

Abstract
Five‐year overall survival (OS) of localized RMS exceeds 70% in children (<18) but is very poor in adult patients. We analyzed the outcome and prognostic factors (PF) of a national series of adult patients with RMS in a large study. The study population consisted of two different cohorts: a retrospective cohort (157 adult patients treated in 13 reference centers between 05/1981 and 02/2010) and the prospective cohort (292 patients with RMS diagnosed and treated between 01/2010 and 12/2014 in France) included in the NetSarc database. A descriptive analysis of patients’ characteristics and prognostic factors was conducted on both series which were compared. In the retrospective series, histological subtypes were embryonal (E‐RMS) for 21% of patients, alveolar (A‐RMS) for 35% of patients, and “adult‐type” P‐RMS (pleomorphic, spindle cell RMS, not otherwise specified) (P) for 44% patients. This distribution significantly differed in the prospective cohort: A‐RMS: 18%; E‐RMS: 17%; and P‐RMS 65%. With a median follow‐up of 8.5 years, 5‐year OS for localized RMS and advanced RMS (with nodes and/or metastases) was 43% and 5%, respectively, (P < 0.0001), and median OS was 51, 33, and 16 months for E‐RMS, A‐RMS, and P‐RMS, respectively, in the retrospective cohort. The median OS was less than 40 months for the prospective nationwide cohort for the entire population. In a multivariate analysis of the retrospective study, independent prognostic factors for OS were A‐RMS, R0 resection, and adjuvant radiotherapy (RT). For localized RMS, age and use of pediatric chemotherapy (CT) regimen are independent prognostic factors. Adult patients with RMS have a poorer overall survival than pediatric patients, and survival varies considerably across histological subtypes.
Keywords: Adult cancer, pediatric, rhabdomyosarcoma
Introduction
Rhabdomyosarcoma (RMS) accounts for <3% of adult soft tissue sarcoma but is the most frequent soft tissue sarcoma histological subtype before age 10 and the 4th most prevalent cancer during childhood 1, 2, 3, 4. Five‐year overall survival (OS) of children has dramatically improved in the last 30 years based on the results of successive studies of large multinational collaborative trials dedicated to children. Currently, the 5‐year OS exceeds 70% for nonmetastatic RMS 5, 6, 7, 8, 9, 10. In contrast, the outcome of adult patients remains poor. Furthermore, given the rarity of adult RMS, limited information is available in the literature, and optimal management and identification of prognostic factors (PF) have not been established in large cohorts, in particular in nationwide studies 11, 12, 13, 14.
The primary aim of the present retrospective study was to describe the treatment, outcome, and prognostic factors for adult patients with RMS. We analyzed data from a retrospective database of patients treated in reference centers as well as a more recent prospective nationwide database.
Patient and Methods
Retrospective study
The retrospective study was conducted using the “Conticabase” (see http://www.conticabase.org), which was described elsewhere (https://www.ncbi.nlm.nih.gov/pubmed/27207359). The data were obtained from 13 reference centers that managed incident cases of adult RMS between May 1981 and February 2010. A total of 157 adult patients (>18 years) with RMS were included. Histological diagnosis was confirmed with a central review by expert pathologists of the French Sarcoma Group (FSG). According to WHO classification, histological subtypes included embryonal RMS (E‐RMS), alveolar RMS (A‐RMS), or “adult‐type” RMS (including pleomorphic, adult spindle cell RMS, and not otherwise specified RMS: P‐RMS). Histological grade was established using the FNCLCC grading.
Prospective study
The data collection process in the prospective study is described elsewhere (see Netsarc.org; https://www.ncbi.nlm.nih.gov/pubmed/26974708). This prospective cohort included 292 adult patients with RMS (>18 years) treated in France in reference centers or outside reference centers between February 2010 and December 31, 2014. Histological diagnosis was systematically reviewed by expert pathologists from the French Sarcoma Group (FSG). The clinical and histological characteristics and relapse‐free, progression‐free, and overall survival are described with a median follow‐up of 26 months.
Treatments
Management adhered to local policy without formal recommendation. Surgical procedures included excision (removal of the tumor without normal tissue around tumor), wide resection (removal of the tumor with normal tissue around the tumor), compartmental resection, or amputation. Resection margins were analyzed (R0 microscopically negative margins, R1 macroscopically resected, or R2 incomplete tumor resection). Radiotherapy (RT) and chemotherapy (CT) administration were administered in a neoadjuvant, adjuvant, or palliative setting as chemotherapy. The nature of chemotherapy was classified as “adult protocol” (mainly adriamycin and/or ifosfamide) or “pediatric protocol” (PP) (mainly based on the combination of ifosfamide, vincristine, and actinomycin +/− maintenance chemotherapy with metronomic cyclophosphamide). In the retrospective cohort, all information was available, while in the prospective cohort, the precise description of chemotherapy and radiotherapy was not available. Response status was obtained after completion of treatment (chemotherapy+/− surgery +/− radiotherapy) for each patient. No information was collected on treatment after relapse. Overall survival was recorded in both databases.
Statistical analysis
Continuous variables were compared using Student's t‐test (two‐tailed) and ANOVA if normally distributed or, if not normally distributed, the Mann–Whitney or Kruskal–Wallis tests. Categorical variables were compared using the chi‐square test or Fisher's exact test if necessary.
Median follow‐up was calculated using the inverse Kaplan–Meier method. OS was calculated as the time from the date of diagnosis to the date of the event (death from any cause) or censored at the date of last visit. For patients with localized RMS, relapse‐free survival was calculated as the time from the date of diagnosis to the first relapse (local and/or metastatic) or censored at the date of last visit. OS and relapse‐free survival curves were calculated using the Kaplan–Meier method to assess the probability of survival without event during the follow‐up period and compared using the log‐rank test. Only parameters with a univariate P‐value <0.20 were introduced into the multivariate Cox regressions. Proportional hazards assumption was verified on final models by means of Schoenfeld residuals.
Patients with localized RMS (without nodes and metastases) and patients with advanced RMS (with nodes and/or metastases) were separately analyzed. Outcome of patients with locoregional nodal involvement and those with metastasis is similar, so we have gathered both in only one group, called “advanced RMS”. Moreover, patients with localized RMS were analyzed separately according to histological subtypes (alveolar RMS: A‐RMS; embryonal RMS: E‐RMS; and “adult‐type” RMS including pleomorphic, adult spindle cell RMS, and not otherwise specified RMS: P‐RMS).
All tests were two‐sided with P < 5% indicating significance. All analyses were performed with SAS 9.3 (Copyright (c) 2002–2010 by SAS Institute Inc., Cary, NC, USA).
