

Abstract
Purpose
Fuchs' endothelial corneal dystrophy (FECD) caused by the CTG triplet repeat expansion in the TCF4 gene (CTG18.1 locus) is the most common repeat expansion disorder. Intergenerational instability of expanded repeats and clinical anticipation are hallmarks of other repeat expansion disorders. In this study, we examine stability of triplet repeat allele length and FECD disease severity in parent–child transmission of the expanded CTG18.1 allele.
Methods
We studied 44 parent–child transmissions of the mutant expanded CTG18.1 allele from 26 FECD families. The CTG18.1 polymorphism was genotyped using short tandem repeat analysis, triplet repeat primed PCR assay, and Southern blot analysis. FECD severity was assessed using modified Krachmer grading (KG) system. Triplet repeat length of mutant allele and KG severity were compared between generations.
Results
Instability of the expanded allele was seen in 14 of 44 (31.8%) parent–child transmissions, and the likelihood of an unstable event increased with the size of the parental allele (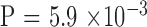 ). A tendency for contraction was seen in transmission of large alleles (repeat length > 120), whereas intermediate alleles (repeat length between 77 and 120) had predilection for further expansion (
). A tendency for contraction was seen in transmission of large alleles (repeat length > 120), whereas intermediate alleles (repeat length between 77 and 120) had predilection for further expansion ( ). Although we noted increased KG severity in the offspring in three pairs, none of these transmissions were associated with allele instability.
). Although we noted increased KG severity in the offspring in three pairs, none of these transmissions were associated with allele instability.
Conclusions
We observed instability of the TCF4 triplet repeat expansion in nearly a third of parent–child transmissions. Large mutant CTG18.1 alleles are prone to contraction, whereas intermediate mutant alleles tend to expand when unstably transmitted. Intergenerational instability of TCF4 repeat expansion has implications on FECD disease inheritance.
Keywords: Fuchs' endothelial corneal dystrophy, triplet repeat expansion, TCF4, genetics
Fuchs' endothelial corneal dystrophy (FECD, MIM 13680) is a bilateral, age-related degenerative disorder affecting 4% of whites over the age of 40 years.1 Confluent central corneal guttae with concomitant loss of endothelial cell density results in corneal edema and scarring. FECD represents the leading indication for corneal transplantation in the United States.2
Trinucleotide repeat expansions at the intronic CTG18.1 locus of the TCF4 gene are found in approximately 70% of FECD cases in whites.3–6 As such, FECD is now considered the most common human repeat expansion disease. Expansions of greater than 40 CTG triplet repeats at this locus confer significant risk for the development of FECD.4,7 The CTG triplet repeat allele length is positively correlated with FECD disease severity.5 Expanded CUG repeat RNA transcripts may be visualized as nuclear foci in FECD endothelial tissue by fluorescent in situ hybridization.8–10 These RNA nuclear foci are thought to exert cellular toxicity by sequestering the splicing factor muscleblind-like 1 (MBNL1) and triggering mis-splicing of genes in endothelium.10,11
More than 20 diseases underlying neuromuscular degenerative disorders are caused by unstable nucleotide repeats within genes.12 These expanded repeats can occur within coding regions, introns, or 3′-untranslated regions and the pathogenic mechanisms linking the molecular mutation to disease are diverse. In these other disorders, the mutant alleles with a repeat number beyond a particular threshold have been found to be unstable as they are passed from one generation to the next. Intergenerational instability of repeat expansions has been found to be related to size of the parental repeat and the sex of the parent transmitting the repeat.13 Noncoding trinucleotide repeat expansions have a different pattern of intergenerational instability compared with coding trinucleotide repeat expansions.13 Clinical anticipation is the phenomenon of greater disease severity or earlier age of onset of symptoms in successive generations of families with repeat expansion disorders.
Before its association with FECD was known, Breschel et al.14 reported that the CTG18.1 triplet repeat polymorphism in TCF4 was present in 3% of individuals in examined white pedigrees and found alleles with >37 CTG repeats to be unstable. An assessment of the intergenerational stability of the TCF4 expansion in the context of FECD is currently lacking in the literature. In this study, we sought to examine the stability of the triplet repeat allele length and FECD disease severity in parent–child transmissions of the expanded CTG18.1 allele.
