For these studies, we used AR constructs with a heterologous nuclear localization signal fused to the N terminus of the AR, due to the potential effect of mutation of K630/K632/K633 on AR nuclear localization. The presence of the heterologous nuclear localization signal resulted in nuclear localization of all AR proteins, even in the absence of hormone (data not shown).
A single acetylation-null mutation (K630A) in polyQ-expanded AR (AR110Q) resulted in both fewer cells with nuclear inclusions (Fig. 9A) and resistance to DHT-dependent death (Fig. 9B). Moreover, the acetylation-null mutation of all three lysines (K630,632,633R) (R3) resulted in an even more substantial reduction in AR110Q nuclear inclusions (Fig. 9A) and also abrogated DHT-dependent death (Fig. 9B). Biochemical analysis revealed a reduction in SDS-insoluble species of AR-K630A-110Q and the near elimination of SDS-insoluble species of AR110Q-R3 (Fig. 9C). Conversely, acetylation mimic (threonine; T3) -expressing cells displayed an increase in the number of cells with AR nuclear inclusions and enhanced toxicity (Fig. 9A,B). Expression of expanded AR with an alternative acetylation-mimic (glutamine; Q3) mutation resulted in the same AR nuclear inclusion load and toxicity as cells expressing lysine-intact AR110Q (Fig. 9A,B). Biochemical analysis revealed increased SDS-insoluble acetylation-mimic AR110Q (T3 and Q3) compared with lysine-intact AR110Q upon DHT treatment (Fig. 9C). Normal Q-length acetylation mutant AR-expressing cells did not form nuclear inclusions (data not shown) nor did they die in response to DHT treatment (Fig. 9B). These data indicate that lysines 630, 632, and 633, which are acetylated in response to DHT (Fu et al., 2003), are critical to the aberrant aggregation and toxicity of polyQ-expanded AR.
Figure 9.

Genetic inhibition of mutant AR acetylation ameliorates polyQ-expanded AR aggregation and toxicity. PC12 cells expressing polyQ-expanded AR110Q with an exogenous nuclear localization signal in triplicate (NLSX3) at the N terminus were used for these studies. Cells expressing a single acetylation-null mutation (AR-K630A–110Q), a triple acetylation-null mutant (AR-R3–110Q), or triple acetylation-mimic mutant (AR-T3–110Q or AR-Q3–110Q) were treated with 10 nm DHT or EtOH for 48 h. A, Cells were immunostained and the percentage of cells (300 cells from triplicate wells) containing nuclear inclusions was graphed. A substantial reduction in the number of cells with nuclear inclusions was observed upon expression of acetylation-null (K630A; R3) expanded AR, while an increase (T3) or a similar inclusion load (Q3) was observed upon expression of acetylation-mimic expanded AR. ANOVA with post hoc Tukey test was performed to determine statistical significance. *p < 0.05 B. Cells were treated for 12 d and evaluated for cell death. Two hundred cells were counted from triplicate wells for each treatment. Normal AR110Q- and acetylation-mimic AR-expressing cells died in response to DHT, while acetylation-null AR-expressing cells remained viable. AR35Q-expressing cells did not form inclusions or die in response to DHT. ANOVA with post hoc Tukey test was performed to determine statistical significance. *p < 0.01 C, Western analysis of AR (AR-H280) revealed equivalent levels of AR between all cell lines. GAPDH served as a loading control. A longer development of the AR immunoblot revealed a substantial reduction in SDS-insoluble polyQ-expanded AR in cells expressing acetylation-null AR, while there was an increase in SDS-insoluble high molecular weight species of AR in cells expressing acetylation-mimic AR.