Results
Patient and tumor characteristics
Table 1 summarizes characteristics of both cohorts. Characteristics differed according to the histological subtype. P‐RMS was more frequently located in the limbs (52/69, 75%) compared with E‐RMS (6/33, 18%) or A‐RMS (18/55, 33%) (P = 0.001). The median age was 26 (range 24–76), 25 (range 20–73), and 51 (range 23–86) years for A‐RMS, E‐RMS, and P‐RMS, respectively (P = 0.001). The median age was 24 (range 20–76), 22 (range 25–71), 45 (range 18–86), and 38 (range 55–76) years for head and neck (HN) primaries, genitourinary primaries, limbs, or other primary sites, respectively (P = 0.001). Lymph nodes (LN) and/or metastases were statistically more frequently noted in A‐RMS (30/55, 54%) compared with E‐RMS (7/33, 21%) or P‐RMS (9/69, 13%) (P = 0.001). Only metastases other than LN were observed in 22 of 157 (14%) patients with 13 of 22 (59%) patients with A‐RMS, two of 22 (9%) patients with E‐RMS, and seven of 22 (32%) patients with P‐RMS. Metastasis or LN involvement was significantly associated with the primary site, occurring in nine of 14 (64%) patients with genitourinary RMS, 16 of 39 (41%) with HN RMS, 10 of 76 (13%) with limb RMS, and 11 of 28 (39%) with other sites (P = 0.001).
Table 1.
Patient and tumor characteristics
| Retrospective 1980–2010 | Prospective 2010–2015 | |
|---|---|---|
| n (%) | n (%) | |
| No of patients | 157 | 292 |
| Gender | ||
| Male | 100 (64) | 168 (57) |
| Female | 57 (36) | 124 (43) |
| Age (years) | ||
| Median (range) | 37 (18–86) | 55 (18–99) |
| 18–25 | 43 (27) | 60 (21) |
| >25 | 114 (73) | 232 (79) |
| Disease spread | ||
| Localized | 111 (71) | 204 (70) |
| Advanced: | 46 (29) | 74 (30) |
| N+M− | 15 | NR |
| N−M+ | 22 | NR |
| N+M+ | 9 | NR |
| Histology | ||
| E‐RMS | 33 (21) | 49 (17) |
| A‐RMS | 55 (35) | 54 (18) |
| P‐RMS | 69 (44) | 189 (65) |
| Site of origin | ||
| Limbs | 76 (48) | 111 (38) |
| Head and Neck | 39 (25) | 65 (22) |
| Parameningeal | 31 | NR |
| No parameningeal | 8 | NR |
| Genitourinary | 14 (9) | NR |
| Vesicoprostatic | 6 | NR |
| No vesicoprostatic | 8 | NR |
| Others | 28 (18) | 116 (40) |
| Size (cm) | ||
| Median (range) | 8 (1–56) | 8 (1–25) |
| <5 | 31 (20) | 74 (25) |
| ≥5 | 103 (66) | 177 (61) |
| NA | 23 (14) | 41 (14) |
N, nodes; M, metastases; E‐RMS, embryonal rhabdomyosarcoma; A‐RMS, alveolar rhabdomyosarcoma; P‐RMS, “adult‐type” rhabdomyosarcoma.
In the prospective cohort, the proportion of metastatic RMS and histotype distribution was similar to the proportion reported in the retrospective cohort.
Treatment in the retrospective cohort
In the retrospective cohort, surgery of the primary tumor was performed in 108 of 157 (69%) patients: 89 of 111 (80%) patients with localized RMS versus 19 of 46 (41%) of metastatic patients (P < 0.00001). Surgery was described as excision, wide resection, compartmental resection, and amputation in 42/157 (27%), 60/157 (38%), 3/157 (2%), and 2/157 (1%) patients, respectively. Among 96 patients whose surgery was evaluable for margins, R0, R1, or R2 resection was achieved in 58/96 (60%), 28/96 (29%), and 10/96 (10%) of patients, respectively. Surgery was performed for the primary tumor in 67 of 76 (88%) patients with limb RMS, 18 of 28 (64%) with other sites RMS, six of 14 (43%) with genitourinary site, and 17 of 39 (43%) with HN RMS (P = 0.0001). Radiotherapy was performed for 107 of 157 (68%) patients including 81 patients with localized RMS. Chemotherapy was administered to 127 of 157 (81%) patients as neoadjuvant (49/157, 31%), adjuvant (37/157, 23%), both (12/157, 8%), or in a palliative situation (29/157, 18%) (Appendix S1). In total, 83 patients who received chemotherapy had localized RMS with the following subtype RMS: 24 of 25 (96%) for A‐RMS, 22 of 26 (84%) for E‐RMS, and 37 of 60 (62%) for P‐RMS (P = 0.002). The histological subtype influenced administration of chemotherapy and type of chemotherapy. Patients with A‐RMS, E‐RMS, and P‐RMS received CT for 53 of 55 (96%), 29 of 33 (88%), and 45 of 69 (62%) patients, respectively (P = 0.0001). Among 122 patients with a known administered regimen, PP was used for 27 of 51 (53%) with A‐RMS, 20 of 29 (69%) with E‐RMS, and five of 42 (12%) patients with P‐RMS (P = 0.0001). Patients older than 25 years received more surgery (P < 0.001), less chemotherapy (P < 0.001), and less combination of treatments (P < 0.003).
Response to treatment and patterns of failure in the retrospective cohort
Overall, complete remission (CR) after initial treatment was obtained for 93 of 153 (59%) of patients (Table 2).
Table 2.
Prognostic factor for complete response (CR) in the retrospective study
| Variable | No CR n = 60 (%) | CR n = 93 (%) | P |
|---|---|---|---|
| Disease spread (N+ or M+) | |||
| No | 29 (48) | 80 (86) | <10−4 |
| Yes | 31 (52) | 13 (14) | |
| Gender | |||
| Female | 24 (40) | 30 (32) | 0.387 |
| Male | 36 (60) | 63 (68) | |
| Subtype RMS | |||
| P‐RMS | 23 (38) | 45 (48) | 0.013 |
| A‐RMS | 29 (49) | 24 (26) | |
| E‐RMS | 8 (13) | 24 (26) | |
| Location | |||
| Limbs | 21 (35) | 53 (57) | 0.008 |
| Head and neck | 14 (23) | 24 (26) | |
| Genitourinary | 9 (15) | 5 (5) | |
| Other | 16 (27) | 11 (12) | |
| Radiotherapy | |||
| No | 24 (40) | 22 (24) | 0.046 |
| Yes | 36 (60) | 71 (76) | |
| Chemotherapy | |||
| No chemotherapy | 8 (13) | 21 (23) | 0.210 |
| Nonpediatric protocol | 31 (52) | 36 (39) | |
| Pediatric protocol | 18 (30) | 34 (36) | |
| NA | 3 (5) | 2 (2) | |
| Surgery | |||
| No surgery | 34 (57) | 13 (14) | <0.001 |
| R > 0 | 14 (23) | 23 (25) | |
| R = 0 | 7 (12) | 50 (54) | |
| R = NA | 5 (8) | 7 (7) | |
| Tumor size | |||
| <50 mm | 5 (8) | 26 (28) | 0.017 |
| ≥50 mm | 41 (68) | 59 (63) | |
| NA | 14 (23) | 8 (9) | |
| Natural size (mm) | 98.7 ± 82.7 | 81.5 ± 54.8 | 0.157 |
| Age | |||
| <25 | 19 (32) | 23 (25) | 0.360 |
| ≥25 | 41 (68) | 70 (75) | |
| Natural age (years) | 41.2 ± 20.7 | 41.2 ± 17.9 | 0.999 |
N, nodes; M, metastases; E‐RMS, embryonal rhabdomyosarcoma; A‐RMS, alveolar rhabdomyosarcoma; P‐RMS, “adult‐type” rhabdomyosarcoma; NA, missing data.