Methods
Study Participants
The study was conducted with the approval of the institutional review board of the University of Texas Southwestern Medical Center (UTSW) and was in compliance with the tenets of the Declaration of Helsinki. All study subjects were recruited from the cornea referral practice at UTSW after informed consent.
The UTSW FECD cohort of 640 individuals belonging to 309 families was reviewed. We identified all parent–child pairs in which the triplet repeat length was known for both parent and child, at least one was affected with FECD, at least one was carrier of the expanded allele, and transmission of the expanded allele from parent to child was ascertainable. An allele was considered expanded and mutant if it contained 40 or more CTG repeats, as we have done in previous reports.4,5,7 Instability was defined as a difference of 10 or more triplet repeats between generations. Parent–child pairs with mosaic alleles were excluded from subsequent instability analysis.
All subjects underwent a complete eye examination including slit-lamp biomicroscopy by a cornea fellowship–trained ophthalmologist (VVM). Inclusion criteria for FECD cases was the presence of slit-lamp examination findings of grade 2 or higher on the modified Krachmer grading (KG) scale15: grade 0, no central guttae; grade 1, up to 12 scattered central guttae; grade 2, ≥12 scattered central guttae; grade 3, 1- to 2-mm confluent central guttae; grade 4, 2- to 5-mm confluent central guttae; grade 5, >5-mm confluent central guttae without stromal edema; grade 6, >5-mm confluent central guttae with stromal edema and/or histopathologic confirmation of the diagnosis after keratoplasty.
CTG18.1 Polymorphism Genotyping
Genomic DNA of subjects was extracted from leukocytes of peripheral blood samples with the NucleonBlood Extraction Kit (Amersham, Biosciences, Buckinghamshire, UK). The CTG18.1 trinucleotide repeat polymorphism was genotyped using a combination of short tandem repeat (STR) assay, triplet repeat primed PCR (TP-PCR) assay, and Southern blot analysis as our group has previously described.4,5
Statistical Analysis
In transmission investigation, parent–offspring pairs were partitioned into three groups with similar sample sizes based on the parental CTG18.1 repeat length of largest allele: small, ≤77 (n = 15); intermediate, 78 to 120 (n = 15); large, >120 (n = 12) repeats. The KG of the more severely affected eye for each subject was reported and used for statistical analysis. Age adjustment of the KG was performed according to the linear regression model: 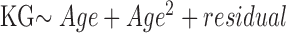 , based on all individuals. Comparisons of the demographic features were performed by exact tests, paired and unpaired t-tests, and the Wilcoxon-Mann-Whitney test, wherever it is proper, as clarified at each table. Associations between the parental CTG18.1 triplet repeat length and the intergenerational stability and length change of the repeats, as well as KG severity change, were examined by the trend test.16 Software R (version 3.3.3) was used for statistical analysis.
, based on all individuals. Comparisons of the demographic features were performed by exact tests, paired and unpaired t-tests, and the Wilcoxon-Mann-Whitney test, wherever it is proper, as clarified at each table. Associations between the parental CTG18.1 triplet repeat length and the intergenerational stability and length change of the repeats, as well as KG severity change, were examined by the trend test.16 Software R (version 3.3.3) was used for statistical analysis.
Results
Demographics of Study Subjects
There were a total of 44 parent–child pairs consisting of 76 individuals from 26 families meeting the inclusion criteria. All families were white except for one black family. The demographic information is summarized in Table 1 and Supplementary Table S1. There were 32 individuals that appeared only in the parent generation, 41 only in the child generation, and 3 in both generations. There were more affected individuals in the parent generation (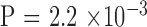 ). There were more females in both generations (
). There were more females in both generations (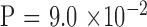 and
and 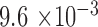 by binomial exact test), without significant difference between the two generations (
by binomial exact test), without significant difference between the two generations (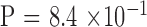 ). There were 3, 11, 10, and 20 male-to-male, male-to-female, female-to-male, and female-to-female pairs, respectively, which indicates no sex-to-sex bias in transmission (
). There were 3, 11, 10, and 20 male-to-male, male-to-female, female-to-male, and female-to-female pairs, respectively, which indicates no sex-to-sex bias in transmission (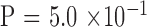 by Fisher's exact test). The expanded mutant CTG18.1 allele appeared in all parents (in this cohort, there was no occurrence of an allele that expanded from <40 repeats to >40 repeats when transmitted). The KG was significantly greater in the parent generation by the paired comparison; however, the difference diminished to insignificance after adjusting for age effect (
by Fisher's exact test). The expanded mutant CTG18.1 allele appeared in all parents (in this cohort, there was no occurrence of an allele that expanded from <40 repeats to >40 repeats when transmitted). The KG was significantly greater in the parent generation by the paired comparison; however, the difference diminished to insignificance after adjusting for age effect (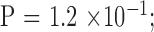 Supplementary Fig. S1).
