

Abstract
Pancreatic cancer has been a life-threatening illness associated with high incidence and mortality rates. Paclitaxel that causes mitotic arrest in cancer cells disrupting microtubule function is used for pancreatic cancer treatment[1]. Nausea, anorexia and abdominal pain are some of the typical dose-limiting toxicity associated gastrointestinal side effects of the drug[2]. Here we present the use of polymeric mixed micelles to enable a targeted delivery of Paclitaxel and to provide additional advantages such as enhanced drug solubility, bioavailability and minimal dose-limiting toxicity. Also, these micelles self-assemble with pancreatic cancer cells-specific phage proteins P38, L1 and with the hydrophobic drug Paclitaxel resolving the issue of complex chemistry efforts normally needed for any conjugation. Our cytotoxicity and binding experiment results in vitro in 2D and 3D models suggested that the phage protein-targeted drug-loaded micelles bind and exhibit higher cell killing over the non-targeted ones.
INTRODUCTION
Pancreatic cancer showed an increasing incidence during the past 15 years with more than 40,000 cases diagnosed each year[3]. Pancreatic cancer is associated with high death rate with 5-year survival rate of 5 %. The challenge for the treatment of pancreatic cancer is the lack of effective treatment options. Surgery is known to cure pancreatic cancer, but less than 20% of cases are operable. Pancreatic tumors are also resistant to chemotherapy. In addition, conventional chemotherapies in cancer therapy have major problems with insufficient therapeutic outcomes and dose-limiting toxicity[4]. Paclitaxel (PCT) is an active treatment for a number of cancers including pancreatic cancer, breast cancer, lung cancer, and ovarian cancer. Although PCT has high potency, its poor solubility causes limited bioavailability and it demonstrates dose-limiting toxicities in clinical uses[5]. So, the development of a suitable delivery system to enhance its solubility and in vivo efficacy has been pursued.
Polymeric micelles offer an efficient system for the tumor-targeted delivery for the hydrophobic therapeutic agents. Loading such therapeutic agents into the hydrophobic core of micelle can dramatically increase drug solubility, half-life, and bioavailability[6].
Earlier, we have identified various tumor-homing peptides from phage-displayed peptide libraries. Recently, we have screened a landscape phage protein bearing a panc-1-specific peptide (P38 and L1) from an 8-mer landscape library (f8/8) via a biopanning protocol against panc-1 cells[7]. The well-known amphiphilic nature of phage fusion coat protein allows its stable incorporation into the micelles. The hydrophobic transmembrane domain of the protein is inserted into the micellar membrane and the hydrophilic terminal peptide sequences are well exposed to water and provide targeting specificity[8, 9] for targeting of micelles loaded with poorly soluble therapeutic agents to tumor cells. The incorporation of the protein into micelles proceed spontaneously and does not require any chemistry to be involved. The protein recognition sequences (surface-exposed peptide fragments) have been demonstrated to be non-immunogenic in nature in earlier studies[9, 10, 11].
In this study, we produced mixed micelles assembled with polyethylene glycol-phosphati-dylethanolamine(PEG-PE) conjugate and panc-1 specific phage protein (P38 and L1) loaded with water-insoluble anticancer drug, PCT, to target panc-1 cells. Unlike traditional immunomicelles that require high cost monoclonal antibodies (mAbs) and chemistry efforts to conjugate mAbs to polymeric micelle, this method is much more straightforward, based on a simple self-assembly process, and does not involve any conjugation chemistry. Binding and cytotoxicity experiments with panc-1 cells in 2D and 3D models revealed that the PCT-loaded targeted phage-micelles bind better with target panc-1 cells and have a significantly improved cytotoxicity compared to non-targeted micelles.
Materials
l,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethyleneglycol)2000] (ammonium salt; PEG2000-PE) and l,2-dimyristoyl-snglycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt, Rho-PE) were purchased from Avanti Polar Lipids Inc. (Alabaster, AL). Paclitaxel (PCT) was from Cedarburg Pharmaceuticals (Beverly, MA). Dimethyl sulfoxide (DMSO), acetonitrile (HPLC grade), and methanol (HPLC grade) were purchased from Fisher Scientific (Pittsburgh, PA). Sodium cholate was from Sigma (St. Louis, MO). Pyrene was purchased from Aldrich Chemical Co., Inc. (Milwaukee, WI). CellTiter-Blue assay kit was from Promega (Madison, WI). Fluor Mounting Medium was from Trevigen Inc. (Gaithersburg, MD). MCF-7 human breast adenocarcinoma (HTB 22) cells, C166-GFP (CRL-2583) and C166 (CRL-2581) mouse yolk sac endothelial cells, as well as NIH3T3 (CRL-1658) mouse fibroblasts were obtained from the ATCC (Manassas, VA). All cells were grown at 37 °C, 5% C02 as recommended by the ATCC. Phage protein, phage selection and phage protein purification have been performed as described by us earlier[7].