Acetylation-null polyQ-expanded AR does not induce DHT-dependent death of motor neurons
We next sought to determine whether AR acetylation is required for the toxic effects of polyQ-expanded AR in the disease-relevant cell type, motor neurons. For these studies, we initiated mixed spinal cord cultures from nontransgenic, wild-type mice and infected motor neurons with AAVs that express either NLSX3-AR111Q (K3; lysine intact) or NLSX3-AR111Q (R3; acetylation-null) to determine whether prevention of AR acetylation at lysines 630, 632, and 633 would protect motor neurons from DHT-dependent death. For these experiments, a heterologous nuclear localization signal was fused to the N terminus of the acetylation mutant AR to ensure nuclear localization of polyQ-expanded AR (Fig. 10A). Motor neurons expressing AR111Q died in response to DHT, whereas motor neurons expressing equivalent levels (Fig. 10B) of acetylation-null AR111Q (AR111Q-R3) remained viable (Fig. 10C). These results further validate a role for lysines 630, 632, and 633 within the AR in DHT-dependent, polyQ-expanded AR motor neuron toxicity.
Figure 10.
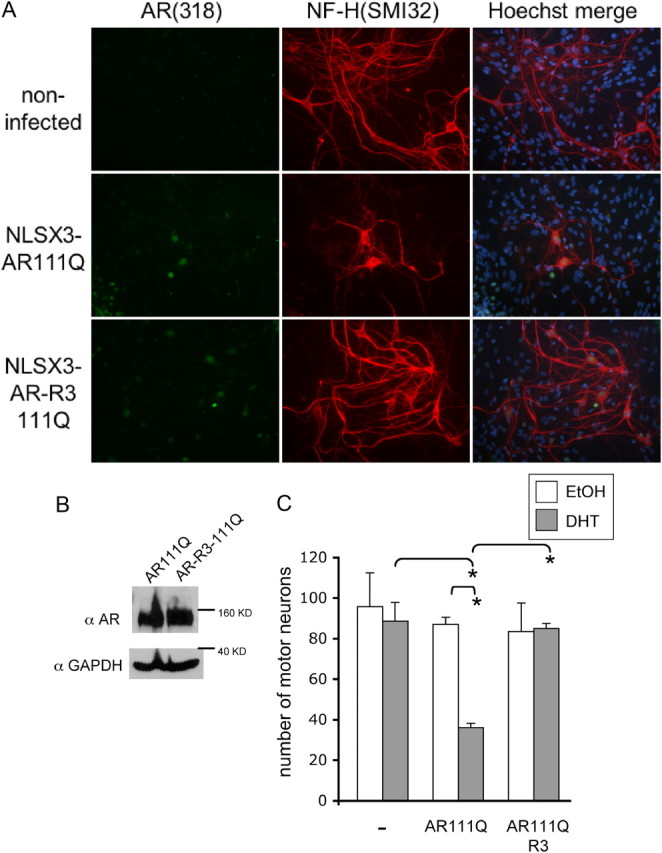
Acetylation-null polyQ-expanded AR does not induce DHT-dependent death of motor neurons. Dissociated spinal cord cultures were initiated from nontransgenic embryonic mice. AAVs were made to express AR111Q or acetylation-null AR111Q-R3 with an exogenous nuclear localization signal in triplicate (NLSX3) at the N terminus. The cultures were infected with these viruses to achieve an equal number of motor neurons expressing AR. After 5 d, cultures were treated with either EtOH or DHT for an additional 7 d. A, Image represents immunofluorescence of DHT-treated cultures that were noninfected or infected with virus expressing AR111Q or AR111Q-R3. Percentage infectivity of motor neurons with AAV = 78% AR111Q, 82% AR111Q-R3. B, Protein lysates harvested from DHT-treated cultures and analyzed by Western blotting using AR-H280 and GAPDH as a loading control. Similar levels of AR111Q and AR111Q-R3 were observed. C, The number of motor neurons expressing AR was graphed following 7 d of EtOH or DHT treatment. Immunofluorescence analysis was performed to reveal motor neurons (with antibody SMI32) and AR (AR-318); Hoechst 33258 was used to stain nuclei. Motor neurons were counted from 10 random fields of cells under 20,000× magnification from triplicate wells for each treatment. DHT treatment significantly reduced the number of motor neurons expressing AR111Q, while DHT had no effect on the number of viable motor neurons expressing acetylation-null AR111Q-R3. ANOVA was performed with post hoc Tukey test to determine significance. *p < 0.005.