Complete remission was obtained in 80 of 111 (72%) patients with localized RMS and 13 of 46 (28%) patients with advanced RMS (P = 0.0001). Predictors for CR are given in Table 2. Evaluation at the end of treatment was CR was obtained at the end of treatment for 50 of 57 (88%) patients who achieved R0 after surgery versus 23 of 37 (62%) patients after R1/R2 resection versus 13 of 47 (27%) patients when surgery was not performed (P = 0.0001). With a median follow‐up of 8.5 years, median OS was 49 months (95% CI: 31–284) versus 9 months (95% CI: 9–16) for patients with, respectively, complete response after treatment or not (P < 0.0001).
Among the 111 of 157 (70%) patients with localized RMS at diagnosis, data for local relapse were available for 109 of 111 (98%) patients, and 37 of 109 (34%) patients relapsed. Metastases occurred for 45 of 111 (40%) patients. In total, 22 of 109 (20%) patients experienced both local relapse and metastases. Median time to relapse was 9.3 months since initial diagnosis (range: 0.4–199). When LN was present at diagnosis, metastases occurred in nine of 15 (60%) patients at a median time of 8.9 months since initial diagnosis (range: 2.7–13.9).
Survival in the retrospective cohort
With a median follow‐up of 8.5 years, 96 of 157 (61%) patients died of disease, including 54 of 111 (49%) with localized RMS and 42 of 46 (91%) with advanced RMS (Tables 3 and 4). Five‐year OS rates for patients with localized RMS and advanced RMS were 43% (median: 40 months, range = 1–337) and 5% (median: 13 months, range 1–297), respectively (P < 0.0001) (Fig. 1). Median OS was 16 (range: 2–227), 33 (range = 1–337), and 51 months (range = 1–297) for A‐RMS, P‐RMS, and E‐RMS, respectively. In patients with localized disease, median OS was 24, 42, and 66 months for A‐RMS, P‐RMS, and E‐RMS, respectively. In patients with metastatic disease at diagnosis, median OS was 9, 13, and 28 months for A‐RMS, P‐RMS, and E‐RMS, respectively. In a multivariate analysis, OS of localized RMS was significantly better in patients with non‐A‐RMS (P = 0.01), younger age (P = 0.004), R0 resection (P < 0.0001), receiving radiotherapy (P = 0.011), and pediatric chemotherapy protocols (P = 0.003) (Fig. 1). For advanced RMS, OS was positively influenced by non‐A‐RMS histological subtypes (P = 0.009), R0 resection (P = 0.04), and use of RT (P = 0.02). In a multivariate analysis, PF for OS in A‐RMS and E‐RMS included gender, location, age, administration of pediatric protocols, and R0 surgery. For P‐RMS, PF included radiotherapy, R0, and age. Predictors for OS are given in Table 3 and Table 4.
Table 3.
Prognostic factor for overall survival stratified by histological subtypes (retrospective study)
| Univariate analysis for all RMS | Stepwise multivariate analysis for all RMS | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Localized RMS | Advanced RMS | Localized RMS | Advanced RMS | ||||||||||
| HR | 95% CI HR | P | HR | 95% CI HR | P | HR | 95% CI HR | P | HR | 95% CI HR | P | ||
| Gender | Male vs. Female | 1.26 | 0.73–2.16 | 0.830 | 0.79 | 0.43–1.46 | 0.453 | 1.72 | 0.94–3.15 | 0.081 | – | – | – |
| Histotype | P‐RMS vs. E‐RMS | 1.06 | 0.56–2.03 | 0.850 | 2.65 | 0.88–7.97 | 0.083 | 0.83 | 0.37–1.88 | 0.653 | 9.57 | 2.24–40.82 | 0.002 |
| A‐RMS vs. E‐RMS | 1.58 | 0.77–3.27 | 0.215 | 3.43 | 1.23–9.55 | 0.018 | 2.83 | 1.25–6.40 | 0.012 | 5.90 | 1.55–22.50 | 0.009 | |
| Site | GU/Other vs. Limbs/HN | 0.77 | 0.40–1.47 | 0.422 | 1.61 | 0.88–2.96 | 0.121 | 2.41 | 1.16–4.97 | 0.018 | |||
| Radiotherapy | Yes vs. No | 0.77 | 0.45–1.32 | 0.342 | 0.67 | 0.36–1.22 | 0.190 | 0.42 | 0.22–0.82 | 0.011 | 0.42 | 0.18–0.98 | 0.021 |
| Chemotherapy | No PP vs. no chemotherapy | 1.23 | 0.67–2.25 | 0.511 | 1.07 | 0.25–4.62 | 0.923 | 0.76 | 0.38–1.54 | 0.455 | – | – | – |
| PP vs. no chemotherapy | 0.71 | 0.35–1.45 | 0.347 | 0.70 | 0.15–3.16 | 0.641 | 0.19 | 0.07–0.57 | 0.003 | – | – | – | |
| Surgery | >R0 vs. No Surgery | 0.59 | 0.28–1.24 | 0.162 | 0.41 | 0.28–1.24 | 0.029 | 0.10 | 0.04–0.30 | <0.0001 | 0.42 | 0.18–0.98 | 0.044 |
| R0 vs. No Surgery | 0.51 | 0.27–0.98 | 0.042 | 0.46 | 0.27–0.98 | 0.160 | 0.06 | 0.02–0.18 | <0.0001 | 0.25 | 0.07–0.94 | 0.041 | |
| Size | <50 vs. ≥50 | 0.44 | 0.22–0.89 | 0.021 | 0.26 | 0.06–1.09 | 0.066 | – | – | – | |||
| Natural age | |||||||||||||
| Age | <25 vs. ≥25 | 0.59 | 0.30–1.16 | 0.126 | 1.03 | 0.56–1.91 | 0.100 | 0.23 | 0.08–0.63 | 0.004 | |||
| Years | >2000 vs. ≤2000 | 0.95 | 0.57–1.60 | 0.850 | 1.46 | 0.78–2.75 | 0.234 | – | – | – | – | – | – |
| Univariate analysis A/E‐RMS | Univariate analysis P‐RMS | Stepwise multivariate analysis P‐RMS | Stepwise multivariate analysis A/E‐RMS | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HR | 95% CI HR | p | HR | 95% CI HR | P | HR | 95% CI HR | P | HR | 95% CI HR | P | ||
| Gender | Male vs. Female | 1.41 | 0.64–3.07 | 0.392 | 1.16 | 0.55–2.47 | 0.692 | 3.63 | 1.07–12.31 | 0.038 | |||
| Histotype | P‐RMS vs. E‐RMS | ||||||||||||
| A‐RMS vs. E‐RMS | |||||||||||||
| Site | GU/Other vs. Limbs/HN | 0.34 | 0.10–1.12 | 0.075 | 1.35 | 0.61–3.03 | 0.460 | 0.11 | 0.01–0.89 | 0.038 | |||
| Radiotherapy | Yes vs. No | 1.60 | 0.61–4.19 | 0.336 | 0.46 | 0.23–0.91 | 0.026 | 0.27 | 0.11–0.71 | 0.008 | – | – | – |
| Chemotherapy | No PP vs. no chemotherapy | 1.38 | 0.39–4.86 | 0.617 | 1.10 | 0.54–2.25 | 0.793 | 1.26 | 0.50–3.21 | 0.623 | – | – | – |
| PP vs. no chemotherapy | 0.65 | 0.18–2.26 | 0.493 | 0.31 | 0.04–2.41 | 0.265 | 0.39 | 0.05–3.34 | 0.393 | 0.19 | 0.06–0.66 | 0.009 | |
| Surgery | >R0 vs. No Surgery | 0.77 | 0.28–2.12 | 0.607 | 0.03 | 0.01–0.28 | 0.003 | 0.06 | 0.01–0.73 | 0.027 | 0.18 | 0.04–0.92 | 0.040 |
| R0 vs. No Surgery | 0.67 | 0.29–1.55 | 0.347 | 0.02 | 0.01–0.23 | 0.001 | 0.03 | 0.01–0.36 | 0.005 | 0.13 | 0.03–0.62 | 0.010 | |
| Size | <50 vs. ≥50 | 0.43 | 0.18–1.06 | 0.067 | 0.37 | 0.11–1.21 | 0.099 | – | – | – | – | – | – |
| Natural age | 1.04 | 1.01–1.06 | 0.004 | 1.04 | 1.01–1.07 | 0.014 | |||||||
| Age | <25 vs. ≥25 | 0.45 | 0.21–0.98 | 0.043 | 0.73 | 0.33–1.60 | 0.429 | 0.24 | 0.07–0.84 | 0.026 | |||
| Years | >2000 vs. ≤2000 | 1.13 | 0.55–2.33 | 0.731 | – | – | – | ||||||
E‐RMS, embryonal rhabdomyosarcoma; A‐RMS, alveolar rhabdomyosarcoma; P‐RMS, “adult‐type” rhabdomyosarcoma.
