

Abstract
Tuberculous meningitis is a serious, life-threatening disease affecting vulnerable populations, including HIV-infected individuals and young children. The US National Institutes of Health convened a workshop to identify knowledge gaps in the molecular and immunopathogenic mechanisms of tuberculous meningitis and to develop a roadmap for basic and translational research that could guide clinical studies.
The World Health Organization (WHO) estimates that 10.4 million new cases and 1.7 million deaths occurred from tuberculosis (TB) in 2016 alone. Tuberculous meningitis (TBM) is a devastating disease, especially among HIV-infected individuals and young children, and accounts for 1–2% of all TB cases. Early diagnosis is challenging; outcomes are poor even with treatment1, and prolonged (12 months) antimicrobial treatments are recommended, but not on the basis of clinical trial evidence. Bacillus Calmette-Guérin vaccine, developed almost 100 years ago, provides limited protection, but only during early childhood. Although immunoinflammatory damage is the critical pathological process in TBM and is amenable to host-directed treatments (HDTs)2, pulmonary TB is still used as the treatment paradigm and limited efforts have been made to utilize preclinical models of TBM to guide clinical studies3. Increasing numbers of multidrug-resistant (MDR) strains of Mycobacterium tuberculosis and limited access to the central nervous system (CNS) by key antimicrobials are additional challenges in combating this disease. Therefore, in May 2017, the US National Institutes of Health convened a two-day workshop to address these knowledge gaps (Table 1) and develop a roadmap for advancing TBM research.
Table 1.
Key gaps in understanding the molecular basis and immunopathogenesis of TBM
| Knowledge gaps | Directions | |
|---|---|---|
| Vaccines | ||
| BCG ineffective in adults/variable efficacy in children Live vaccine avoided in HIV-infected individuals |
Mechanisms of protective immunity and microbial targets poorly characterized | Studies to understand Hostmicrobial interactions Mechanism(s) of infection Microbial determinants of disease Developing subunit- and TBM-specific vaccines |
| Host-directed treatmentsx | ||
| HDTs improve survival in the short term but are ineffective against neurological deficits | Poor understanding of immunopathogenesis Dynamics of immune responses and lesion formation remain poorly characterized |
Discovery of new pathways Thalidomide and its analogs PARP1/DAMP inhibitors Role of inflammation vs. bacterial killing in treatment response |
| Antimicrobial treatments | ||
| Limited CNS penetration of key antimicrobials CNS tissues difficult to sample |
Data on intralesional drug concentrations in infected tissues lacking Spatial and temporal changes in drug penetration over time unknown |
Intralesional biodistribution Post-mortem studies in animals/fresh tissue analysis Live in vivo imaging (for example, PET) Developing TBM-specific antimicrobial regimens |
| Diagnostic/biomarkers | ||
| Paucity of bacteria in CSF leading to poor diagnostic sensitivity | Lack of a gold standard | Post-mortem studies in humans Host transcriptomics Serum/CSF biomarkers Diagnostic imaging (for example, MR spectroscopy, PET) |
BCG, bacillus Calmette-Guérin; PARP1, poly(ADP-ribose) polymerase 1 (also known as ARTD1); DAMPs, damage-associated molecular pattern; CNS, central nervous system; PET, positron emission tomography; CSF, cerebrospinal fluid; MR, magnetic resonance.
Pathogenesis and molecular mechanisms
Studies performed by Arnold Rich in the 1930s remain the current basis for TBM pathogenesis4. After initial infection of the lung, bacteria replicate and disseminate via the bloodstream. ‘Rich foci’ develop around bacteria deposited in the meninges and brain parenchyma during this initial bacteremia (Fig. 1). Months to years later, these foci rupture, with bacterial dissemination into the subarachnoid space, causing inflammatory meningitis. Although late-onset TBM occurs in adults and older children, meningitis in young children is not due to reactivation and often is concurrent with disseminated TB. ‘Paradoxical reactions’, that is, delayed development or worsening of brain lesions, can occur several months after the initiation of TB treatments, even in HIV-negative individuals5. Although the reasons for these delayed immunological responses in TBM are poorly understood, intracranially injected mycobacteria in rats escape immune recognition for months and delayed CNS lesions develop only after peripheral sensitization in these animals6. HIV-infected patients with TB can also develop immune reconstitution inflammatory syndrome (IRIS) after initiating antiretroviral treatments, and upregulation of certain innate signaling pathways that can predict IRIS has been identified recently. Overall, the dynamics of M. tuberculosis–induced immune responses and the development of granuloma-like lesions remain poorly characterized.