Isolation of phage fusion protein with panc-1 specificity
Isolation of major coat protein ETPPSWGG (P38) and EPSQSWSM (L1) were carried out according to the described protocol[7]. Briefly, p38 or L1 phage were destroyed by incubation in 80Mm cholate in the presence of chloroform and rotated in a 37 °C incubator overnight. Phage major coat protein was purified by size-exclusion chromatography through a Sepharose 6 resin with 10 Mm cholate/10Mm Tris-HCL/0.2 Mm EDTA, pH 8.0 running buffer. Flow rate was set at 0.5ml/min and the fractions were collected every 5 min. The structure class of protein was predicted according to Alpha Deleage &Roux Modification of Nishikawa & Ooi 1987.
Preparation of phage-PEG-PE micelles
For studies of cancer cell viability, PCT in chloroform was added into 5mM of PEG2000-PE in chloroform at 1.5:100 drug-to-lipid weight ratios, and evaporate to remove organic solvent to form the lipid film, which was hydrated with panc-1 specific phage protein P38 or L1 dissolved in 10mM of sodium cholate at protein to polymer ratio 1:200 by weight, followed by vortexing and overnight dialysis against PBS (pH 7.4) to remove sodium cholate. Non-incorporated PCT was excluded by filtration of the micellar suspension through a polycarbonate membrane with molecular weight cut-off (0.2 μΜ). As controls, PCT-loading plain PEG-PE micelles were prepared using the similar procedure.
For FACS and fluorescence microscopy study, micelles were prepared with additional traces of rhodamine-PE added in chloroform into the formulations using the similar procedure.
Characterization of phage-PEG-PE micelles
Size and size distribution
Size and size distribution of the micellar preparations was measured by the dynamic light scattering. Briefly, samples were diluted in PBS, pH 7.4 and measured using a Beckman Coulter N4 Plus Particle analyzer (Beckman Coulter, Fullerton, CA) with a scattering angle of 90° and particle size range measurements of 1–1000 nm. The measurements were run three times.
Zeta potential
Zeta-potential of the micellar preparations was analyzed with a ZetaPLUS apparatus (Brookhaven, Holtsville, NY) in triplicate.
Critical Micelle Concentration (CMC)
Critical Micelle Concentration of the micelle preparations was determined using pyrene as a hydrophobic probe with fluorescent properties[12]. Briefly, 1 mL of pyrene dissolved in chloroform at a concentration of 8 ×10−5 M was added to each test tube, followed by the removal of chloroform under vacuum. Subsequently, the crystalline pyrene was incubated with varying concentrations (ranging from 1×10−2 M to 5×10−9 M) of plain micelles or phage micelles overnight on a Lab Line Environ shaker (Lab Recyclers Inc., USA) at room temperature. The insoluble pyrene was removed by filtration through the 0.2 μΜ polycarbonate membrane, followed by the determination of the pyrene amount solubilized in the micelles using an F-2000 fluorescence spectroscopy (Hitachi, Japan) with the excitation wavelength (λex) of 373nm and an emission wavelength (λem) of 394 nm. The intensity ratio (I394/I373) was calculated and plotted against the logarithm of the polymer concentration. The CMC value corresponds to the concentration of micelles at which there is a sharp increase in the fluorescence of the solution (as the crossover point of the two tangents of the curves) due to the formation of micelles and partial pyrene solubilization.
Drug loading
Drug loading into the micelle preparations was determined by the ratio of drug content to polymer (PEG-PE) content within micellar formulations. The amount of PCT in the micelle formulation was detected using a reversed phase D-7000 HPLC system equipped with a diode array and Spherisorb ODS2 column (Hitachi, Japan). The mobile phase consisted of water and acetonitrile with volume ratio 60:40 (v/v). The elution was performed at a rate of 1.0 mL/ min. PCT was detected from injected sample (50 μL) at 227 nm. The PEG-PE content of the micellar formulations was detected using a colorimetric assay [13]
Phage protein content
Final protein content in the formulation was detected using the micro BCA ™ protein assay kit (Pierce, Rockford, IL).
Stability
Storage stability of the micelles was assessed by the examination of the changes of particle sizes of micellar formulations under accelerated conditions (such as 37 °C with shaking) and a normal storage condition (4°C).
Serum stability
Stability of the micelles in serum was assessed by examination of the serum-protein adsorption/interaction with the nanoparticles. Briefly, micelles were incubated with the normal FBS at a ratio of 1 to 1.5 (v/v) at 37 °C. The particle size was analyzed by DLS on a Coulter® N4-Plus submicron particle sizer at predetermined time points (0, 1,4,8,24,48 and 72h). Control groups included serum alone and micelles in PBS.
In vitro drug release
The in vitro drug release from the different micellar formulations was detected using dialysis method. Briefly, 0.3 ml of the different micellar preparations including non-modified plain micelles, p38 phage micelles and L1 phage micelles was dialyzed at 37 °C using membrane with molecular weight cut off of 2000 Da [12]against 40 mL of 1 M sodium salicylate solution, which set up the sink condition and mimicked an in vivo condition. The media outside of dialyzed bag were sampled at predetermined time points, including 0, 1, 2, 3, 4, 5, 6, 8, 24, 48 and 72h. The PCT in the outside media was determined by RP-HPLC according to a method described above.