Table 4.
Prognostic factors for relapse in localized RMS from the retrospective series (n = 111)
| Variable | Univariate analysis | Multivariate analysis | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI HR | P | HR | 95% CI HR | P | |
| Gender | ||||||
| Male vs. Female | 1.17 | 0.65–2.09 | 0.582 | 2.02 | 0.97–4.19 | 0.059 |
| Histotype | ||||||
| P‐RMS vs. E‐RMS | 1.57 | 0.74–3.33 | 0.230 | 0.93 | 0.34–2.55 | 0.888 |
| A‐RMS vs. E‐RMS | 2.07 | 0.90–4.75 | 0.085 | 3.58 | 1.15–11.11 | 0.028 |
| Site | ||||||
| Genitourinary/Other vs. Limbs/Head and Neck | 0.50 | 0.23–1.11 | 0.088 | 0.89 | 0.35–2.26 | 0.809 |
| Radiotherapy | ||||||
| Yes vs. No | 0.69 | 0.38–1.25 | 0.220 | 0.31 | 0.13–0.74 | 0.008 |
| Chemotherapy | ||||||
| Nonpediatric protocol vs. no chemotherapy | 0.81 | 0.45–1.50 | 0.515 | 0.60 | 0.26–1.35 | 0.216 |
| Pediatric protocol vs. no chemotherapy | 0.28 | 0.12–0.64 | 0.003 | 0.11 | 0.03–0.38 | 0.001 |
| Surgery | ||||||
| >R0 vs. No Surgery | 1.28 | 0.52–3.18 | 0.596 | 0.69 | 0.22–2.15 | 0.520 |
| R = 0 vs. No Surgery | 1.05 | 0.45–2.42 | 0.913 | 0.26 | 0.08–0.81 | 0.021 |
| Size | ||||||
| <50 vs. ≥50 | 0.61 | 0.31–1.19 | 0.149 | 0.74 | 0.30–1.84 | 0.517 |
| Age at diagnosis | ||||||
| <25 vs. ≥25 | 0.53 | 0.25–1.13 | 0.101 | 0.96 | 0.31–2.97 | 0.939 |
| Year | ||||||
| >2000 vs. ≤2000 | 0.49 | 0.27–0.90 | 0.021 | 0.58 | 0.28–1.21 | 0.149 |
E‐RMS, embryonal rhabdomyosarcoma; A‐RMS, alveolar rhabdomyosarcoma; P‐RMS, “adult‐type” rhabdomyosarcoma.
Figure 1.
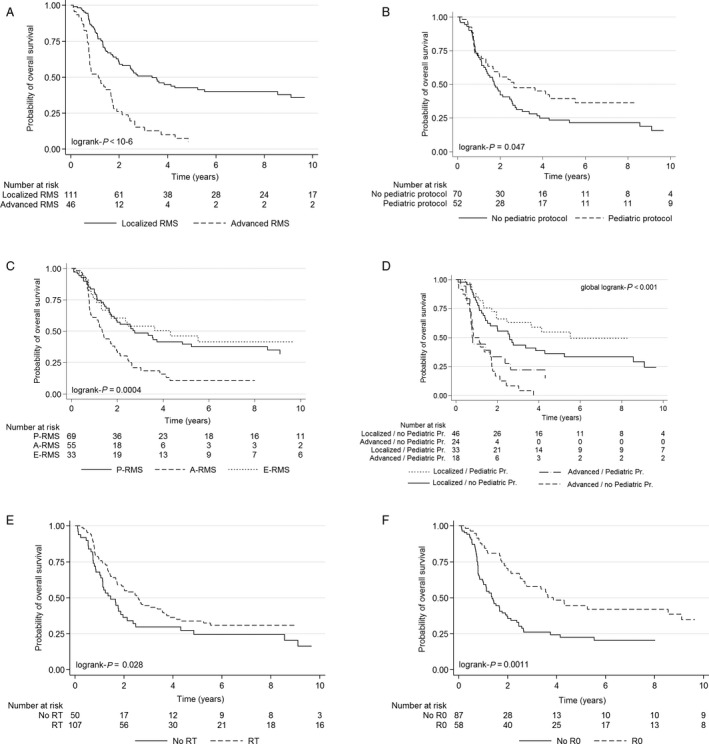
- (A) Overall survival for localized and advanced RMS; (B) overall survival and treatment: administration of pediatric protocol; (C) overall survival and histological subtype; (D) overall survival and treatment: administration of pediatric protocol according to stage disease; (E) overall survival and treatment: administration radiotherapy; (F) overall survival and treatment: R0 surgery (R0 versus no R0: R>0 + no surgery).
In a multivariate analysis, PFS of localized RMS was positively correlated with female gender, non‐A‐RMS histologies, use of RT, use of pediatric protocols, and R0 surgery (Table 4).
Characteristics and outcome in the prospective nationwide cohort
This is the nationwide series of patients with RMS treated in France between 2010 and 2014 and differs therefore from the retrospective series obtained from a selected panel of large centers. As shown in Table 1, an overall higher proportion of patients with P‐RMS was noted in this prospective series.
Median OS was 35 months and 37 months and not reached for A‐RMS, P‐RMS, and E‐RMS, respectively. Median PFS was 19, 11, and 27 months for A‐RMS, P‐RMS, and E‐RMS, respectively. The OS and PFS of E‐RMS were superior in the localized phase (nonsignificant) but was worse compared with patients treated in the 13 reference centers in the previous period for both OS and PFS, with a median PFS < 20 months and median OS < 40 months (Fig. 2). Although the outcome of patients treated in these centers was marginally better (not shown), this finding highlights the severe prognosis of adult RMS in unselected patient populations from both reference and nonreference centers.