Acetylation of poly-Q expanded AR enhances stabilization of the protein by DHT
To begin to understand the mechanism by which acetylation of polyQ-expanded AR is involved in its aggregation and toxicity, we assessed DHT-dependent stabilization of the AR with mutations at lysines 630, 632, and 633. Unlike other nuclear receptors (for review, see (Stanisić et al., 2010), the AR is stabilized upon hormone binding (Wright et al., 1981; Kemppainen et al., 1992). As acetylation of the AR occurs in response to hormone binding, we questioned whether the acetylation state of the polyQ-expanded AR contributes to its stabilization. We evaluated the effect of mutation of lysines 630, 632, and 633 on the stabilization of AR110Q in the presence of DHT. As shown in Figure 9C, all acetylation mutant forms of AR110Q were stabilized by DHT. To more precisely analyze the effect of AR acetylation on its stabilization in response to hormone binding, we used conditions of minimal aggregation and inhibition of new AR synthesis. We took advantage of the inducible nature of AR in Tet-On PC12 cells to manipulate the production of AR and treated Tet-On PC12 cells with doxycycline to express AR110Q with either intact lysines (K3), or with acetylation-null (K630A and R3) or acetylation-mimic (T3 and Q3) mutations. We then inhibited AR production by washing out doxycycline and treated cells with DHT to compare AR levels before and after DHT treatment. Under these conditions, a significant stabilization of AR110Q protein was revealed upon DHT treatment, as expected (Fig. 11). However, mutation of lysine 630 to an acetylation-null alanine or mutation of lysines 630, 632, and 633 to acetylation-null arginines reduced the stabilization of AR by DHT (Fig. 11). There was, however, a significant stabilization of AR when these lysines were mutated to either of the acetylation-mimic amino acids threonine (T3) or glutamine (Q3). In fact, the DHT-induced T3-mutant AR was substantially more stabilized than was lysine-intact AR110Q (Fig. 11). This finding may explain the higher propensity of this mutant AR to aggregate and induce toxicity in PC12 cells (Fig. 9A,B). We thus conclude that acetylation of polyQ-expanded AR enhances its stabilization in the presence of DHT. These data suggest that one mechanism by which the acetylation state of polyQ-expanded AR impacts its aggregation and toxicity is by regulating the steady-state levels of the mutant protein in the presence of DHT.
Figure 11.

Acetylation site mutation alters the stabilization of AR by DHT. PC12 cells expressing polyQ-expanded AR110Q with an exogenous nuclear localization signal in triplicate (NLSX3) at the N terminus were used for these studies. Cells expressing lysine-intact AR (AR-110Q), AR with a single acetylation-null mutation (AR-K630A–110Q), a triple acetylation-null mutant AR (AR-R3–110Q), or triple acetylation-mimic AR mutants (AR-T3–110Q or AR-Q3–110Q) were treated with doxycycline (DOX) to express equivalent levels of AR for 48 h, then DHT samples were treated for an additional 36 h with 10 nm DHT in the absence of DOX. Western analysis of AR (AR-H280) reveals reduced DHT-stabilization of acetylation-null mutant AR110Q. Statistical analysis was performed to determine differences between samples with or without DHT for each cell line. Additionally, the percentage increase in AR levels in the presence of DHT was compared between cell lines. Note that the T3 (acetylation-mimic) AR110Q AR is stabilized substantially more than lysine-intact (K3) AR110Q. Student's t test was used to determine statistical significance.
Discussion
The understanding of a role for protein posttranslational modifications in the pathogenesis of several neurodegenerative diseases has fueled research into the validation of such modifications as “druggable” therapeutic targets. Such transient protein modification has the potential to alter protein metabolism and function and, in the case of disease-causing proteins, can significantly impact proteotoxicity.