Supplementary Fig. S1).
Table 1.
Demographic Characteristics of 76 Individuals Comprising 44 Parent–Child Pairs Stratified by Generations

Mutant CTG18.1 Allele Length in Parent–Child Transmission
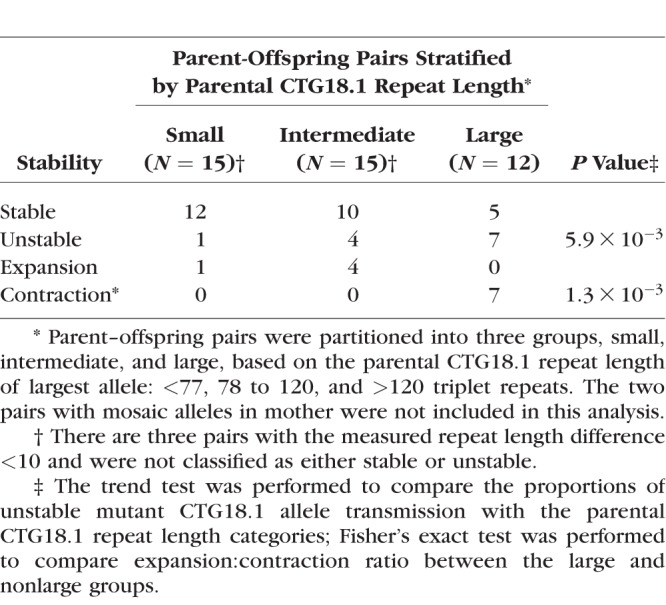
Instability of the expanded mutant CTG18.1 allele was noted in 14 of 44 (31.8%) parent–child pair transmissions (Table 2; Fig. 1; Supplementary Table S1). There were five pairs with further expansion of the mutant allele, of which four were mother–offspring pairs (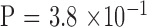 by binomial exact test). One mother was present in three of these five pairs. There were seven pairs with contraction of the mutant allele, of which four were father–offspring pairs (
by binomial exact test). One mother was present in three of these five pairs. There were seven pairs with contraction of the mutant allele, of which four were father–offspring pairs (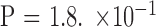 by Fisher's exact test compared with the noncontraction groups). One father was present in two of these seven pairs. There were three pairs with a difference of <10 CTG repeats between generations. There were two instances of somatic mosaicism or variation in the repeat allele length of parent: one pair wherein the mother carried one allele with mosaic repeat length of 100 to 500 and the daughter inherited an allele with repeat length of 130; another pair wherein the mother carried one allele with mosaic repeat length of 100 to 800 and the daughter inherited an allele with repeat length of 160. Comparisons between the groups are summarized in Table 2.
by Fisher's exact test compared with the noncontraction groups). One father was present in two of these seven pairs. There were three pairs with a difference of <10 CTG repeats between generations. There were two instances of somatic mosaicism or variation in the repeat allele length of parent: one pair wherein the mother carried one allele with mosaic repeat length of 100 to 500 and the daughter inherited an allele with repeat length of 130; another pair wherein the mother carried one allele with mosaic repeat length of 100 to 800 and the daughter inherited an allele with repeat length of 160. Comparisons between the groups are summarized in Table 2.
Table 2.
Comparison Between Groups With Unstable Transmission of the Mutant CTG18.1 Allele

Figure 1.

Correlation of CTG18.1 allele length difference between generations and the parental allele length. Parent–offspring pairs were partitioned into three groups, small, intermediate, and large. with similar sample sizes based on the parental CTG18.1 repeat length of largest allele: <77 (n = 15), 78 to 120 (n = 15), and >120 (n = 12) triplet repeats. Association was examined by a trend test (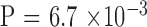 ).
).