Figure 2.
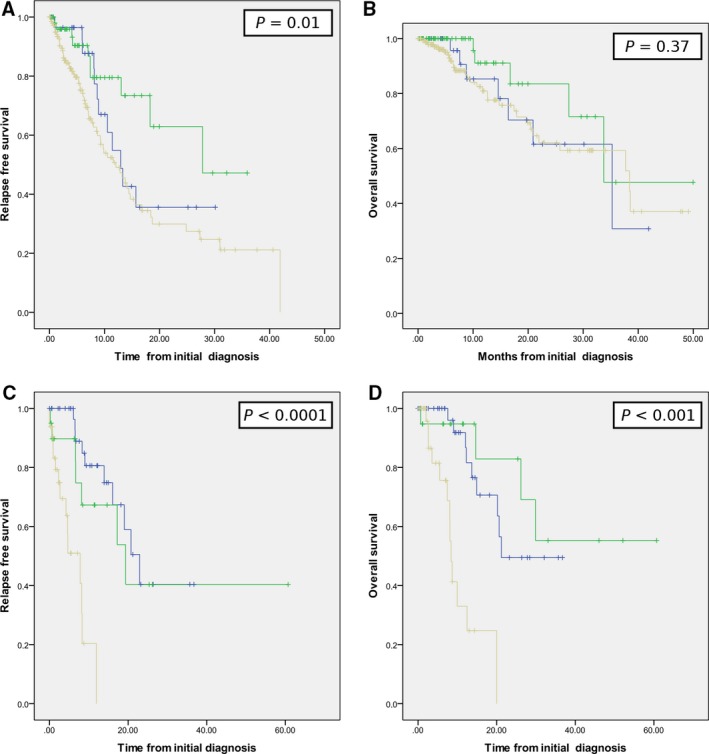
- (A) RFS of localized RMS. (B) Overall survival of localized RMS; (C) RFS of metastatic RMS. (D) Overall survival of metastatic RMS (green: E‐RMS; blue: A‐RMS; and yellow: P‐RMS).
Discussion
Rhabdomyosarcoma is a rare tumor in adult patients. In the prospective cohort, close to exhaustive, there were 292 incident cases of adult RMS in 5 years in this country of 65 million inhabitants for an estimated incidence of 0.9/106/year.
Adult RMS is a difficult‐to‐treat cancer because of its rarity and its heterogeneity. We usually attempt the treated adult A/E‐RMS according to the pediatric guidelines. The present study underlines that pleomorphic RMS is more likely treated as other adult soft tissue sarcomas, with more heterogeneity.
Nevertheless, to improve the standard of care, a better understanding of treatment as performed in routine setting is a logical starting point.
The FSG reports one of the largest recent retrospective studies performed in a multicenter setting at a national level for adult RMS. These results are consistent with those described in the literature. RMS occurs frequently in men and young adults: 37 versus 26 years in other studies and increased by twofold in P‐RMS 11, 12, 13, 14, 15, 16, 17. HN and extremities tumors represent the main primary site in this study and previous reports 11, 12, 16, 17. Synchronous metastases were observed in 20% versus 17–44% in previous studies 11, 13, 14, 15, 16, 17. LN involvement was uncommon (14%) compared with other series (33–46%) 11, 16, 17. This finding is likely related to the prevalence of P‐RMS: 44% versus 9–20% with the exception of the Little series focused on localized RMS 12 (Table 5). In this present work, the clinical presentation is variable according to the histological subtype and is generally worse than that of the pediatric population with RMS.
Table 5.
Present study and data
| Auteur | N | Age | Time period | Histological subtype (%) | M+ (%) | OS 5 (%) | OS5 (median) months | OS5 (%) | OS5 (%) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Embryonal | Alveolar | Pleomorphic NOS | Cohort | Cohort | Localized | M | |||||
| Lloyd,83 (MSKCC) | 54 | 40 (20–73) | 1950–1978 | 100 | 11 | 21 | 17 | ||||
| La Quaglia,94 (MSKCC) | 290 | 1970–1991 | 77 | 14 | 9 | 23 | 56 | ||||
| Hawkins,01 (MSKCC) | 84 | 23 (16–76) | 1982–1999 | 53 | 30 | 17 | 44 | 35 | 22 | ||
| Esnaola, 01 (Dana Farber) | 39 | 26 (16–82) | 1973–1996 | 18 | 56 | 26 | 33 | 31 | |||
| Little,02 (MDA) | 82 | 27 (17–84) | 1960–1998 | 34 | 23 | 43 | 0 | 44 | |||
| Ferrari,03 (Milan) | 171 | 27 (19–83) | 1975–2001 | 33 | 34 | 32 | 18 | 40 | 38 | 4 | |
| Gerber,13 (MDA) | 148 | 27 (19–83) | 1990–2011 | 54 | 33 | 14 | 36 | 34 | 45 | ||
| Dumont, 13 (MSKCC) | 239 | 10–102 (19) | 1957–2003 | 38 | 23 | 37 | 32 | 44 | 18 | ||
| Present study (retrospective series) | 157 | 37 (18–86) | 1980–2010 | 33 | 55 | 69 | 46 | 31 | 24 | 43 | 5 |
| Present study (prospective series) | 292 | 55 (18–99) | 2010–2015 | 49 | 54 | 189 | 74 | 40 | |||
The 5‐year OS is 30% for our population and confirms the poor prognosis of adult RMS (Table 5). For localized RMS, OS is similar to that reported by the MDA and MSKCC experience 14, 15. Importantly, the outcome of patients initially treated in the reference center in the retrospective cohort (Conticabase series) is superior to that of the nationwide and more recent series, possibly explaining the excellence of the reference center compared with other centers 18. The relapse rate is consistent with that reported in the literature: 33–57% local relapses 11, 12, 13, 14, 19 and up to 48–68% metastatic relapse 14, 16, 19. It is observed often during the first year, leading to death in 6–12 months 11, 12, 14, 15, 16, 17, 19, with median survival of 9, 8, and 7 months for patients with local relapse, metastatic relapse, or both in the present work. This outcome is notably inferior to that achieved in other ASTS of adults in 2016. The present report is one of the largest studies and confirms the poor prognosis of adult RMS treated in reference centers or in the general population of patients with RMS.
The univariate analysis and multivariate analysis of clinical and therapeutic PF performed only in the Conticabase series enable us to conclude that adults with RMS share the same PF in terms of disease characteristics compared with pediatric patients. For therapeutic parameters, surgery, radiotherapy, and chemotherapy are identified as PF for OS. But in our study, patients older than 25 years received significantly less chemotherapy and numerous treatments. Surgical results were consistent with those previously reported 11, 12, 13, 15, 16, 17, 18, 19. The absence of surgery and incomplete resection were deleterious for OS. Adjuvant RT and PP were also associated with a better outcome. This finding is consistent with the nationwide French NetSarc reference work that observed a twofold increased R2 surgery outside the NetSarc center and threefold more secondary resection after primary surgery performed without multidisciplinary tumor board in other sarcoma types 18. In the present study, local surgery had a favorable impact on survival even for patients with metastatic RMS which is consistent with the MSKCC and French pediatric experience. These observations could change this approach, and advanced disease surgery should be debated whenever possible 15, 20. Radiotherapy also had a favorable impact on OS for localized and metastatic disease addressing the issue of local treatment in a metastatic setting.
