

Abstract
We demonstrate that changing the backbone between peptides, peptoids and the underexplored dual N/Cα-substituted peptoids analogues allows for control over the preferred conformation of the intrinsically disordered biomimetic oligomers. The conformation tunability is directly probed by Electron Paramagnetic Resonance (EPR), and is shown to manifest itself in differences in the nanoparticle-oligomer hybridization propensity.
Graphical Abstract
Conformational space of peptides can be fine-tuned by chemical modifications of the backbone.
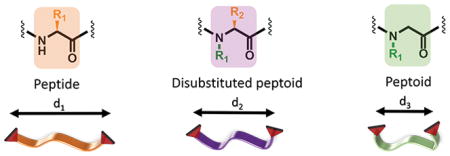
Structured proteins and peptides adopt well-defined functional conformations that are determined by their primary amino acid sequence. In contrast, non-structured peptides exist in a multitude of different conformations span what is known as their conformational space, determined by the primary sequence.1 Peptides are also essential building blocks in composite and bio materials, in which the ability to control the peptide conformation is crucial for the rational design and control of their function.2,3 Here, approaches to achieve tunability over the peptide conformational space without changing the sequence of the chemical side chain groups displayed is desirable for biomedical to biotechnological applications.4 For example, Reches and Alemán have recently demonstrated through single molecule force spectroscopy, molecular simulations, and alanine scanning that the interactions between the peptide sequence QPASSRY and flat inorganic mica surfaces are dominantly dictated by the conformation of the peptide.3 The conformational space of an unstructured peptide is largely defined by the constraints imposed by its backbone. One possible solution for expanding the conformational space, without affecting the desired side-chain chemistry is the use of peptidomimetics, such as the peptoids (poly-N-substituted glycines) class.5 Peptoids are excellent examples of peptidomimetics with an alternative, more flexible, backbone that, in addition, possess promising biochemical properties, such as protease resistance and increased cell permeability.6–10 Despite obvious benefits, peptoids suffer from the lack of rigidity and chirality that can reduce their affinity and specificity towards biological targets.11 Here we demonstrate that a fine control over the oligomer conformational space can be achieved with a new class of N/Cα-disubstituted (DS) peptoids, where both NH and Cα positions are functionalized (Fig 1) to controllably increase the rigidity, and reintroduce chirality.12–16 The DS-peptoid units are mildly nature-modified in terms of their length, compared to β-peptides or γ-peptides (where α-, β- and γ-carbons constitute the peptide backbone), and maintain their chirality in contrast to peptoids or D-peptides, while offering a platform to achieve enriched chemical diversity through their side-chain sequence. In addition, we demonstrate as a proof of concept, that these differences in the conformational space have implications in the propensity of the three oligomer types to either stabilize or promote aggregation of gold nanoparticles (AuNPs). We examined the conformational space of an octapeptide sequence, Phe-Glu-Phe-Abu-Phe-Glu-Phe-Dap and its peptidomimetic analogues (Fig 2). The sequence was chosen to contain both hydrophobic and hydrophilic side chain residues, and lack secondary structure. For all three oligomer classes, the order and identity of the side chains were kept identical, with their position changed from Cα in peptides (Fig 2A) and to NH in peptoids (Fig 2B), while in the case of DS-peptoids, methyl groups were inserted alternatively at the Cα positions of the peptoid sequence to yield chiral DS-peptoid repeat units (Fig 2C). To fully understand the impact of oligomer length, trimer and pentamer oligomers consisting of a sub-sequence of the octamer series were also synthesized. The oligomers were synthesized using solid-phase peptide synthesis. Fmoc monomers were initially synthesized by reductive amination or aza-Michael addition reactions starting from glycine and alanine amino acids derivatives, respectively, followed by Fmoc protection. The monomers were reacted using different reaction conditions, depending on the number of substituents per monomer. A strong reagent such as triphosgene was utilized for the sterically demanding DS-peptoid monomers (see SI for detailed synthesis procedures).17 All nine constructs were functionalized with a nitroxide radical spin-label on both termini. The nitroxide spin label was chosen to be minimally disturbing: it is small compared to commonly used fluorescent labels, and is chemically benign given the lack of charge and its “neutral” chemical property that is neither strongly hydrophobic nor hydrophilic. Here, the nitroxide label allows for dual comparative EPR studies of the conformational space of peptidomimetics and AuNPs aggregation studies.