Fig. 1. Knowledge gaps in molecular mechanisms and immunopathogenesis.

TBM continues to be a serious, life-threatening disease affecting vulnerable populations, including HIV-infected individuals and young children. Key gaps exist in understanding the molecular basis, immunopathogenesis and mechanisms of bacterial invasion. New basic and translational research is required to develop novel therapeutic strategies for TBM. a, Cartoon of the brain highlighting multiple immunopathological processes. M. tuberculosis are shown as small yellow bacilli, and exudative meningitis is shown in pink. b, Enlarged view of a TBM lesion in the brain parenchyma demonstrating interaction between immune cells and bacteria. c, Details of the specialized barriers that separate the CNS from the systemic circulation are shown. CSF, cerebrospinal fluid; DAMPs, damage-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; APC, antigen-presenting cell; PMNs, polymorphonuclear leukocytes.
The CNS is protected from systemic circulation by tightly apposed brain microvascular endothelial and epithelial cells forming the blood–brain barrier (BBB) and blood–cerebrospinal fluid barrier (BCSFB), respectively. Although these barriers are impermeable to many large, hydrophilic molecules and circulating pathogens, many neuroinvasive bacteria, including M. tuberculosis, can invade the CNS, aided by specific microbial virulence determinants7. Although the mechanism(s) of M. tuberculosis traversal are poorly understood, endothelial cell invasion observed in vitro8 and in animal models3 and the extensive endovasculitis associated in humans with TBM4 suggest that ‘extracellular’ bacteria can invade the CNS. Infected dendritic cells can also disseminate bacteria, and entry of intracellular M. tuberculosis through the choroid plexus (BCSFB) via infected monocytes (‘Trojan horses’) was also presented in mouse models. Understanding the molecular mechanism(s) of CNS bacterial invasion and identification of key microbial determinants would be helpful in developing novel therapeutic strategies for TBM9.
Immune mechanisms and host-directed treatment strategies
Inflammation in response to bacterial infection in TBM causes endovasculitis, exudative meningitis, infarction, hydrocephalus and neuronal damage (Fig. 1). Experimentally induced intracisternal or intracerebral mycobacterial infections in mice and rabbits have demonstrated interferon-γ (IFN-γ)-dominated immune responses, microglial activation and elevated concentrations of proinflammatory mediators, including damage-associated molecular patterns (DAMPs), in the CSF3. Microglia are also critical for neurodevelopment, making the immature brain of young children uniquely vulnerable to injury. Host genetics may also have a key role. There is evidence that a variant of LTA4H, which encodes leukotriene A4 hydrolase, affects TBM-related mortality in multiple Vietnamese cohorts10, although a study in Indonesia did not find this association11. Analysis of CSF and serum metabolites in TBM has also identified an association between tryptophan metabolism and survival, likely mediated by host responses12.
Adjunctive corticosteroids improve survival by 25% in patients with TBM in the short term2 but are ineffective against neurological deficits, at least in adults. Therefore, development of improved HDTs specifically for TBM is a critical need. Glutathione is an important antioxidant with potential anti-mycobacterial and immune-enhancing properties. Augmenting glutathione abundance in macrophages derived from HIV-infected individuals resulted in improved control of infection13. Thalidomide and its analogs effect several immune functions, and adjunctive thalidomide improved survival in a rabbit model of TBM14. However, a trial evaluating thalidomide (24 mg/kg) in children with TBM was discontinued prematurely because of serious adverse events and deaths, although subsequently, lower doses (4 mg/kg) of thalidomide have benefited individuals with refractory CNS masses, optic neuritis and steroid-resistant paradoxical reactions associated with TBM. Recently, a phase 2 trial in HIV-negative adults suggested that addition of aspirin to dexamethasone could reduce new infarcts or deaths in microbiologically confirmed TBM15.