The acetylation of polyQ-expanded AR represents a candidate posttranslational AR modification for evaluation due to its dependence on AR hormone binding, a critical feature of disease in SBMA (Katsuno et al., 2002; Takeyama et al., 2002; Chevalier-Larsen et al., 2004). Sites of acetylation on the AR are clustered in the KLKK motif located in the hinge region at positions 630/632/633 (Fu et al., 2000). Regulation of AR acetylation occurs through the combined activities of the nuclear type A acetyltransferases CBP/p300, PCAF, Tip60 (Fu et al., 2000, 2003; Gaughan et al., 2002; Gong et al., 2006) and the deacetylases HDAC1 (Gaughan et al., 2002) and SIRT1 (Fu et al., 2006).
SIRT1, while better known for its effects on longevity and calorie restriction-induced cellular protection, gained recent attention for its protective role in neurodegenerative diseases (for review, see Haigis and Sinclair, 2010; Huber and Superti-Furga, 2011). Additional studies have revealed the ability of SIRT1 to deacetylate a host of proteins, including several involved in neurodegenerative disorders. We thus reasoned that SIRT1 might also be neuroprotective in SBMA.
We found that SIRT1 reduced DHT-dependent nuclear polyQ-expanded AR aggregation and toxicity in cell models of SBMA. Moreover, the deacetylase activity of SIRT1 was crucial for this effect, as deacetylase-deficient SIRT1(H363Y) did not prevent toxicity or decrease AR aggregation, but rather increased these disease features, perhaps by preventing the binding of endogenous SIRT1 to the mutant AR. Consistent with this interpretation is the result of our studies in which SIRT1 protein knockdown (Fig. 2), or inhibition with Sirtinol (data not shown), increased expanded AR aggregation. Our finding that the deacetylase activity of SIRT1 was critical to its ability to reduce polyQ-expanded AR aggregation complements our finding of decreased AR aggregation in the presence of the acetyltransferase inhibitor anacardic acid and suggested that SIRT1 may function to decrease polyQ-AR aggregation and toxicity directly, through its deacetylation of the mutant AR. However, as SIRT1 has broad neuroprotective effects on various cellular pathways (Bouras et al., 2005; Rodgers et al., 2005; Lee et al., 2008; Westerheide et al., 2009), we thought it also possible that its effects on AR toxicity might be indirect. To our surprise, the neuroprotective effect of SIRT1 on motor neurons was completely dependent upon this direct ability of SIRT1 to deacetylate AR.
The effect of SIRT1 on both AR acetylation and its aggregation and toxicity suggested that AR acetylation at K630/K632/K633 plays a central role in the aggregation and toxicity of polyQ-expanded AR. In fact, a role for these lysines in AR cytoplasmic/nuclear trafficking and folding homeostasis was previously informed by Lieberman's group (Thomas et al., 2004), which motivated us to evaluate the role of nuclear polyQ-expanded AR acetylation in disease. Confirmation of our hypothesis that AR acetylation is a modifier of expanded AR proteotoxicity was provided by genetic studies in which the acetylation-mimic substitution of threonine for lysine enhanced the aggregation and toxicity of polyQ-expanded AR. Moreover, mutation of K630/K632/K633 to an acetylation-null amino acid (arginine) almost entirely prevented expanded AR aggregation and completely abolished toxicity, fully validating a role for acetylation of these lysines in the altered metabolism and toxicity of the mutant AR in cell models of SBMA.
Although our studies reveal that deacetylation of K630/632/633 by SIRT1 is the mechanism by which SIRT1 protects motor neurons from polyQ-expanded AR toxicity, it is not completely clear how the acetylation state of the mutant AR impacts its toxicity. In addressing this question, we discovered that acetylation of K630/632/633 enhances the stabilization of polyQ-expanded AR by DHT. As polyQ-expanded protein levels correlate with aggregation and toxicity, even a modest disruption in mutant protein turnover could substantially impact these components of disease. However, modulating AR expression to levels similar to those observed upon DHT binding of the acetylation-null AR mutant fails to mimic the dramatic effect of this acetylation site mutation on AR aggregation and toxicity (data not shown), suggesting that additional factors are important in mediating the effects of AR acetylation on its metabolism and toxicity.