Fig 1.
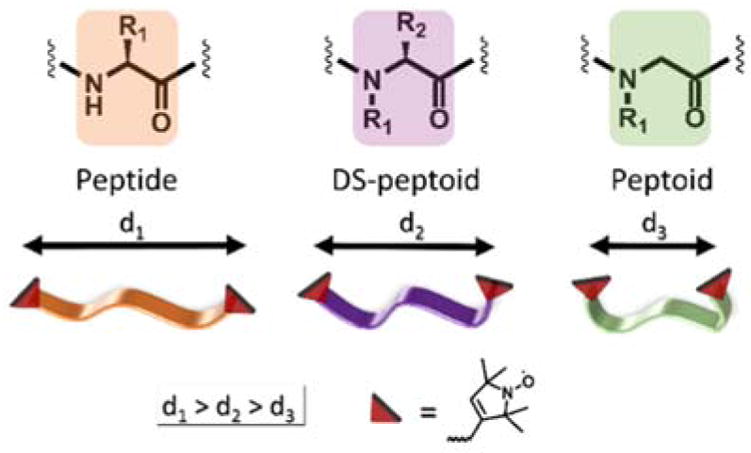
Analogous series of peptide, peptoid, and DS-peptoid backbones and their relative mean distance distributions reflecting the overall conformations.
Fig 2.

Analogous series of trimer, pentamer, and octamer double spin-labeled (A) peptides, (B) peptoids, (C) and N/Cα-disubstituted (DS) peptoids oligomers. Monomer color legend: Orange; peptide, green; peptoid, purple; DS-peptoids.
In order to characterize the differences in conformational space adopted by these molecules in solution, we carried out pulsed EPR DEER measurements. DEER measurements are performed on rapidly frozen samples at 85 K. Well-vitrified samples present a snapshot of the conformations adopted by the molecules in solution at room temperature, as was verified by comparison between X-ray and NMR techniques.18,19 Here, a 1:1 (v:v) mixture of H2O/DMSO was chosen as the solvent, as all oligomers were fully soluble in this composition, and since this solvent mixture is known to vitrify efficiently.20,21 (Further experimental details are available in the SI.) For each molecule, a distance distribution was reconstructed from the DEER data, with the mean of the distance distribution reflecting the overall extension of the molecules in solution, and width of this distribution reflecting the conformational flexibility. Fig 3A shows the obtained distance distributions from the peptide (orange), peptoid (green) and DS-peptoid (purple) octamer samples. The data shows that the peptoid adopted the most compact assembly with a mean distance of d = 20.5 Å and the narrowest distance distribution of σ = 8.3 Å. The peptide displayed the largest mean distance of d = 23.0 Å and the broadest distance distribution of σ = 11.0 Å, while the DS-peptoid showed intermediate behavior with a mean distance of d = 22.0 Å and distance distribution of σ = 9.0 Å. Notably, all three distance distributions are broad compared to literature reports for oligomers with well-defined secondary structure, confirming that all three octamers are intrinsically disordered.22,23 Since the side chains and their order were kept identical, the observed variations in the experimental end to end distance probabilities solely originate from differences in the nature of the backbone. In general, oligomers with a higher tendency for trans configuration around the amide bonds adopted more extended conformations, as is the case with peptides and DS-peptoids. In contrast, the peptoid octamer adopts a more compact conformation, likely due to a relative increase in cis configurations around the amide bonds.14,15 The DS-peptoid octamer falls in between, as both cis/trans options are viable given its alternating sequence. The comparison of the different conformational space adopted by the different classes of peptidomimetics presents a fundamental evaluation of their different backbones, which can be utilized to tune their functional properties in different applications.