Next, we examined the association between parental CTG18.1 repeat length and its transmission stability (Table 3). The two pairs with mosaic alleles in mother were excluded. The rate of instability increased with the length of parental repeat length (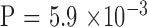 ). It is of interest to note all seven unstable transmissions in the large group were contractions, whereas all five unstable transmissions in the nonlarge group were expansions (
). It is of interest to note all seven unstable transmissions in the large group were contractions, whereas all five unstable transmissions in the nonlarge group were expansions (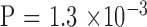 ). There is no significant difference of CTG18.1 expansion length between the two generations by a crude comparison (Table 1); however, there is a clear trend of contraction when parental CTG18.1 triplet repeat length is >120 (Fig. 1;
). There is no significant difference of CTG18.1 expansion length between the two generations by a crude comparison (Table 1); however, there is a clear trend of contraction when parental CTG18.1 triplet repeat length is >120 (Fig. 1; 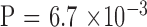 ).
).
Table 3.
Stability of Mutant CTG18.1 Allele in Parent-Offspring Transmission
FECD Severity in Parent–Child Transmission of the Mutant CTG18.1 Allele
There are three pairs in which the KG was greater in the child compared with the parent. However, the mutant repeat allele length was stable in all of them. We found no association between KG disease severity difference of a parent–offspring pair and parental repeats length after age adjustment (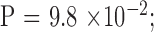 Fig. 2).
Fig. 2).
Figure 2.

Correlation of age-adjusted KG difference between generations and the parental allele length. Age adjustment of the KG was performed according to the linear regression model: 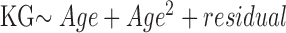 . Parent–offspring pairs were partitioned into three groups, small, intermediate, and large, with similar sample sizes based on the parental CTG18.1 repeat length of largest allele: <77 (n = 15), 78 to 120 (n = 15), and >120 (n = 12) triplet repeats. Association was examined by a trend test (
. Parent–offspring pairs were partitioned into three groups, small, intermediate, and large, with similar sample sizes based on the parental CTG18.1 repeat length of largest allele: <77 (n = 15), 78 to 120 (n = 15), and >120 (n = 12) triplet repeats. Association was examined by a trend test (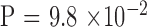 ).
).
Discussion
In this study, we noted instability of the expanded TCF4 CTG18.1 allele in nearly a third of the parent–child transmissions in families with FECD. Additionally, the likelihood of an unstable event increased with the size of the mutant parental allele. We found intermediate-sized mutant CTG18.1 alleles have a tendency for further expansion in contrast to large mutant alleles that have a tendency for contraction in unstable transmissions from parent to child. These patterns of intergenerational instability of the TCF4 triplet repeat expansion may directly impact FECD inheritance because disease severity is positively correlated to repeat length of the mutant allele.5
Because we are at an early stage of understanding FECD molecular pathogenesis and disease inheritance mediated by the TCF4 triplet repeat expansion, it is important for us to learn lessons from other repeat disorders. Parent–child transmission of noncoding repeat expansions is best characterized for myotonic dystrophy type 1 (DM1) caused by a CTG expansion in the 3′ untranslated region of the DMPK gene. There are important parallels between CTG triplet repeat expansions in the TCF4 and DMPK genes; we recently reported an increased risk for FECD in subjects with DM1 via a similar accumulation of toxic CUG repeat RNA nuclear foci in corneal endothelium.6,8
DNA repair and replication mechanisms have been implicated in trinucleotide repeat instability. Trinucleotide repeat expansion occurs in various stages of human germ cell development and is sensitive to the sex of the transmitting parent.13 Six of seven expansions in our cohort were seen with maternal transmission of the mutant TCF4 allele. Although not statistically significant, our observation in FECD parallels that of other noncoding CTG trinucleotide repeat disorders including DM1, Huntington's disease-like type 2 type, and spinocerebellar ataxia 8, in which expansions occur almost exclusively through maternal transmission.13 In DM1, expansion has been noted at the two-cell stage in preimplantation embryos, and the length of expansion is related to maternal age; both of these observations support the notion that expansion occurs in quiescent oocytes involving DNA repair mechanisms rather than DNA replication mechanisms.13
Five of eight contractions in our FECD cohort occurred with paternal transmission of the mutant TCF4 repeat allele. In the sperm of DM1 males, large trinucleotide repeat expansions contract, and the frequency of the contraction in male transmission is positively correlated to repeat length.13
Our study was adequately powered to detect the instability of the CTG18.1 repeat locus in parent–child transmissions with increasing sizes of parental allele. However, a limitation of this study was that the small sample size precluded a rigorous evaluation of role of sex. Studies on larger FECD cohorts are certainly warranted to assess the role of sex in the transmission of expanded repeat. We speculate that a shared pattern of repeat mutation may emerge in patients with FECD and DM1 with noncoding CTG repeat expansions.