Much is known about the function of AR acetylation. Acetylation of K630/632/633 stimulates the expression of AR target genes by enhancing binding to coactivators, including p300/CBP, whereas unacetylated AR recruits AR corepressors such as N-Cor (Fu et al., 2000, 2004). Whether components of the AR transcriptional machinery modulate AR metabolism and toxicity in response to its acetylation state is under investigation. In addition, a relationship exists between AR acetylation and its phosphorylation, as acetylation-site mutants exhibit reduced total AR phosphorylation (Fu et al., 2004), another posttranslational modification of the AR that contributes to disease (Palazzolo et al., 2007, 2009). Acetylation of the AR may also impact the amino-carboxyl interaction, a feature essential for mutant AR aggregation and toxicity (Orr et al., 2010). In addition, the acetylation status of the AR is known to alter the activation of specific target genes (Fu et al., 2003) and to affect MEKK1-dependent apoptosis (Fu et al., 2002). Thus, it may be that altered transcriptional activation by the AR mutants used here contributed to the distinct cellular phenotypes of survival and death.
In addition to achieving a better mechanistic understanding of the role of polyQ-expanded AR acetylation in SBMA, it is important to determine the basis of polyQ-expanded AR hyperacetylation. PolyQ-expanded AR was aberrantly acetylated in the absence of hormone in a cell model of SBMA (Lieberman et al., 2002). In our current studies, in the presence of DHT, polyQ-expansion was associated with AR hyperacetylation. The cause of this enhanced acetylation is currently unclear. Hyperacetylation of the mutant AR may result from the increased addition or the decreased removal of acetyl groups. The increased addition of acetyl groups to the AR may result from an increased association of polyQ-expanded AR with histone acetyltransferases. For instance, CBP, an acetyltransferase of the AR (Fu et al., 2000), is sequestered into mutant AR nuclear inclusions (McCampbell et al., 2000); perhaps an enhanced colocalization of CBP with misfolded polyQ-expanded AR increases the transfer of acetyl groups to the AR. Alternatively, there may be decreased removal of acetyl groups from polyQ-expanded AR, resulting from reduced interaction of mutant AR with a deacetylase. Our finding that increased expression of the AR deacetylase SIRT1 reduced the acetylation of polyQ-expanded AR indicates that the mutant AR is capable of being deacetylated. Finally, polyQ-expanded AR hyperacetylation may represent an un-degraded species of the protein, consistent with our studies revealing enhanced DHT-stabilization of acetylated AR.
Acetylation affects other polyQ-expanded proteins through disparate roles in pathogenesis. Acetylation of lysine 444 of the huntingtin protein enhances its turnover by the autophagic pathway, highlighting acetylation of huntingtin as a protective modification (Jeong et al., 2009). However, in models of SCA-7, the acetylation of polyQ-expanded ataxin-7 decreases its turnover, leading to accumulation of a toxic fragment of polyQ-expanded ataxin-7 (Mookerjee et al., 2009). Our studies indicate that one mechanism by which acetylation of polyQ-expanded AR alters its metabolism and toxicity is through its impact on protein turnover. Thus, it appears that the differential roles of acetylation, with regard to the metabolism of the mutant protein and its neuronal toxicity, depend upon the protein within which the polyQ expansion is contained. This highlights the significance of protein context and the importance of understanding the normal metabolism and function of polyQ-expanded proteins when investigating mechanisms of disease.
In addition to direct effects on polyQ proteins, acetylation can have indirect effects on polyQ protein toxicity. For example, increasing CBP/p300 levels in a Drosophila model of polyQ disease reduced aggregation of a polyQ peptide (Taylor et al., 2003), while in our studies, inhibition of CBP/p300 with anacardic acid reduced polyQ expanded AR aggregation. It may be that the pathogenic pathways in these models differ, due to either differences in the polyQ proteins themselves or to the in vivo versus in vitro settings. Our studies of AR acetylation in mouse models of SBMA should clarify this discrepancy.