FACS evaluation of cell association with the micelles
Target panc-1 cells and non-target MCF-7 breast cancer cells and non-cancer cells C166 or NIH3T3 were seeded and grown in completed DMEM supplemented with 10% FBS and 1% antibiotics until 70–80% confluence. Cells were incubated with 75 μM rhodamine-labeled plain micelles or phage-micelles for 2 h. After removing drug-containing medium followed by 3 times cold PBS washing (pH 7.4), cells were detached and collected by centrifugation. The cell pellets were re-suspended in 200 μL of sheath fluid, and analyzed by flow cytometry. A right shift on the X-axis of the histogram plot indicated the cellular association of the rhodamine-labeled micelles.
Fluorescence microscopy evaluation of cell association with the micelles
Panc-1 cells were grown on 6-well plates with a density of 2×105 cells/well for 24h at 37°C, and then incubated with 75 μM rhodamine-labeled plain micelles or phage-micelles in DMEM with 10% serum for 30min at 37°C. Followed by washing 3 times with PBS, the coverslip was placed onto a glass slide over the fluorescence-mounting medium. The images were acquired by a fluorescence microscope (Nikon, Japan) 40X magnification with FITC or TRITC filter.
Apoptosis assay
Apoptosis of the target panc-1 cells was determined by the examination of induction of caspase 3/7 in the response of different micellar formulations. Target panc-1 cells were seeded into 96 well microplates at a density of 4 × 103 cells/well, respectively. After growth to 50–60% confluence, cells were treated with 750ng/ml of free PCT in DMSO, PCT-loaded plain micelles, PCT-loaded micelles modified with panc-1-targeted phage protein p38 or L1 (PCT-loaded p38-targeted phage-micelles). After 24h treatment, caspase 3/7 activities were determined according to protocol of Apo-one homogeneous caspase 3/7assay.
Tumor cell viability as tested in the monolayer cell culture
Target panc-1 cells were seeded into 96 well microplates at a density of 5 × 103 cells/well, respectively. After growth to 50–60% confluence, cells were treated in completed DMEM for 72 h with varying concentration of PCT-loaded plain micelles, PCT-loaded micelles modified with pac-1-targeted phage protein p38 or L1. Cells were then washed once with PBS, pH 7.4, and incubated with fresh complete medium (100 μL/well) along with the CellTiter-Blue assay reagent (20 μL/well) for 2 h at 37°C. The fluorescence intensity was measured using a multi-detection microplate reader (Bio-Tek, Winooski, VT) with 525/590 nm excitation/emission wavelengths.
Establishment of panc-1 multicellular 3D spheroids
Panc-1 multicellular spheroids were formed according to the method[14, 15, 16]. Briefly, panc-1 cells were grown in completed DMEM supplemented with 1% antibiotic and 10% FBS at 37 °C. The 96-well plates were coated with 1.5% agarose in DMEM to prevent cell adhesion and seeded with 1 × 104 cells per well. The plates were then centrifuged at 405 × g for 15 min, and the multicellular aggregates were maintained at 37 °C for spheroid formation. The spheroid was identified by its size and shape. The 6-d spheroids with a diameter of 700–900 μm were used as the in vitro tumor model.
Tumor cell viability tested in 3D spheroid
The tumor cell viabilities were further tested in 3D spheroids. Different micellar nano-formulations were used to treat 6-day panc-1 spheroids at the varied PCT concentrations, including 1.875, 3.75 and 15 μg/mL The spheroids were maintained in the 96-well plate with 100μL of complete growth media. The additional 50μL of fresh media was added to compensate for evaporation when the treatment was given. On day 12, the LDH release was measured with a Cytotox 96 Non-Radioactive Cytotoxicity kit to evaluate the cytotoxicity of the formulations on spheroids. The LDH release of spheroids was normalized to total LDH content from both spheroids and their growing media.
Statistical analysis
The statistical significance of the results was analyzed using SPSS (version 16). Differences between experimental groups were compared using ANOVA followed by a Bonferroni post hoc test. The results were considered statistically significant if the p value was less than 0.05.
RESULTS
Micelle formulation
To physically characterize phage-micelles, critical micelle concentration (CMC), zeta-potential and sizes were determined. The CMC of micelles was estimated as 6.6 × 10−7 M (p38 phage micelles) and 6.5× 10−7 M (L1 phage micelles), which were within a similar range of the CMC of the plain PEG2000-PE-based micelles (6.6 × 10 −7 M)[17, 18](Figure 1(A)). The low CMC of the nanopreparations could ensure the in vitro and in vivo stability of these core-shell/micellar structures against dilution effect[18, 19]. The size of the panc-1-targeted phage-micelles by dynamic light scattering measurement was narrow distribution and within a 13.3–20.5 nm interval (Figure 1(B)), consistent with that of typical PEG-PE micelles. These micellar particles had negative charge (Figure 1(C)), which contributed to the physical stability of colloidal formulations against aggregation of nanoparticles due to electrical repulsion.