We noted two examples of unstable transmissions in the setting of somatic mosaicism of the parental allele. Both somatic and germline instability of repeat DNA are thought to contribute to unstable intergenerational transmission and disease.13
Anticipation and instability studies of Huntington disease, DM1, and other repeat expansion disorders have used genomic DNA extracted from peripheral blood leukocytes.17–19 Studies on DM1 leukocytes over a 5-year period suggest that the repeat length in patient's blood cells may continue to expand with time due to somatic mutation.20 Instability of repeats in leukocytes during the lifetime of an individual may contribute to larger alleles in the parental generation. We did not examine the repeat number in endothelial tissue in this study and such experiments are warranted to understand how somatic instability in endothelial tissue impacts disease.
Clinical anticipation is a hallmark of the genetics of DM1 and other repeat disorders characterized by increasing severity of disease and/or earlier onset of disease in subsequent generations. This phenomenon in DM1 has been attributed to the instability of the CTG repeat expansion in the DMPK gene and its tendency to expand in successive generations. However, anticipation was a rare event in our cohort of FECD subjects with the CTG repeat expansion in the TCF4 gene. We found three examples in which the KG of FECD disease severity was greater in the child compared with their parent but did not see further expansion in the transmitted mutant allele. Although clinical anticipation is generally associated with expansions of unstable repeats, examples of anticipation have been noted even with contractions of the CTG repeat in DM1.21 After adjusting for age at its first and second order, there was no significant difference between KG residuals between the parental and children generation. Additionally, we found no association between KG difference of a parent–offspring pair and parental repeats length.
Earlier age of disease onset in subsequent generations is another feature of clinical anticipation. A limitation of our study was that age of onset of symptoms was not examined. Given the insidious onset of symptoms in FECD, one may not be able to rely on patient history alone to ascertain age of disease onset. Meticulous, longitudinal familial studies may be required to determine whether onset of disease occurs at an earlier age in successive generations as evidence of clinical anticipation in FECD mediated by the triplet repeat expansion in TCF4.
We noted instability of the expanded TCF4 triplet repeat expansions in nearly a third of parent–child transmissions in FECD families. Large mutant CTG18.1 alleles are prone to contraction, whereas intermediate mutant alleles tend to expand when unstably transmitted from parent to child. Intergenerational instability of the TCF4 triplet repeat expansion has implications on FECD disease inheritance.
Supplementary Material
Acknowledgments
The authors thank the patients for their participation in this study, the study coordinator efforts of Aimee Tilley and Bryan Gallerson, and the collaborating corneal specialists Wayne R. Bowman, James P. McCulley, H. Dwight Cavanagh, Steven Verity, Brad Bowman, and Walter Beebe.