Fig 3.
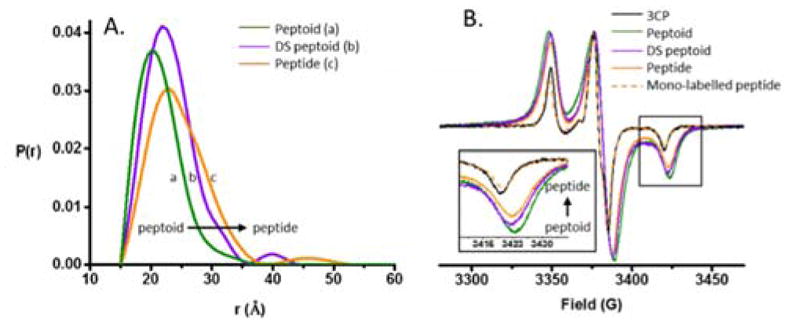
(A) DEER distance distributions obtained for the peptide, peptoid, and the DS-peptoid octamers. (B) Low temperature CW EPR of the trimer series overlaid with the mono-labeled peptide and 3CP free radical. Insets: Zoom in of high-field region.
To confirm the observed trend, the trimer sub-series was also examined. For such short oligomers DEER experiment is not applicable, however short interspin distances (d < 17 Å) manifest themselves in the CW EPR spectra of vitrified samples displaying characteristic dipolar broadening. The extent of the dipolar broadening depends on the distances present in the sample, with shorter distances resulting in larger spectral broadening.24 The spectra for all three trimers are significantly broadened as compared to the 3CP free radical and the mono-labeled peptide (Fig 3B). The observed trend in spectral broadening correlates with the trend for trimer distances in the order of peptide > DS-peptoid > peptoid, in agreement with the trend observed for the octamer oligomers. Note, that the spectra of the mono-labeled peptide is comparable to that of the 3CP radical, confirming that the samples are well dispersed and no aggregation apparent.
(The detailed analysis of the CW EPR spectra is available in the SI). The expected distances for the pentamer series fall in between the applicability of DEER and CW-EPR methodologies, complicating their evaluation. The analysis of the pentamer series data is presented in the SI (Fig. S6), and is still consistent with the observations drawn for the trimer and octamer series. In order to understand how the differences in the preferred conformation for these oligomers manifests themselves in their function, their propensity to stabilize or induce aggregation of gold nanoparticles (AuNPs) was explored. Nitroxides have been demonstrated to preferentially adsorb to AuNPs upon exchange with the weaker citrate ligand, and the spin-spin exchange interaction between the radical’s unpaired electron and the conduction-band electrons of the AuNPs proposed to mediate this binding.25–27 We used the AuNP’s localized surface plasmon resonance (LSPR) as a sensitive tool28 to track particle-particle proximity by using UV-vis spectroscopy.29,30 Nitroxide labelled oligomers can be treated as bivalent ligands that can either promote AuNPs aggregation or stabilize individual particles. Previously, it was shown that multi-valent pyridyl small molecules with varying geometries induce different levels of aggregation of AuNPs that correspond to the binding site accessibility.30 In a similar way, the different backbone conformation may affect the nitroxide termini accessibility, and hence the propensity to stabilize individual particles or promote AuNPs aggregation via crosslinking.
AuNPs were synthesized by the Turkevich method leading to citrate-capped AuNPs in aqueous solutions (d=12.1 ± 1.1 nm, (see SI for details). The dual-nitroxide-labeled oligomers (dissolved in acetonitrile) were added to an aqueous solution of AuNPs. Upon reaction, the intensity of the single LSPR absorption band at λmax=518 nm was reduced, while the intensity of the coupled LSPR band at λmax=600–645 nm was increased, congruent with the formation of particle-particle assemblies (Fig 4, A–C). Interestingly, a similar trend for aggregation propensity was observed for all oligomer lengths: peptoids < DS-peptoids < peptides, with no aggregation propensity detected with the pentamer and octamer peptoids. Notably, no significant change in the LSPR absorption was observed when only acetonitrile was added or when mono-labeled peptide trimers were mixed with AuNPs (Fig 4A, and SI). We hypothesize that for the pentamer and octamer oligomer series, the more rigid and extended peptide conformation gives rise to higher accessibility of the two nitroxide termini to substrates, resulting in a more efficient cross-linking between different AuNPs and leading to their aggregation. In contrast, the flexibility of the peptoids results in more compact conformations that are less efficient in facilitating the cross-linking between particles, perhaps by adopting a more entropically favorable bivalent binding to a single AuNP. Similar effects of promoting single AuNP stabilization was observed by others for highly-flexible peptides with high-Glycine content.31 The contribution of additional stabilization by π interaction with the AuNP surface is plausible.32,33
Fig 4.