Figure 1.
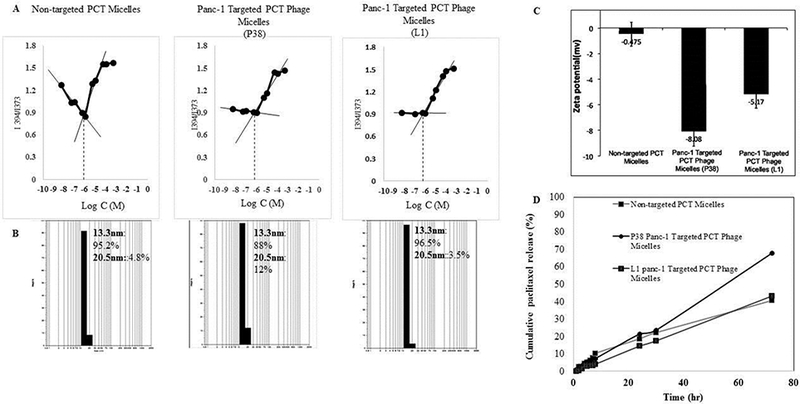
Characterization of the Micelle Formations. (A) Critical micelle concentration (CMC) determination; (B) Size; (C) Zeta potential; (D) Drug release profiles.
To chemically characterize phage-micelles, PEG content, phage protein density and drug loading were determined. Incorporation of phage protein into micellar formulations did not significantly influence the loading efficiency of PCT (PCT/PEG (% wt) with 2.84±0.14 for non-modified plain micelles, 3.21±0.16 for P38 phage micelles and 2.86 ± 0.14 for L1 phage micelles. Protein densities, defined as the ratio of phage protein to PEG content by weight, were 0.43 for both p38 and L1 phage micelles.
The in vitro drug release study showed that PCT loaded into PEG-PE micellar formulations followed a zero-order kinetics of release with the delivery of drug at a constant rate, providing a predictable bioavailability status. The incorporation of P38 phage protein into PEG-PE micelles facilitated PCT release, while the inclusion of L1 phage protein maintained the similar release kinetics as non-modified plain micellar formulations (Figure 1(D)).
Stability of the phage micelles
The particle size of the nanopreparations was commonly-used as an indicator of the stability of the micellar nanoparticle[18, 19]. After the incubation of the micellar formulations at 37°C for 10 days, there was no significant change in the size of the nanopreparation (Figure 2). Longterm storage at 4 °C for 12 months, showed no phase separation. These data indicated that the formed micelles were quite stable in the aqueous buffer.
Figure 2.
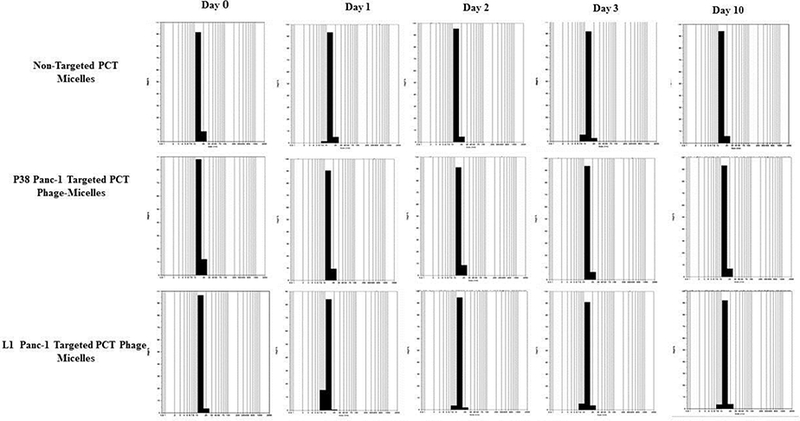
Stability testing of phage-micelles in PBS under accelerated conditions such as shaking and elevated temperature of 37°C as opposed to normal storage at 4°C.
To examine the possibility of in vivo blood protein adsorption/interaction, micellar nanoparticles were incubated and shaken with FBS at 37oC up to 72h in a shaker. While 100% non-modified PCT micelles aggregated after 48h at 37oC, P38 and L1 modified micelles showed a slower and lesser protein adsorption or interaction in FBS within 48 h with only 0.8% and 2.8% aggregates detected. When the incubation time was prolonged for up to 72h, both the P38 panc-1-targeted PCT phage-micelles and L1 panc-1-targeted PCT phage-micelles produced 100% aggregates. The control groups (both serum alone and formulations tested in PBS) had no detectable size change under the time frame validating that the aggregation was due to protein adsorption/interaction in FBS rather than the properties of formulation or serum on its own (Figure 3). The results revealed that the panc-1-targeted PCT-loaded phage-micelles decreased the tendency for opsonization in vivo and had a higher possibility to escape the immune cell capture that indicated their potential for functioning as long-circulation nanocarriers for drug targeting.
Figure 3.