We have demonstrated that the ability of SIRT1 to deacetylate polyQ-expanded AR is central to its neuroprotection in cell culture models of SBMA. The finding that SIRT1 failed to protect motor neurons from DHT-dependent toxicity of a polyQ-expanded AR that was refractory to SIRT1 deacetylation was somewhat surprising, given the effect of SIRT1 on several neuroprotective pathways in models of other neurodegenerative diseases. Whether the ability of SIRT1 to protect from polyQ-expanded AR toxicity in vivo is similarly dependent on its ability to deacetylate the AR is under investigation. Nonetheless, the discovery of the mechanistic basis for the neuroprotection by SIRT1 in SBMA motor neurons highlights both the vital role of AR acetylation in disease and the therapeutic potential of targeting both SIRT1 and AR acetylation in SBMA.
Footnotes
This work was supported by NIH Grants R01 NS047381, R01 NS32214 (D.E.M.), and DK07705 (H.L.M.) and by the Kennedy's Disease Association (H.L.M). We thank Yuhong Liu, M.S., for technical assistance with dissociated spinal cord cultures and Michael Powell for thoughtful discussions.
The authors declare no competing financial interests.
References
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Bouras T, Fu M, Sauve AA, Wang F, Quong AA, Perkins ND, Hay RT, Gu W, Pestell RG. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J Biol Chem. 2005;280:10264–10276. doi: 10.1074/jbc.M408748200. [DOI] [PubMed] [Google Scholar]
- Chevalier-Larsen ES, O'Brien CJ, Wang H, Jenkins SC, Holder L, Lieberman AP, Merry DE. Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J Neurosci. 2004;24:4778–4786. doi: 10.1523/JNEUROSCI.0808-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M, Wang C, Reutens AT, Wang J, Angeletti RH, Siconolfi-Baez L, Ogryzko V, Avantaggiati ML, Pestell RG. p300 and p300/cAMP-response element-binding protein-associated factor acetylate the androgen receptor at sites governing hormone-dependent transactivation. J Biol Chem. 2000;275:20853–20860. doi: 10.1074/jbc.M000660200. [DOI] [PubMed] [Google Scholar]
- Fu M, Wang C, Wang J, Zhang X, Sakamaki T, Yeung YG, Chang C, Hopp T, Fuqua SA, Jaffray E, Hay RT, Palvimo JJ, Jänne OA, Pestell RG. Androgen receptor acetylation governs trans activation and MEKK1-induced apoptosis without affecting in vitro sumoylation and trans-repression function. Mol Cell Biol. 2002;22:3373–3388. doi: 10.1128/MCB.22.10.3373-3388.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M, Rao M, Wang C, Sakamaki T, Wang J, Di Vizio D, Zhang X, Albanese C, Balk S, Chang C, Fan S, Rosen E, Palvimo JJ, Jänne OA, Muratoglu S, Avantaggiati ML, Pestell RG. Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Mol Cell Biol. 2003;23:8563–8575. doi: 10.1128/MCB.23.23.8563-8575.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M, Rao M, Wu K, Wang C, Zhang X, Hessien M, Yeung YG, Gioeli D, Weber MJ, Pestell RG. The androgen receptor acetylation site regulates cAMP and AKT but not ERK-induced activity. J Biol Chem. 2004;279:29436–29449. doi: 10.1074/jbc.M313466200. [DOI] [PubMed] [Google Scholar]
- Fu M, Liu M, Sauve AA, Jiao X, Zhang X, Wu X, Powell MJ, Yang T, Gu W, Avantaggiati ML, Pattabiraman N, Pestell TG, Wang F, Quong AA, Wang C, Pestell RG. Hormonal control of androgen receptor function through SIRT1. Mol Cell Biol. 2006;26:8122–8135. doi: 10.1128/MCB.00289-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W. Androgen receptor as a therapeutic target. Adv Drug Deliv Rev. 2010;62:1277–1284. doi: 10.1016/j.addr.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Gaughan L, Logan IR, Cook S, Neal DE, Robson CN. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J Biol Chem. 2002;277:25904–25913. doi: 10.1074/jbc.M203423200. [DOI] [PubMed] [Google Scholar]