A univariate analysis and multivariate analysis were also performed for each histological subtype in localized RMS. For A‐RMS and E‐RMS, age, location, surgery R0, and PP are relevant. These factors are similar to those reported in the pediatric patients. The positive impact of PP for A‐RMS and E‐RMS on OS is also reported by MSKCC and MDA experience 14, 15 and Ferrari who described better survival with administration of chemotherapy per current guidelines for pediatric RMS 11. This finding suggests that PP should be considered for adult patients with A‐RMS and E‐RMS. Conversely, for nonmetastatic P‐RMS, chemotherapy was not associated with an improved OS in our study. The data presented do not support the use of pediatric regimen in P‐RMS but clearly cannot allow to draw a definitive conclusion on this question, which could be obtained only from a prospective study. It is still important to highlight that P‐RMS are not as responsive (in terms of CR/PR rates) as A/E‐RMS to CT. The best combination of CT for P‐RMS is not defined. There are now a large number of adult series all showing results achieved with doxorubicin‐containing regimens, and because this is the largest evidence in terms of patient numbers, it is reasonable to infer that doxorubicin remains the standard of care. The situation is different for A/E adult RMS, while their management is similar to the pediatric populations for most patients, the level of evidence for adult patient population is not so clear (as for pediatric population), in the absence again of any randomized trial. Considering the toxicity observed in this retrospective series, firm recommendations cannot be proposed; if considered feasible by the adult physician, pediatric regimens and strategies are certainly a relevant option, in particular for young adults, but may be not applicable to older patients, where adult type of regimens (AI based or A can be safely applied). Prospective and homogeneous management need to be implemented and analyzed to identify how adult can be treated with A/E‐RMS if pediatric protocol is not feasible (specifically after 30 years old).
However, the recent ISG/Eurosarc suggested an improved survival achieved with neoadjuvant CT in high‐risk patients 21, 22, 23, 24, 25, 26, 27. Chemotherapy is still a matter of debate and must be discussed in a dedicated multidisciplinary committee with an individual approach basis 28. For all adult sarcoma in a localized phase, R0 surgery remains the cornerstone of treatment 29, 30, 31, 32, 33, 34, 35. The present study underlines the heterogeneity of adult RMS management. This heterogeneity is a study limitation, but at the end is helpful to identify factors favorably influence the outcome. Another study limitation is that we are not able to analyze the prognostic value of the presence (or absence) of specific translocation; nevertheless, the vast majority of adult RMS are undifferentiated ones without specific translocation. We aim to use the prospective cohort to validate the prognostic factors identified in the retrospective series; nevertheless, this requires more mature data and longer follow‐up.
Why are adult patients with A‐RMS and E‐RMS at higher risk of relapse than children affected with the same disease? Since 1972, within IRS group trials, 5‐year OS of children has improved from 55%, 63%, and 69% to 73% in an IRS IV study, representing a threefold increase over these 30 years 6, 7, 8, 9, 36. This improvement did not translate to adult patients, and several hypotheses are suggested involving age, clinical presentation, and intensity/duration of treatment. Ferrari et al., and more recently, the MDA study, reported that only the oldest patients treated with a non‐PP had less favorable survival 11, 14. Clinical presentation is also less favorable. All retrospective studies note more aggressive disease with more unfavorable sites 1, 36, 37, 38, 39, more LN, more metastasis 9, 36, 40, 41, and more A‐RMS. Nevertheless, all parameters being equal, the outcome of adult patients with RMS is less favorable. Beyond the obvious difference in therapeutic approaches and management, the genomics of A‐RMS and E‐RMS should be compared between adult and children, similar to that performed for synovial sarcoma 42. Sharing the same classification would allow easier comparison between clinical presentation and behavior in adults and children. Finally, specific chemotherapy for A‐RMS and E‐RMS is an independent prognostic value in our study and others 2, 6, 7, 8, 9, 11, 14, 15, 38, 39, 40, 41, 43, 44, 45, 46, 47. North American and European pediatric groups have identified several risk factors (favorable location, IRSG stage, IRSG group, and age) to define the clinical group and deliver a regimen adapted to the risk group. Thus, reflection should be engaged for common protocols, and PP should be adjusted for risk groups using vincristine, dactinomycin, and alkylating agents 6, 7, 8, 9, 10, 48, 49, 50, 51, 52, 53, as anthracycline is always controversial in PP. For patients up to 50 years old, toxicity in PP is unacceptable with age, as demonstrated through OS or Ewing protocols.
Conclusion
This is the largest study analyzing all factors in univariate analysis and multivariate analysis for localized and metastatic disease and for each histological subtype. Specific management for A‐RMS and E‐RMS using a pediatric protocol chemotherapy and carcinologic surgery is the cornerstone to improving survival. The FSG experience emphasizes the urgent need to build a worldwide clinical trial using these rare entities that exhibit a dismal prognosis.
Conflict of Interest
None declared.
Supporting information
Appendix S1. Regimens of chemotherapy.
Acknowledgments
Supported by Association DAM's, Ensemble contre Le GIST, Eurosarc (FP7‐278742), la Fondation ARC, Infosarcome, InterSARC (INCA), LabEx DEvweCAN (ANR‐10‐LABX‐0061), Ligue de L'Ain contre le Cancer, LYric (DGOS‐INCa‐4664), NetSARC (INCA) and RREPS (INCA).