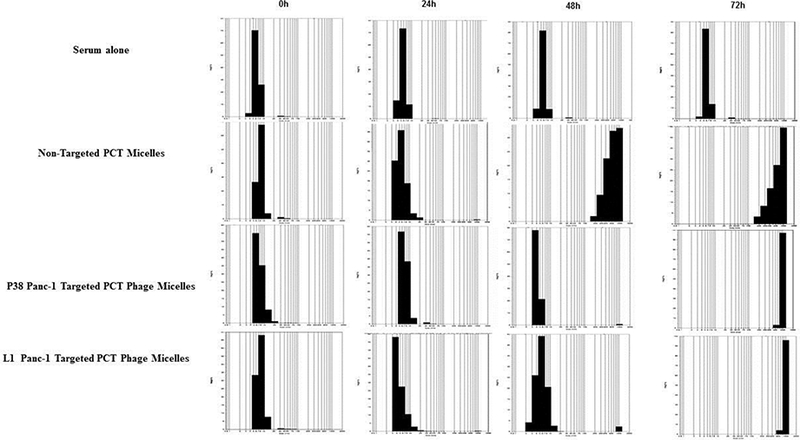
Stability testing using FBS. P38 and L1 Modified micelles had slower and lesser protein adsorption / interaction in FBS within 48h at 37 °C.
Cell association of the phage-micelles
Uptake of panc-1-targeted phage-micelles by cancer and non-cancer cells for 2h was determined using FACS analysis in comparison with the control plain micelles. Both the P38 and L1 phage-micelles showed a stronger uptake by target panc-1 cells than non-modified plain micelles, as was indicated by a stronger right shift along the X-axis of the histogram. P38 phage-micelles showed a weak binding to the non-target MCF-7 cancer cells as well as the non-target, non-cancerous NIH3T3 and C166 cells to a similar extent as the non-modified plain micelles, whereas L1 phage micelles had a negligible association with the non-target MCF-7 cancer cells and non-specific uptake by non-target, non-cancerous C166 cells when compared to the non-modified plain micelles (Figure 4,5)[20].
Figure 4.
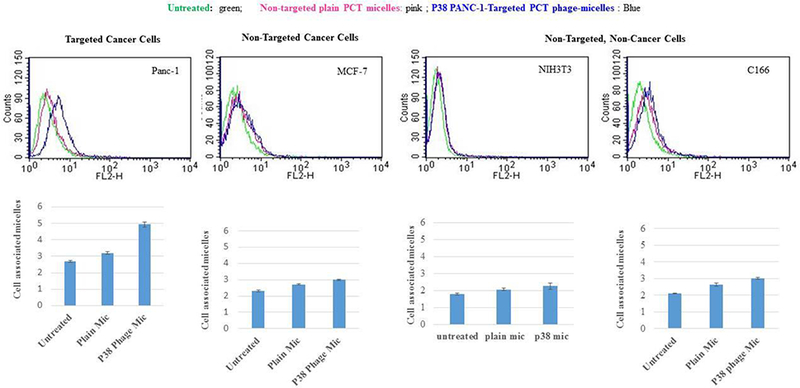
Cellular uptake by P38 phage micelles.
Figure 5.
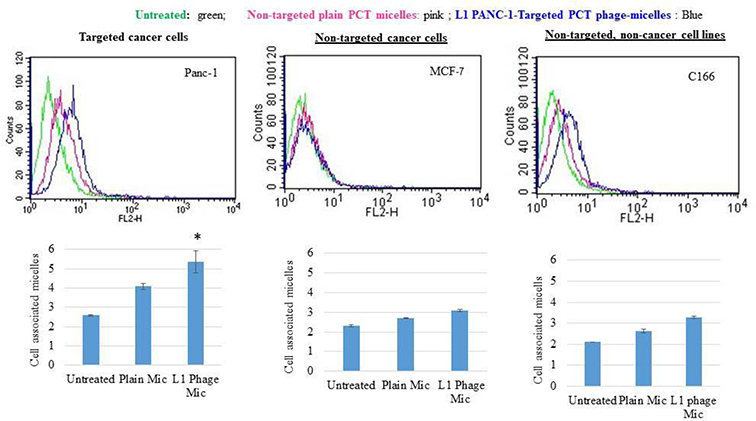
Cellular uptake by L1 phage micelles.
The enhanced binding of the phage-micelles to the target panc-1 cells compared to non-modified plain micelle was further confirmed using the fluorescence microscopy. Based on the red fluorescence of rhodamine-PE, those panc-1 cells bound with the micelles were distinguished from cells that had no binding as visualized under a fluorescence microscope. The two treatments with different rhodamine-labeled panc-1-targeted phage-micelles (one with p38 and the other with L1) showed much higher binding of the phage-micelles to panc-1 cells in comparison to the non-modified plain micelles. Control plain micelles showed weak binding to panc-1 cells indicating that the phage protein mediated the improved targeting of the phage-micelles to panc-1 cells. The images were captured using three different fields for each of the three formulations (Figure 6).
Figure 6.