- Gong J, Zhu J, Goodman OB, Jr, Pestell RG, Schlegel PN, Nanus DM, Shen R. Activation of p300 histone acetyltransferase activity and acetylation of the androgen receptor by bombesin in prostate cancer cells. Oncogene. 2006;25:2011–2021. doi: 10.1038/sj.onc.1209231. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber K, Superti-Furga G. After the grape rush: Sirtuins as epigenetic drug targets in neurodegenerative disorders. Bioorg Med Chem. 2011;19:3616–3624. doi: 10.1016/j.bmc.2011.01.018. [DOI] [PubMed] [Google Scholar]
- Jeong H, Then F, Melia TJ, Jr, Mazzulli JR, Cui L, Savas JN, Voisine C, Paganetti P, Tanese N, Hart AC, Yamamoto A, Krainc D. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell. 2009;137:60–72. doi: 10.1016/j.cell.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Sang C, Kobayashi Y, Doyu M, Sobue G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. 2002;35:843–854. doi: 10.1016/s0896-6273(02)00834-6. [DOI] [PubMed] [Google Scholar]
- Kemppainen JA, Lane MV, Sar M, Wilson EM. Androgen receptor phosphorylation, turnover, nuclear transport and transcriptional activation: specificity for steroids and antihormones. J Biol Chem. 1992;267:968–974. [PubMed] [Google Scholar]
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Miwa S, Kobayashi Y, Merry DE, Yamamoto M, Tanaka F, Doyu M, Hashizume Y, Fischbeck KH, Sobue G. Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol. 1998;44:249–254. doi: 10.1002/ana.410440216. [DOI] [PubMed] [Google Scholar]
- Lieberman AP, Harmison G, Strand AD, Olson JM, Fischbeck KH. Altered transcriptional regulation in cells expressing the expanded polyglutamine androgen receptor. Hum Mol Genet. 2002;11:1967–1976. doi: 10.1093/hmg/11.17.1967. [DOI] [PubMed] [Google Scholar]
- McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, Merry D, Chai Y, Paulson H, Sobue G, Fischbeck KH. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet. 2000;9:2197–2202. doi: 10.1093/hmg/9.14.2197. [DOI] [PubMed] [Google Scholar]
- Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, Meyers D, Cole PA, Ott M, Gan L. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67:953–966. doi: 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojsilovic-Petrovic J, Nedelsky N, Boccitto M, Mano I, Georgiades SN, Zhou W, Liu Y, Neve RL, Taylor JP, Driscoll M, Clardy J, Merry D, Kalb RG. FOXO3a is broadly neuroprotective in vitro and in vivo against insults implicated in motor neuron diseases. J Neurosci. 2009;29:8236–8247. doi: 10.1523/JNEUROSCI.1805-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montie HL, Cho MS, Holder L, Liu Y, Tsvetkov AS, Finkbeiner S, Merry DE. Cytoplasmic retention of polyglutamine-expanded androgen receptor ameliorates disease via autophagy in a mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2009;18:1937–1950. doi: 10.1093/hmg/ddp115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mookerjee S, Papanikolaou T, Guyenet SJ, Sampath V, Lin A, Vitelli C, DeGiacomo F, Sopher BL, Chen SF, La Spada AR, Ellerby LM. Posttranslational modification of ataxin-7 at lysine 257 prevents autophagy-mediated turnover of an N-terminal caspase-7 cleavage fragment. J Neurosci. 2009;29:15134–15144. doi: 10.1523/JNEUROSCI.4720-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelsky NB, Pennuto M, Smith RB, Palazzolo I, Moore J, Nie Z, Neale G, Taylor JP. Native functions of the androgen receptor are essential to pathogenesis in a Drosophila model of spinal and bulbar muscular atrophy. Neuron. 2010;67:936–952. doi: 10.1016/j.neuron.2010.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr CR, Montie HL, Liu Y, Bolzoni E, Jenkins SC, Wilson EM, Joseph JD, McDonnell DP, Merry DE. An interdomain interaction of the androgen receptor is required for its aggregation and toxicity in spinal and bulbar muscular atrophy. J Biol Chem. 2010;285:35567–35577. doi: 10.1074/jbc.M110.146845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzolo I, Burnett BG, Young JE, Brenne PL, La Spada AR, Fischbeck KH, Howell BW, Pennuto M. Akt blocks ligand binding and protects against expanded polyglutamine androgen receptor toxicity. Hum Mol Genet. 2007;16:1593–1603. doi: 10.1093/hmg/ddm109. [DOI] [PubMed] [Google Scholar]