Cancer Medicine 2018; 7(8): 4023–4035
References
- 1. Breitfeld, P. P. , and Meyer W. H.. 2005. Rhabdomyosarcoma: new windows of opportunity. Oncologist 10:518–527. [DOI] [PubMed] [Google Scholar]
- 2. Dantonello, T. M. , Int‐Veen C., Winkler P., Leuschner I., Schuck A., Schmidt B. F., et al. 2008. Initial patient characteristics can predict pattern and risk of relapse in localized rhabdomyosarcoma. J. Clin. Oncol. 26:406–413. [DOI] [PubMed] [Google Scholar]
- 3. Raney, R. B. , Maurer H. M., Anderson J. R., Andrassy R. J., Donaldson S. S., Qualman S. J., et al. 2001. The Intergroup Rhabdomyosarcoma Study Group (IRSG): major lessons from the IRS‐I through IRS‐IV studies as background for the current IRS‐V treatment protocols. Sarcoma 5:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ducimetiere, F. , Lurkin A., Ranchere‐Vince D., Decouvelaere A. V., Peoc'h M., Istier L., et al. 2011. Incidence of sarcoma histotypes and molecular subtypes in a prospective. PLoS One 6:e20294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Walterhouse, D. , and Watson A.. 2007. Optimal management strategies for rhabdomyosarcoma in children. Paediatr. Drugs 9:391–400. [DOI] [PubMed] [Google Scholar]
- 6. Maurer, H. M. , Beltangady M., Gehan E. A., Crist W., Hammond D., Hays D. M., et al. 1988. The intergroup rhabdomyosarcoma study‐I. A final report. Cancer 61:209–220. [DOI] [PubMed] [Google Scholar]
- 7. Maurer, H. M. , Gehan E. A., Beltangady M., Crist W., Dickman P. S., Donaldson S. S., et al. 1993. The intergroup rhabdomyosarcoma study‐II. Cancer 71:1904–1922. [DOI] [PubMed] [Google Scholar]
- 8. Crist, W. , Gehan E. A., Ragab A. H., Dickman P. S., Donaldson S. S., Fryer C., et al. 1995. The third intergroup rhabdomyosarcoma study. J. Clin. Oncol. 13:610–630. [DOI] [PubMed] [Google Scholar]
- 9. Crist, W. M. , Anderson J. R., Meza J. L., Fryer C., Raney R. B., Ruymann F. B., et al. 2001. Intergroup rhabdomyosarcoma study‐IV: results for patients with nonmetastatic disease. J. Clin. Oncol. 19:3091–3102. [DOI] [PubMed] [Google Scholar]
- 10. Stevens, M. C. , Rey A., Bouvet N., Ellershaw C., Flamant F., Habrand J. L., et al. 2005. Treatment of nonmetastatic rhabdomyosarcoma in childhood and adolescence: third study of the International Society of Paediatric Oncology–SIOP Malignant Mesenchymal Tumor 89. J. Clin. Oncol. 23:2618–2628. [DOI] [PubMed] [Google Scholar]
- 11. Ferrari, A. , Dileo P., Casanova M., Bertulli R., Meazza C., Gandola L., et al. 2003. Rhabdomyosarcoma in adults. A retrospective analysis of 171 patients treated at a single institution. Cancer 98:571–580. [DOI] [PubMed] [Google Scholar]
- 12. Little, D. J. , Ballo M. T., Zagars G. K., Pisters P. W., Patel S. R., El‐Naggar A. K., et al. 2002. Adult rhabdomyosarcoma: outcome following multimodality treatment. Cancer 95:377–388. [DOI] [PubMed] [Google Scholar]
- 13. Hawkins, W. G. , Hoos A., Antonescu C. R., Urist M. J., Leung D. H., Gold J. S., et al. 2001. Clinicopathologic analysis of patients with adult rhabdomyosarcoma. Cancer 91:794–803. [PubMed] [Google Scholar]
- 14. Gerber, N. K. , Wexler L. H., Singer S., Alektiar K. M., Keohan M. L., Shi W., et al. 2013. Adult rhabdomyosarcoma survival improved with treatment on multimodality. Int. J. Radiat. Oncol. Biol. Phys. 86:58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dumont, S. N. , Araujo D. M., Munsell M. F., Salganick J. A., Dumont A. G., Raymond K. A., et al. 2013. Management and outcome of 239 adolescent and adult rhabdomyosarcoma patients. Cancer Med. 2:553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Esnaola, N. F. , Rubin B. P., Baldini E. H., Vasudevan N., Demetri G. D., Fletcher C. D., et al. 2001. Response to chemotherapy and predictors of survival in adult rhabdomyosarcoma. Ann. Surg. 234:215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. La Quaglia, M. P. , Heller G., Ghavimi F., Casper E. S., Vlamis V., Hajdu S., et al. 1994. The effect of age at diagnosis on outcome in rhabdomyosarcoma. Cancer 73:109–117. [DOI] [PubMed] [Google Scholar]
- 18. Blay, J. Y. , Soibinet P., Penel N., Bompas E., Duffaud F., Stoeckle E., et al. 2017. Improved survival using specialized multidisciplinary board in sarcoma patients. Ann. Oncol. 28:2852–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lloyd, R. V. , Hajdu S. I., and Knapper W. H.. 1983. Embryonal rhabdomyosarcoma in adults. Cancer 51:557–565. [DOI] [PubMed] [Google Scholar]
- 20. Ben Arush, M. , Minard‐Colin V., Mosseri V., Defachelles A. S., Bergeron C., Algret N., et al. 2015. Does aggressive local treatment have an impact on survival in children with metastatic rhabdomyosarcoma? Eur. J. Cancer 51:193–201. [DOI] [PubMed] [Google Scholar]
- 21. Gronchi, A. LBA6_PR – Full‐dose neoadjuvant anthracycline + ifosfamide chemotherapy is associated with a relapse free survival (RFS) and overall survival (OS) benefit in localized high‐risk adult soft tissue sarcomas (STS) of the extremities and trunk wall: Interim analysis of a prospective randomized trial. ESMO 2016.
- 22. Italiano, A. , Delva F., Mathoulin‐Pelissier S., Le Cesne A., Bonvalot S., Terrier P., et al. 2010. Effect of adjuvant chemotherapy on survival in FNCLCC grade 3 soft tissue sarcomas: a multivariate analysis of the French Sarcoma Group Database. Ann. Oncol. 21:2436–2441. [DOI] [PubMed] [Google Scholar]
- 23.[No authors listed]. 1997. Adjuvant chemotherapy for localised resectable soft‐tissue sarcoma of adults: meta‐analysis of individual data. Sarcoma Meta‐analysis Collaboration. Lancet 350:1647–1654. [PubMed] [Google Scholar]
- 24. Pervaiz, N. , Colterjohn N., Farrokhyar F., Tozer R., Figueredo A., and Ghert M.. 2008. A systematic meta‐analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft‐tissue sarcoma. Cancer 113:573–581. [DOI] [PubMed] [Google Scholar]
- 25. Woll, P. J. , Reichardt P., Le Cesne A., Bonvalot S., Azzarelli A., Hoekstra H. J., et al. 2012. Adjuvant chemotherapy with doxorubicin, ifosfamide, and lenograstim for resected soft‐tissue sarcoma (EORTC 62931): a multicentre randomised controlled trial. Lancet Oncol 13:1045–1054. [DOI] [PubMed] [Google Scholar]
- 26. Casali, P. G. , and Blay J. Y.. 2010. ESMO/CONTICANET/EUROBONET Consensus Panel of experts. Soft tissue sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann. Oncol. 21(Suppl. 5):v198–v203. [DOI] [PubMed] [Google Scholar]
- 27. Blay, J. Y. , and Le Cesne A.. 2009. Adjuvant chemotherapy in localized soft tissue sarcomas: still not proven. Oncologist 14:1013–1020. [DOI] [PubMed] [Google Scholar]
- 28. ESMO/European Sarcoma Network Working Group . 2014. Soft tissue and visceral sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann. Oncol. 25 (Suppl 3):iii102–iii112. [DOI] [PubMed] [Google Scholar]
- 29. Rodeberg, D. A. , Paidas C. N., Lobe T. L., Brown K., Andrassy R. J., Crist W. M., et al. 2002. Surgical principles for children/adolescents with newly diagnosed rhabdomyosarcoma: a report from the soft tissue sarcoma committee of the children's oncology group. Sarcoma 6:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Andrassy, R. J. , Corpron C. A., Hays D., Raney R. B., Wiener E. S., Lawrence W., et al. 1996. Extremity sarcomas: an analysis of prognostic factors from the Intergroup Rhabdomyosarcoma Study III. J. Pediatr. Surg. 31:191–196. [DOI] [PubMed] [Google Scholar]
- 31. Bonvalot, S. , Raut C. P., Pollock R. E., Rutkowski P., Strauss D. C., Hayes A. J., et al. 2012. Technical considerations in surgery for retroperitoneal sarcomas: position paper from E‐Surge, a master class in sarcoma surgery, and EORTC‐STBSG. Ann. Surg. Oncol. 19:2981–2991. [DOI] [PubMed] [Google Scholar]
- 32. Rossi, C. R. , Vecchiato A., Mastrangelo G., Montesco M. C., Russano F., Mocellin S., et al. 2013. Adherence to treatment guidelines for primary sarcomas affects patient survival: a side study of the European CONnective TIssue CAncer NETwork (CONTICANET). Ann. Oncol. 24:1685–1691. [DOI] [PubMed] [Google Scholar]
- 33. Stoeckle, E. , Gardet H., Coindre J. M., Kantor G., Bonichon F., Milbéo Y., et al. 2006. Prospective evaluation of quality of surgery in soft tissue sarcoma. Eur. J. Surg. Oncol. 32:1242–1248. [DOI] [PubMed] [Google Scholar]
- 34. Derbel, O. , Cropet C., Meeus C., Gilly F.‐N., Vaz G., Thiesse P., et al. 2012. Adhesion to clinical practices guidelines (CPGs) and role on survival for soft tissue sarcoma patients. Analysis of a population based cohort from Rhone‐Alpes region.