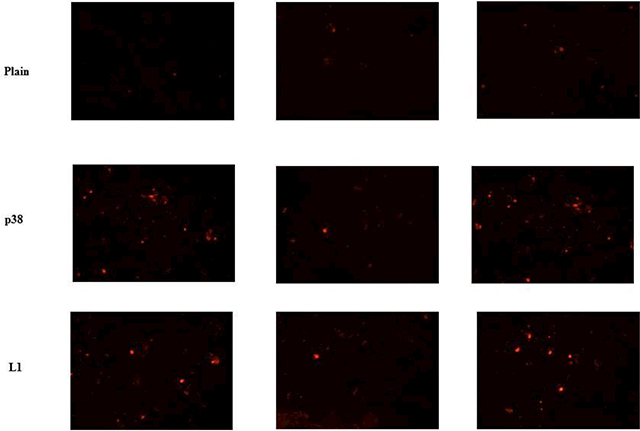
Fluorescence microscopy study the association of micellar nanoparticle with targeted panc-1 cells, showing improved targeted cell binding of p38 and L1 phage micelles when compared to non-modified plain micelles.
Cell viability tested on monolayer panc-1 cancer cells
As a result of the improved targeted cellular uptake, 72 h cytotoxicity study showed higher potency of panc-1-targeted PCT phage micelles in induction of panc-1 cell death compared to non-targeted PCT micelles at various concentrations of 187.5 ng/ml, 750 ng/ml, 1500 ng/ml and 2570 ng/ml (Figure 4). Targeted PCT phage micelles showed significantly higher cell-death at the concentrations of 0.750 mg/ml, 1.5mg/ml and 2.570 mg/ml. While the non-targeted plain PCT-loaded micelles caused 33% cell death, P38 and L1 phage-targeted PCT-loaded micelles killed 42% and 45% tumor cells at the same 750ng/ml concentration. Significant cytotoxicity of panc-1-targeted PCT-loaded phage-micelles compared to non-targeted PCT-loaded micelles was also found at the concentrations of 1500 ng/ml and 2570 ng/ml suggesting the higher cytotoxicity of panc-1-targeted phage-micelles towards panc-1 cells than the for the non-targeted plain PCT-containing micelles (Figure 7(A)).
Figure 7.
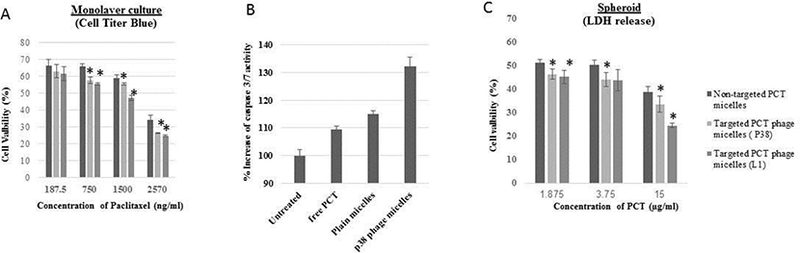
In vitro antitumor activity. (A) Cell viability tested on monolayer panc-1 cancer cells; (B) In vitro apoptosis-inducing ability; (C) In vitro apoptosis-inducing ability
In vitro apoptosis-inducing ability of phage-micelles on the monolayer of panc-1 cancer cells
To compare the in vitro apoptosis-inducing ability of phage-micelles, we tested the activity of caspase-3 (the biomarker of apoptosis)[21] in panc-1 cells that were treated by free PCT, non-modified PCT-containing plain micelles and phage-micelles. While both, free PCT and non-modified plain micelles activated the caspase-3 activity, P38 phage-micelles resulted in significantly enhanced apoptosis (Figure 7(B)).
Cell viability tested on 3D panc-1 cancer spheroids
The two-dimension monolayer cell culture model could not fully capture the in vivo characteristic microenvironments of solid tumors, such as heterogeneous tumors, nutrient and oxygen gradients, cell-cell interactions, matrix deposition, and gene expression profiles. In vitro results may not be correlated with the in vivo drug responses[22]. Here we developed the in vitro panc-1 multicellular 3D spheroid culture to better reflect the tumor microenvironments. The response of panc-1 cancer cells to phage-micelles was assessed by the use of the in vitro spheroid model, and two phage-containing micellar formulations at three different PCT concentrations tested produced higher cytotoxicity with increased lactate dehydrogenase (LDH) release compared to non-modified plain micelles (Figure 7(C)).
DISCUSSION
Here we suggested the development of a suitable targeted drug delivery system for pancreatic cancer therapy. Pancreatic cancer is hard to treat and the conventional therapeutic options have problems associated with serious side effects, reduced bioavailability and half-lives. PCT has been used in the treatment plan for the disease, however, has severe side effects and poor solubility. We suggested the preparation of polymeric phage-modified PEG-PE micelles that have the capability to self-assemble with the hydrophobic PCT and the phage proteins P-38 and L-1. The micellar preparation provides improved PCT solubility and also reduces side effects from the drug. Our task in this study was to achieve targeted drug delivery to pancreatic cancer cells for enhanced therapy using phage coat proteins from the phages selected based on their specific recognition of pancreatic cancer cells as targeting moieties. From this point of view, the most important property of the selected phages was their strong specific binding with target cells. This is why the identification of the exact component/domain on the cancer cell surface represents a separate task and remained out of the scope of this research.