- Palazzolo I, Stack C, Kong L, Musaro A, Adachi H, Katsuno M, Sobue G, Taylor JP, Sumner CJ, Fischbeck KH, Pennuto M. Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron. 2009;63:316–328. doi: 10.1016/j.neuron.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Zhao W, Thiyagarajan M, MacGrogan D, Rodgers JT, Puigserver P, Sadoshima J, Deng H, Pedrini S, Gandy S, Sauve AA, Pasinetti GM. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem. 2006;281:21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Roy J, Minotti S, Dong L, Figlewicz DA, Durham HD. Glutamate potentiates the toxicity of mutant Cu/Zn-superoxide dismutase in motor neurons by postsynaptic calcium-dependent mechanisms. J Neurosci. 1998;18:9673–9684. doi: 10.1523/JNEUROSCI.18-23-09673.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota M, Yokomizo A, Masubuchi D, Tada Y, Inokuchi J, Eto M, Uchiumi T, Fujimoto N, Naito S. Tip60 promotes prostate cancer cell proliferation by translocation of androgen receptor into the nucleus. Prostate. 2010;70:540–554. doi: 10.1002/pros.21088. [DOI] [PubMed] [Google Scholar]
- Simental JA, Sar M, Lane MV, French FS, Wilson EM. Transcriptional activation and nuclear targeting signals of the human androgen receptor. J Biol Chem. 1991;266:510–518. [PubMed] [Google Scholar]
- Stanisić V, Lonard DM, O'Malley BW. Modulation of steroid hormone receptor activity. Prog Brain Res. 2010;181:153–176. doi: 10.1016/S0079-6123(08)81009-6. [DOI] [PubMed] [Google Scholar]
- Takeyama K, Ito S, Yamamoto A, Tanimoto H, Furutani T, Kanuka H, Miura M, Tabata T, Kato S. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron. 2002;35:855–864. doi: 10.1016/s0896-6273(02)00875-9. [DOI] [PubMed] [Google Scholar]
- Tanny JC, Dowd GJ, Huang J, Hilz H, Moazed D. An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell. 1999;99:735–745. doi: 10.1016/s0092-8674(00)81671-2. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Taye AA, Campbell C, Kazemi-Esfarjani P, Fischbeck KH, Min KT. Aberrant histone acetylation, altered transcription, and retinal degeneration in a Drosophila model of polyglutamine disease are rescued by REB-binding protein. Genes Dev. 2003;17:1463–1468. doi: 10.1101/gad.1087503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, Dadgar N, Aphale A, Harrell JM, Kunkel R, Pratt WB, Lieberman AP. Androgen receptor acetylation site mutations cuase trafficking defects, misfolding, and aggregation similar to expanded glutamine tracts. J Biol Chem. 2004;279:8389–8395. doi: 10.1074/jbc.M311761200. [DOI] [PubMed] [Google Scholar]
- Walcott JL, Merry DE. Ligand promotes intranuclear inclusions in a novel cell model of spinal and bulbar muscular atrophy. J Biol Chem. 2002;277:50855–50859. doi: 10.1074/jbc.M209466200. [DOI] [PubMed] [Google Scholar]
- Westerheide SD, Anckar J, Stevens SM, Jr, Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323:1063–1066. doi: 10.1126/science.1165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright W, Chan K, Sundaram K, Bardin CW. New observation on androgen action: androgen receptor stabilization and antisteroid effects of LHRH agonists. Adv Exp Med Biol. 1981;138:325–336. doi: 10.1007/978-1-4615-7192-6_19. [DOI] [PubMed] [Google Scholar]
- Zhou ZX, Sar M, Simental JA, Lane MV, Wilson EM. A ligand-dependent bipartite nuclear targeting signal in the human androgen receptor. J Biol Chem. 1994;269:13115–13123. [PubMed] [Google Scholar]