Pathogen-associated molecular patterns (PAMPs) are microbial products recognized by host cell receptors that activate innate immune responses. DAMPs such as high-mobility-group box 1 (HMGB1) and S100B are released by damaged host cells and react with PAMPs, causing a vicious cycle of inflammation and cell death. Host poly(ADP-ribose) polymerase 1 (PARP1; also known as ARTD1) activates microglial cells to release HMGB1 and potentiates the effects of S100B, resulting in brain injury. Inactivation of PARP1 in leukocytes diminishes inflammation and the migration of these cells across the BBB. Current US Food and Drug Administration (FDA)-approved inhibitors of DAMP, PARP116 and other innate immune pathways, for example, microbial phosphodiesterase subverting the stimulator of interferon genes (STING), are therefore promising HDT candidates for TBM. Paradoxical reactions and TB-IRIS would especially benefit from HDTs.
Antimicrobial treatment
TBM is complicated by the limited CNS penetration of several key antimicrobials, particularly rifamycins. Compartmental modeling for solute exchange and drug delivery to the brain has been attempted, but lack of data on intralesional drug concentrations in infected tissues where M. tuberculosis reside17 has limited these efforts. In a phase 2 trial, reduction in mortality was noted in patients with TBM treated with an intensified regimen using ‘high-dose’ intravenous rifampin (13 mg/kg) during the first two weeks18, although a subsequent larger randomized trial using oral rifampin (15 mg/kg) did not show this benefit19. These differences in clinical outcome may be due to suboptimal CNS exposures with the oral regimen; alternatively, outcomes could be more strongly associated with changes in intracerebral inflammation than with bacterial killing. Linezolid, an oxazolidinone, is a potent sterilizing antimicrobial with excellent CSF penetration, and retrospective studies from China suggest that addition of linezolid to the background TB regimen benefits patients with life-threatening TBM20. Prolonged antimicrobial treatments are recommended for TBM, but these recommendations are not based on clinical trial data and a shorter regimen (6 months) may be as effective, at least in HIV-negative individuals21. The CNS penetration and biodistribution of new or repurposed drugs (for example, bedaquiline, clofazimine and pretomanid), active against MDR-TB, are not well established. Developing TBM-specific antimicrobial regimens, for example, ones using high-dose rifampin and/or new drugs, is a priority and needs to be addressed in preclinical models, and ultimately in clinical studies. Finally, supportive care and neurosurgical interventions also have crucial roles in the outcomes for TBM and need to be optimized.
Modeling
The interaction of neuroinvasive bacteria, including M. tuberculosis, with in vitro cellular barriers has been investigated previously7,8, and efforts to develop an in vitro granuloma model using primary murine cells were also presented. Animal models of TBM have been summarized3, and several models using intracerebral or intracisternal infections have been described3,14. Recently, M. tuberculosis infections in young rabbits were performed to mimic TBM in children, highlighting the role of microglial activation22. Although intracisternal and intracerebral infections are useful for studying pathogenesis and evaluating novel therapeutics, they do not simulate natural disease, caused by hematogenous dissemination of M. tuberculosis. Murine and guinea pig models using intravenous or aerosol infections have been used to identify microbial factors associated with TBM3 and to develop novel vaccine strategies9. Zebrafish afford optical transparency for live in vivo imaging using Mycobacterium marinum3 and have been used to study the dynamics of granuloma formation and bacterial dissemination and to identify a role for LTA4H-dependent inflammation23. Live imaging using novel positron emission tomography (PET) tracers has also been applied in mammalian species to noninvasively measure inflammation22 and rifampin biodistribution in infected tissues in animals17 and patients with TB and for specific detection of bacteria24. These data suggested heterogeneous intralesional physiology and antimicrobial concentrations in infected tissues, which are different from those measured in clinical samples, for example, serum or CSF. Although PET may not be widely available in the developing world, it could enable research studies (for example, phase 0 studies with 10–20 patients) or be of clinical use at major referral centers. Finally, although not able to productively model all aspects of human disease, animal and other preclinical models can provide cellular and molecular insights into the pathogenesis of TBM in ways not possible in humans, as well as allow for screening and initial evaluation of therapeutics (Fig. 2).
Fig. 2. Advancing preclinical and translational research.

Preclinical models have not been fully used to guide TBM clinical studies and could provide important mechanistic insights as well as a platform for screening and initial evaluation of therapeutics. Substantially increased support for sustained basic and translational research to guide clinical studies is needed to advance the management of TBM. HDTs, host-directed treatments; MR, magnetic resonance; PET, positron emission tomography; CSF, cerebrospinal fluid; CNS, central nervous system; BBB, blood–brain barrier; BCSFB, blood–cerebrospinal fluid barrier.