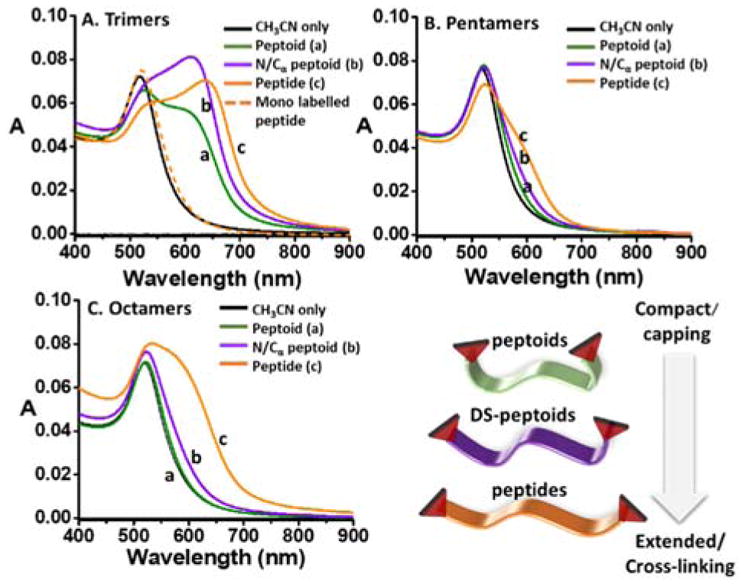
UV-vis absorption spectra of AuNPs solution with trimers, pentamers, and octamers dual-labeled oligomers. Spectra were recorded 5 hours after peptoids (green, a), DS-peptoids (purple, b), peptides (orange, c), mono-labeled peptide trimer (dashed orange), or only acetonitrile (black) were added to the AuNPs aqueous solution. Bottom right: graphical illustration of the oligomers relative conformational space.
Interestingly, similarly to what was observed with oligomer conformations, DS-peptoids also display intermediate aggregation propensity. The fact that DS-peptoids induce some aggregation, but lack H-bonds donors along the backbone (as opposed to peptides), suggests that the backbone conformation is a dominant factor in controlling the stabilization/aggregation trends. As for the trimer series, the sequences are too short to substantially fold or render the nitroxide radical inaccessible, resulting in significant aggregation with all three oligomers.30,32,33 Despite the widespread use of peptidomimetics for a variety of applications, only a few studies have examined the potential of using peptoid-inorganic NP hybrids.34 Zuckermann and Robinson demonstrated the stabilization of AuNPs for DNA co-adsoprbtion,33 with Maayan and Liu reporting on the controlled assembly of AgNPs using peptoids.35,36 Further exploration and tunability is needed in this area—this study presents one possible strategy for this purpose.
In summary, the potential of tuning the peptides and their composite properties was demonstrated by changing the peptidomimetic backbone. The results showed that peptoids are more compact, and their peptide counterparts more extended. The DS-peptoid class yields intermediate properties between that of peptides and peptoids. We demonstrated how these fundamental differences manifest themselves in the propensity for gold nanoparticles to aggregate. Furthermore, the distinction found in the hybridization behaviors of peptide and peptidomimetic oligomers provides an elegant approach that can be utilized for designing bio-inorgainc hybrids for biotechnological applications using different sequences and/or inorganic surfaces.