The micellar preparation was extensively characterized based on the physical parameters, such as size, zeta potential, Critical Micelle Concentration, serum and storage stability, and in vitro drug release. Low CMC values were comparable with the plain PEG-PE micelles suggesting that the formulation should be quite stable under both, in vitro and in vivo conditions. These polymeric micelles with low CMCs remain stable even with low polymer concentrations and resist any dilution effect as opposed to surfactant micelles. Zeta potential characterization supported the aspect of structural stability with the negative charge on particles suggesting lesser aggregation chances for the nanoparticles in solution.
A predictable bioavailability pattern was provided by the in vitro drug release study that showed zero order drug release kinetics over time. Stable formulation characteristics such as absence of precipitation despite storage for a month and stable size were achieved. The P-38 and L-1 modified micellar preparations gave very low blood protein adsorption in vitro looking as ideal nanocarriers with long circulating characteristics and with lower possibilities for attack by reticulo-endothelial system. Moreover, the ideal micelle formulation with the targeting ligands revealed specific in vitro uptake in pancreatic cancer cell lines and negligible association with the non-specific cell lines. This is the most important aspect of the formulation that could offset any undesirable toxicity resulting from the non-specificity. The specific binding of the micelle with the P-38 and L-1 phage proteins on to the panc-1 cells was further confirmed by the fluorescence microscopy.
Apoptosis and cytotoxicity experiment data showed significantly enhanced cancer cell killing with the targeted micellar preparation as opposed to the non-targeted ones, again supporting the specificity of the formulation in executing the desirable effects. Lower concentrations of the targeted formulation (in the nanogram scale) producing these desirable effects as opposed to the non-modified PCT loaded micelles in vitro points at the suitability of the micelles in eliminating unwanted side-effects due to the toxicity at higher concentrations of the drug in formulation. The in vitro spheroid model that resembles the in vivo tumor architecture and surrounding very closely, has also demonstrated improved cytotoxicity results with higher Lactate Dehydrogenase release confirming the possibility of the successful use of the micelle formulation for in vivo applications.
CONCLUSION
The amphiphilic character of the panc-1 cancer cell-specific phage protein was utilized to develop targeted micellar formulation modified with phage protein for the delivery of poorly water soluble antitumor agent to tumors. The phage-micelles have improved PCT solubility, showed zero-order drug release kinetics and improved stability in the blood and upon storage, while also producing an enhanced panc-1 tumor cell killing tested on both 2D monolayer and 3D spheroid as well as an extensive apoptosis as a result of the improved panc-1 targeted delivery. The absence of hepatotoxicity and major histological changes in tissue sections of vital organs has been demonstrated in earlier work using the surface-exposed target-specific terminal peptides of phage coat proteins as targeting agents on the micellar and liposomal nanocarriers[11]. Since the non-immunogenic nature of the terminal peptides (of different sequences) is well-established, the micelles developed in this study are promising candidates for in vivo use and demonstrating it would be the next experimental step in this direction.
Acknowledgments
Grant Support
This work was supported by NIH grant # R01 CA125063–01 and the Animal Health and Disease Research grant 2006–9, College of Veterinary Medicine, Auburn University to Valery A. Petrenko and by NIH grant #1U54CA151881 to Vladimir P. Torchilin.
REFERENCES
- 1.Reynolds RB, Folloder J. Clinical Management of Pancreatic Cancer. Journal of the advanced practitioner in oncology. 2014. Sep-Oct;5(5):356–64. PubMed PMID: ; PubMed Central PMCID: PMC4457174. [PMC free article] [PubMed] [Google Scholar]
- 2.Boussios S, Pentheroudakis G, Katsanos K, et al. Systemic treatment-induced gastrointestinal toxicity: incidence, clinical presentation and management. Annals of gastroenterology. 2012;25(2):106–118. PubMed PMID: ; PubMed Central PMCID: PMC3959393. [PMC free article] [PubMed] [Google Scholar]
- 3.Lowery MA, O’Reilly EM. Pancreatic cancer: the role of molecular markers in diagnosis and management. Clinical advances in hematology & oncology : H&O. 2011. December;9(12):900–8. PubMed PMID: . [PubMed] [Google Scholar]