- 35. Ray‐Coquard, I. , Thiesse P., Ranchère‐Vince D., Chauvin F., Bobin J. Y., Sunyach M. P., et al. 2004. Conformity to clinical practice guidelines, multidisciplinary management and outcome of treatment for soft tissue sarcomas. Ann Oncol 15:307–315. [DOI] [PubMed] [Google Scholar]
- 36. Arndt, C. A. , and Crist W. M.. 1999. Common musculoskeletal tumors of childhood and adolescence. N. Engl. J. Med. 341:342–352. [DOI] [PubMed] [Google Scholar]
- 37. Raney, B. , Anderson J., Breneman J., Donaldson S. S., Huh W., Maurer H., et al. 2008. Results in patients with cranial parameningeal sarcoma and metastases (Stage 4) treated on intergroup rhabdomyosarcoma study group (IRSG) protocols II‐IV, 1978–1997: report from the children's oncology group. Pediatr. Blood Cancer 51:17–22. [DOI] [PubMed] [Google Scholar]
- 38. Neville, H. L. , Andrassy R. J., Lobe T. E., Bagwell C. E., Anderson J. R., Womer R. B., et al. 2000. Preoperative staging, prognostic factors, and outcome for extremity rhabdomyosarcoma: a preliminary report from the Intergroup Rhabdomyosarcoma Study IV (1991–1997). J. Pediatr. Surg. 35:317–321. [DOI] [PubMed] [Google Scholar]
- 39. Rodeberg, D. A. , Garcia‐Henriquez N., Lyden E. R., Davicioni E., Parham D. M., Skapek S. X., et al. 2011. Prognostic significance and tumor biology of regional lymph node disease in patients with rhabdomyosarcoma: a report from the Children's Oncology Group. J. Clin. Oncol. 29:1304–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oberlin, O. , Rey A., Lyden E., Bisogno G., Stevens M. C. G., Meyer W. H., et al. 2008. Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European cooperative groups. J. Clin. Oncol. 26:2384–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Breneman, J. C. , Lyden E., Pappo A. S., Link M. P., Anderson J. R., Parham D. M., et al. 2003. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma–a report from the Intergroup Rhabdomyosarcoma Study IV. J. Clin. Oncol. 21:78–84. [DOI] [PubMed] [Google Scholar]
- 42. Lagarde, P. , Przybyl J., Brulard C., Pérot G., Pierron G., Delattre O., et al. 2013. Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J. Clin. Oncol. 31:608–615. [DOI] [PubMed] [Google Scholar]
- 43. Newton, W. A. , Soule E. H., Hamoudi A. B., Reiman H. M., Shimada H., Beltangady M., et al. 1988. Histopathology of childhood sarcomas, Intergroup Rhabdomyosarcoma Studies I and II: clinicopathologic correlation. J. Clin. Oncol. 6:67–75. [DOI] [PubMed] [Google Scholar]
- 44. Newton, W. A. , Gehan E. A., Webber B. L., Marsden H. B., van Unnik A. J., Hamoudi A. B., et al. 1995. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification–an Intergroup Rhabdomyosarcoma Study. Cancer 76:1073–1085. [DOI] [PubMed] [Google Scholar]
- 45. Meza, J. L. , Anderson J., Pappo A. S., and Meyer W. H.. 2006. Children's Oncology Group. Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: the Children's Oncology Group. J. Clin. Oncol. 24:3844–3851. [DOI] [PubMed] [Google Scholar]
- 46. Dantonello, T. M. , Int‐Veen C., Harms D., Leuschner I., Schmidt B. F., Herbst M., et al. 2009. Cooperative trial CWS‐91 for localized soft tissue sarcoma in children, adolescents, and young adults. J. Clin. Oncol. 27:1446–1455. [DOI] [PubMed] [Google Scholar]
- 47. Joshi, D. , Anderson J. R., Paidas C., Breneman J., Parham D. M., and Crist W., Soft Tissue Sarcoma Committee of the Children's Oncology Group . 2004. Age is an independent prognostic factor in rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. Pediatr. Blood Cancer 42:64–73. [DOI] [PubMed] [Google Scholar]
- 48. Malempati, S. , and Hawkins D. S.. 2012. Rhabdomyosarcoma: review of the Children's Oncology Group (COG) Soft‐Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr. Blood Cancer 59:5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Arndt, C. A. S. 2013. Risk stratification of rhabdomyosarcoma. Am Soc Clin Oncol Educ Book 2013:415–419. [DOI] [PubMed] [Google Scholar]
- 50. Raney, R. B. , Walterhouse D. O., Meza J. L., Andrassy R. J., Breneman J. C., Crist W. M., et al. 2011. Results of the Intergroup Rhabdomyosarcoma Study Group D9602 protocol, using vincristine and dactinomycin with or without cyclophosphamide and radiation therapy, for newly diagnosed patients with low‐risk embryonal rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. J. Clin. Oncol. 29:1312–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Oberlin, O. , Rey A., Sanchez de Toledo J., Martelli H., Jenney M. E. M., Scopinaro M., et al. 2012. Randomized comparison of intensified six‐drug versus standard three‐drug chemotherapy for high‐risk nonmetastatic rhabdomyosarcoma and other chemotherapy‐sensitive childhood soft tissue sarcomas: long‐term results from the International Society of Pediatric Oncology MMT95 study. J. Clin. Oncol. 30:2457–2465. [DOI] [PubMed] [Google Scholar]
- 52. Bergeron, C. , Thiesse P., Rey A., Orbach D., Boutard P., Thomas C., et al. 2008. Revisiting the role of doxorubicin in the treatment of rhabdomyosarcoma: an up‐front window study in newly diagnosed children with high‐risk metastatic disease. Eur. J. Cancer 44:427–431. [DOI] [PubMed] [Google Scholar]
- 53. van Dalen, E. C. , Raphaël M. F., Caron H. N., and Kremer L. C.. 2009. Treatment including anthracyclines versus treatment not including anthracyclines for childhood cancer. Cochrane Database Syst. Rev. CD006647. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Regimens of chemotherapy.