Supplementary Material
Acknowledgments
We acknowledge use of Materials Research Laboratory (MRL) Central Facilities supported by the National Science Foundation (NSF) through the Materials Research Science and Engineering Centers under Grant DMR 1720256. The authors thank the Defense Advanced Research Projects Agency (DARPA) #N66001-14-2-4055 Encode-Sort-Decode (ESD): Integrated System for Discovery of Non-Natural Affinity Reagents program for funding of this work. I.K. acknowledges the long-term postdoctoral fellowship awarded by the Human Frontier Science Program and W.R.G. thanks the NIH for a postdoctoral fellowship (F32GM108323). R.K. is an awardee of the Weizmann Institute of Science-National Postdoctoral Award for Advancing Women in Science, and also the recipient of the California NanoSystems Institute’s Elings Prize Fellowship in Experimental Science.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Conflicts of Interest
There are no conflicts to declare.
Notes and references
- 1.Beck DAC, Alonso DOV, Inoyama D, Daggett V. Proc Natl Acad Sci. 2008;105:12259–12264. doi: 10.1073/pnas.0706527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Skelton AA, Liang T, Walsh TR. ACS Appl Mater Interfaces. 2009;1:1482–1491. doi: 10.1021/am9001666. [DOI] [PubMed] [Google Scholar]
- 3.Maity S, Zanuy D, Razvag Y, Das P, Alemán C, Reches M. Phys Chem Chem Phys. 2015;17:15305–15315. doi: 10.1039/c5cp00088b. [DOI] [PubMed] [Google Scholar]
- 4.Gellman SH. Acc Chem Res. 1998;31:173–180. [Google Scholar]
- 5.Yoo B, Kirshenbaum K. Curr Opin Chem Biol. 2008;12:714–721. doi: 10.1016/j.cbpa.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 6.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. Proc Natl Acad Sci. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peretto I, Sanchez-Martin RM, Wang X, Ellard J, Mittoo S, Bradley M. Chem Commun. 2003:2312. doi: 10.1039/b306438g. [DOI] [PubMed] [Google Scholar]
- 8.Sun J, Zuckermann RN. ACS Nano. 2013;7:4715–4732. doi: 10.1021/nn4015714. [DOI] [PubMed] [Google Scholar]
- 9.Wu H, Mousseau G, Mediouni S, Valente ST, Kodadek T. Angew Chem Int Ed. 2016;55:12637–12642. doi: 10.1002/anie.201605745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuller AA, Holmes CA, Seidl FJ. Pept Sci. 2013;100:380–386. doi: 10.1002/bip.22242. [DOI] [PubMed] [Google Scholar]
- 11.Kodadek T, McEnaney PJ. Chem Commun. doi: 10.1039/C6CC00617E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao Y, Kodadek T. Chem Biol. 2013;20:360–369. doi: 10.1016/j.chembiol.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chatterjee J, Rechenmacher F, Kessler H. Angew Chem Int Ed. 2013;52:254–269. doi: 10.1002/anie.201205674. [DOI] [PubMed] [Google Scholar]
- 14.Butterfoss GL, Yoo B, Jaworski JN, Chorny I, Dill KA, Zuckermann RN, Bonneau R, Kirshenbaum K, Voelz VA. Proc Natl Acad Sci. 2012;109:14320–14325. doi: 10.1073/pnas.1209945109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sui Q, Borchardt D, Rabenstein DL. J Am Chem Soc. 2007;129:12042–12048. doi: 10.1021/ja0740925. [DOI] [PubMed] [Google Scholar]