- 4.Rossi ML, Rehman AA, Gondi CS. Therapeutic options for the management of pancreatic cancer. World journal of gastroenterology. 2014. August 28;20(32):11142–59. doi: 10.3748/wjg.v20.i32.11142. PubMed PMID: ; PubMed Central PMCID: PMC4145755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Z, Zhang F, Koh GY, et al. Cytotoxic and antiangiogenic paclitaxel solubilized and permeation-enhanced by natural product nanoparticles. Anti-cancer drugs. 2015 Feb;26(2):167–79. doi: 10.1097/CAD.0000000000000173. PubMed PMID: ; PubMed Central PMCID: PMC4272611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu W, Ling P, Zhang T. Polymeric micelles, a promising drug delivery system to enhance bioavailability of poorly water-soluble drugs. Journal of drug delivery. 2013;2013:340315. doi: 10.1155/2013/340315. PubMed PMID: ; PubMed Central PMCID: PMC3712247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bedi D, Gillespie JW, Petrenko VA. Selection of pancreatic cancer cell-binding landscape phages and their use in development of anticancer nanomedicines. Protein engineering, design & selection : PEDS. 2014. July;27(7):235–43. doi: 10.1093/protein/gzu020. PubMed PMID: ; PubMed Central PMCID: PMC4064708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petrenko VA, Jayanna PK. Phage protein-targeted cancer nanomedicines. FEBS Lett. 2014. January 21;588(2):341–9. doi: 10.1016/j.febslet.2013.11.011. PubMed PMID: ; PubMed Central PMCID: PMCPMC4557960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang T, D’Souza GG, Bedi D, et al. Enhanced binding and killing of target tumor cells by drug-loaded liposomes modified with tumor-specific phage fusion coat protein. Nanomedicine (Lond). 2010 Jun;5(4):563–74. doi: 10.2217/nnm.10.30. PubMed PMID: ; PubMed Central PMCID: PMCPMC2914609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang T, Yang S, Mei LA, et al. Paclitaxel-loaded PEG-PE-based micellar nanopreparations targeted with tumor-specific landscape phage fusion protein enhance apoptosis and efficiently reduce tumors. Mol Cancer Ther. 2014. December;13(12):2864–75. doi: 10.1158/1535-7163.MCT-14-0052. PubMed PMID: ; PubMed Central PMCID: PMCPMC4258532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang T, Hartner WC, Gillespie JW, et al. Enhanced tumor delivery and antitumor activity in vivo of liposomal doxorubicin modified with MCF-7-specific phage fusion protein. Nanomedicine : nanotechnology, biology, and medicine. 2014. February;10(2):421–30. doi: 10.1016/j.nano.2013.08.009. PubMed PMID: ; PubMed Central PMCID: PMC3946195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu L, Perche F, Wang T, et al. Matrix metalloproteinase 2-sensitive multifunctional polymeric micelles for tumor-specific co-delivery of siRNA and hydrophobic drugs. Biomaterials. 2014. April;35(13):4213–22. doi: 10.1016/j.biomaterials.2014.01.060. PubMed PMID: ; PubMed Central PMCID: PMC3981970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nag A, Mitra G, Ghosh PC. A colorimetric assay for estimation of polyethylene glycol and polyethylene glycolated protein using ammonium ferrothiocyanate. Analytical biochemistry. 1996. June 01;237(2):224–31. doi: 10.1006/abio.1996.0233. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 14.Wen Z, Liao Q, Hu Y, et al. A spheroid-based 3-D culture model for pancreatic cancer drug testing, using the acid phosphatase assay. Brazilian journal of medical and biological research = Revista brasileira de pesquisas medicas e biologicas. 2013. July;46(7):634–42. doi: 10.1590/1414-431X20132647. PubMed PMID: ; PubMed Central PMCID: PMC3859338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Longati P, Jia X, Eimer J, et al. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC cancer. 2013. February 27;13:95. doi: 10.1186/1471-2407-13-95. PubMed PMID: ; PubMed Central PMCID: PMC3617005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu X, Farach-Carson MC, Jia X. Three-dimensional in vitro tumor models for cancer research and drug evaluation. Biotechnology advances. 2014. November 15;32(7):1256–68. doi: 10.1016/j.biotechadv.2014.07.009. PubMed PMID: ; PubMed Central PMCID: PMC4171250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lukyanov AN, Torchilin VP. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Advanced drug delivery reviews. 2004. May 07;56(9):1273–89. doi: 10.1016/j.addr.2003.12.004. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 18.Zhu L, Wang T, Perche F, et al. Enhanced anticancer activity of nanopreparation containing an MMP2-sensitive PEG-drug conjugate and cell-penetrating moiety. Proceedings of the National Academy of Sciences of the United States of America. 2013. October 15;110(42):17047–52. doi: 10.1073/pnas.1304987110. PubMed PMID: ; PubMed Central PMCID: PMC3801051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Owen SC, Chan DP, Shoichet MS. Polymeric micelle stability. Nano Today. 2012;7(1):53–65. [Google Scholar]
- 20.Zou W, Sarisozen C, Torchilin VP. The reversal of multidrug resistance in ovarian carcinoma cells by co-application of tariquidar and paclitaxel in transferrin-targeted polymeric micelles. Journal of drug targeting. 2017. March;25(3):225–234. doi: 10.1080/1061186X.2016.1236113. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 21.Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell death and differentiation. 1999. February;6(2):99–104. doi: 10.1038/sj.cdd.4400476. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 22.Hirschhaeuser F, Menne H, Dittfeld C, et al. Multicellular tumor spheroids: an underestimated tool is catching up again. Journal of biotechnology. 2010. July 01;148(1):3–15. doi: 10.1016/j.jbiotec.2010.01.012. PubMed PMID: . [DOI] [PubMed] [Google Scholar]