Supported by National Eye Institute Grants R01EY022161 (VVM) and P30EY020799, an unrestricted grant from Research to Prevent Blindness, New York, and National Institutes of Health Grants U54AR068791 and UL1TR001105 (CX). The National Eye Institute and Research to Prevent Blindness had no role in the following: design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure: J.S. Saade, None; C. Xing, None; X. Gong, None; Z. Zhou, None; V.V. Mootha, None
References
- 1.Lorenzetti DW, Uotila MH, Parikh N, Kaufman HE. Central cornea guttata. Incidence in the general population. Am J Ophthalmol. 1967;64:1155–1158. [PubMed] [Google Scholar]
- 2.Eye Bank Association of America. 2015 Eye Banking Statisical Report. Washington, DC: Eye Bank Association of America; 2016. [Google Scholar]
- 3.Wieben ED, Aleff RA, Tosakulwong N, et al. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs corneal dystrophy. PLoS One. 2012;7:e49083. doi: 10.1371/journal.pone.0049083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mootha VV, Gong X, Ku HC, Xing C. Association and familial segregation of CTG18.1 trinucleotide repeat expansion of TCF4 gene in Fuchs' endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2014;55:33–42. doi: 10.1167/iovs.13-12611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soliman AZ, Xing C, Radwan SH, Gong X, Mootha VV. Correlation of severity of fuchs endothelial corneal dystrophy with triplet repeat expansion in TCF4. JAMA Ophthalmol. 2015;133:1386–1391. doi: 10.1001/jamaophthalmol.2015.3430. [DOI] [PubMed] [Google Scholar]
- 6.Mootha VV, Hansen B, Rong Z, et al. Fuchs' endothelial corneal dystrophy and RNA foci in patients with myotonic dystrophy. Invest Ophthalmol Vis Sci. 2017;58:4579–4585. doi: 10.1167/iovs.17-22350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xing C, Gong X, Hussain I, et al. Transethnic replication of association of CTG18.1 repeat expansion of TCF4 gene with Fuchs' corneal dystrophy in Chinese implies common causal variant. Invest Ophthalmol Vis Sci. 2014;55:7073–7078. doi: 10.1167/iovs.14-15390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mootha VV, Hussain I, Cunnusamy K, et al. TCF4 triplet repeat expansion and nuclear RNA foci in Fuchs' endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2015;56:2003–2011. doi: 10.1167/iovs.14-16222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu J, Rong Z, Gong X, et al. Oligonucleotides targeting TCF4 triplet repeat expansion inhibit RNA foci and mis-splicing in Fuchs' dystrophy. Hum Molec Genet. 2018;27:1015–1026. doi: 10.1093/hmg/ddy018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Du J, Aleff RA, Soragni E, et al. RNA toxicity and missplicing in the common eye disease Fuchs endothelial corneal dystrophy. J Biol Chem. 2015;290:5979–5990. doi: 10.1074/jbc.M114.621607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wieben ED, Aleff RA, Tang X, et al. Trinucleotide repeat expansion in the transcription factor 4 (TCF4) gene leads to widespread mRNA splicing changes in Fuchs' endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2017;58:343–352. doi: 10.1167/iovs.16-20900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rohilla KJ, Gagnon KT. RNA biology of disease-associated microsatellite repeat expansions. Acta Neuropathol Commun. 2017;5:63. doi: 10.1186/s40478-017-0468-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nature Rev Genet. 2010;11:786–799. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Breschel TS, McInnis MG, Margolis RL, et al. A novel, heritable, expanding CTG repeat in an intron of the SEF2-1 gene on chromosome 18q21.1. Hum Molec Genet. 1997;6:1855–1863. doi: 10.1093/hmg/6.11.1855. [DOI] [PubMed] [Google Scholar]
- 15.Krachmer JH, Purcell JJ, Jr,, Young CW, Bucher KD. Corneal endothelial dystrophy. A study of 64 families. Arch Ophthalmol. 1978;96:2036–2039. doi: 10.1001/archopht.1978.03910060424004. [DOI] [PubMed] [Google Scholar]
- 16.Hothorn T, Hornik K, van de Wiel MA, Zeileis A. A lego system for conditional inference. Am Stat. 2006;60:257–263. [Google Scholar]
- 17.Ranen NG, Stine OC, Abbott MH, et al. Anticipation and instability of IT-15 (CAG)n repeats in parent-offspring pairs with Huntington disease. Am J Hum Genet. 1995;57:593–602. [PMC free article] [PubMed] [Google Scholar]
- 18.Pratte A, Prevost C, Puymirat J, Mathieu J. Anticipation in myotonic dystrophy type 1 parents with small CTG expansions. Am J Med Genet Part A. 2015;167A:708–714. doi: 10.1002/ajmg.a.36950. [DOI] [PubMed] [Google Scholar]
- 19.Matsuura T, Fang P, Lin X, et al. Somatic and germline instability of the ATTCT repeat in spinocerebellar ataxia type 10. Am J Hum Genet. 2004;74:1216–1224. doi: 10.1086/421526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martorell L, Martinez JM, Carey N, Johnson K, Baiget M. Comparison of CTG repeat length expansion and clinical progression of myotonic dystrophy over a five year period. J Med Genet. 1995;32:593–596. doi: 10.1136/jmg.32.8.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashizawa T, Anvret M, Baiget M, et al. Characteristics of intergenerational contractions of the CTG repeat in myotonic dystrophy. Am J Hum Genet. 1994;54:414–423. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.