- 16.Shi Y, Teng P, Sang P, She F, Wei L, Cai J. Acc Chem Res. 2016;49:428–441. doi: 10.1021/acs.accounts.5b00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernández-Llamazares AI, Spengler J, Albericio F. Pept Sci. 2015;104:435–452. doi: 10.1002/bip.22696. [DOI] [PubMed] [Google Scholar]
- 18.Pornsuwan S, Bird G, Schafmeister CE, Saxena S. J Am Chem Soc. 2006;128:3876–3877. doi: 10.1021/ja058143e. [DOI] [PubMed] [Google Scholar]
- 19.Luca Garbuio, Bartosz Lewandowski, Patrick Wilhelm, Ludmila Ziegler, Maxim Yulikov, Helma Wennemers, Gunnar Jeschke. Chem – Eur J. 2015;21:10747–10753. [Google Scholar]
- 20.Jeschke G. Annu Rev Phys Chem. 2012;63:419–446. doi: 10.1146/annurev-physchem-032511-143716. [DOI] [PubMed] [Google Scholar]
- 21.Rasmussen DH, Mackenzie AP. Nature. 1968;220:1315–1317. doi: 10.1038/2201315a0. [DOI] [PubMed] [Google Scholar]
- 22.Fafarman AT, Borbat PP, Freed JH, Kirshenbaum K. Chem Commun. 2007:377–379. doi: 10.1039/b612198e. [DOI] [PubMed] [Google Scholar]
- 23.Banham JE, Baker CM, Ceola S, Day IJ, Grant GH, Groenen EJJ, Rodgers CT, Jeschke G, Timmel CR. J Magn Reson. 2008;191:202–218. doi: 10.1016/j.jmr.2007.11.023. [DOI] [PubMed] [Google Scholar]
- 24.Abragam A. The Principles of Nuclear Magnetism. Clarendon Press; 1961. [Google Scholar]
- 25.Zhang Z, Berg A, Levanon H, Fessenden RW, Meisel D. J Am Chem Soc. 2003;125:7959–7963. doi: 10.1021/ja034830z. [DOI] [PubMed] [Google Scholar]
- 26.Liu CP, Wu TH, Liu CY, Cheng HJ, Lin SY. J Mater Chem B. 2015;3:191–197. doi: 10.1039/c4tb01657b. [DOI] [PubMed] [Google Scholar]
- 27.Swiech O, Hrynkiewicz-Sudnik N, Palys B, Kaim A, Bilewicz R. J Phys Chem C. 2011;115:7347–7354. [Google Scholar]
- 28.Shipway AN, Katz E, Willner I. ChemPhysChem. 2000;1:18–52. doi: 10.1002/1439-7641(20000804)1:1<18::AID-CPHC18>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 29.Frankamp BL, Boal AK, Rotello VM. J Am Chem Soc. 2002;124:15146–15147. doi: 10.1021/ja0280426. [DOI] [PubMed] [Google Scholar]
- 30.Kaminker R, Lahav M, Motiei L, Vartanian M, Popovitz-Biro R, Iron MA, van der Boom ME. Angew Chem Int Ed. 2010;49:1218–1221. doi: 10.1002/anie.200906636. [DOI] [PubMed] [Google Scholar]
- 31.Krpetić Ž, Nativo P, Porta F, Brust M. Bioconjug Chem. 2009;20:619–624. doi: 10.1021/bc8003028. [DOI] [PubMed] [Google Scholar]
- 32.Goldmann C, Lazzari R, Paquez X, Boissière C, Ribot F, Sanchez C, Chanéac C, Portehault D. ACS Nano. 2015;9:7572–7582. doi: 10.1021/acsnano.5b02864. [DOI] [PubMed] [Google Scholar]
- 33.Robinson DB, Buffleben GM, Langham ME, Zuckermann RN. Pept Sci. 2011;96:669–678. doi: 10.1002/bip.21588. [DOI] [PubMed] [Google Scholar]
- 34.Tigger-Zaborov H, Maayan G. Org Biomol Chem. 2015;13:8978–8992. doi: 10.1039/c5ob01093d. [DOI] [PubMed] [Google Scholar]
- 35.Tigger-Zaborov H, Maayan G. J Colloid Interface Sci. 2017;508:56–64. doi: 10.1016/j.jcis.2017.08.039. [DOI] [PubMed] [Google Scholar]
- 36.Maayan G, Liu LK. Pept Sci. 2011;96:679–687. doi: 10.1002/bip.21632. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.