Diagnostics and imaging
Poor outcomes in TBM are related to diagnostic delay, and bacteriological testing is not sensitive. Assays based on nucleic acid amplification, such as Xpert MTB/RIF Ultra (Xpert Ultra), were found to be promising for diagnosing TBM in a small cohort of HIV-infected adults25. Xpert Ultra is now recommended by the WHO, especially for specimens with a low pathogen burden, such as CSF. Development of sensitive and specific lateral flow assays for CSF, which can occur at point of care and are inexpensive (for example, urine lipoarabinomannan), could also improve diagnosis, but current tests fall substantially short of this goal, possibly because of the low pathogen burden in CSF. An ELISA to measure CSF HMGB1 was also found to be promising for diagnosing TBM. It should be noted that, in comparison with a comprehensive ‘shotgun’ approach, a stepwise decision analysis has demonstrated better diagnostic yield and cost-effectiveness. In children with TBM, markers of neuronal and astroglial injury, neuron-specific enolase (NSE), S100B and glial fibrillary acidic protein (GFAP) have been found to be elevated in ventricular but not lumbar CSF or serum. Lastly, the field of TBM diagnostics suffers from the lack of a gold standard, and post-mortem studies could help determine the true sensitivity of diagnostics.
Imaging techniques such as computed tomography (CT) and magnetic resonance imaging (MRI) are increasingly available in developing countries and, when available, are used in the evaluation and management of TBM (Fig. 3). In children with TBM in South Africa, angiographic abnormalities and infarcts were commonly noted on MRI5. MR spectroscopy, PET and pathogen-directed imaging could provide novel insights into pathophysiology and help in the management of TBM.
Fig. 3. Paradoxical reaction in a young child with TBM.
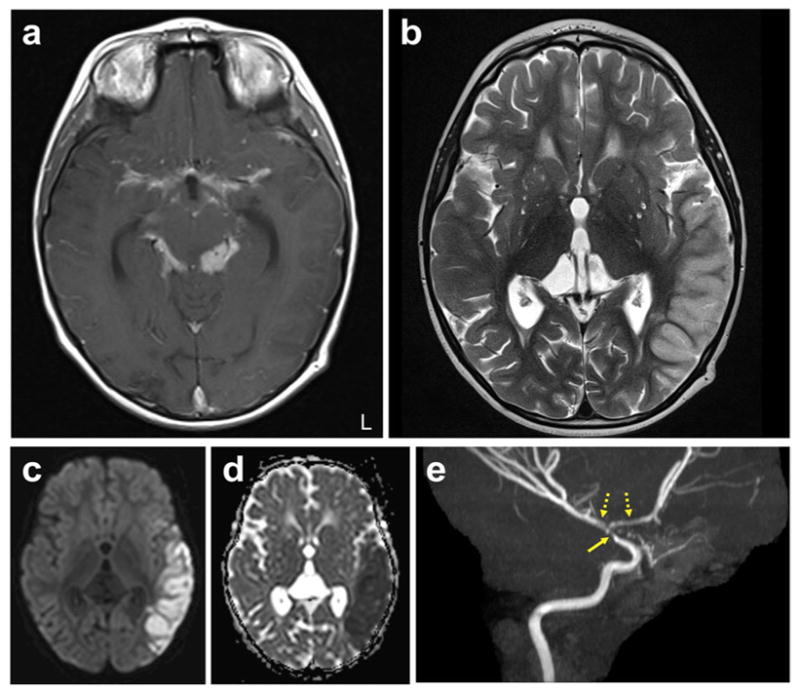
Brain MRI of a four-year-old, HIV-negative child with microbiologically confirmed TBM presenting with paradoxical reaction ten weeks after initiation of TB treatment. a–e, Representative enhanced T1-weighted (a) and T2-weighted (b) images, diffuse weighted imaging (DWI) (c), ADC (apparent diffusion coefficient) map (d) and MR angiography (maximum-intensity projection) (e) are shown. T2 hyperintensity involving the left temporal cortex is seen, with corresponding increased signal on DWI and decreased signal on ADC, consistent with left middle cerebral artery distribution acute stroke. The enhanced T1-weighted image shows extensive thick basilar meningeal enhancement. Narrowing of the distal left internal carotid artery (solid arrow) with irregularity and narrowing of the M1 and A1 segments (dashed arrows) are noted on the angiogram. Corticosteroids were reinitiated at this time, and the child improved considerably, with sustained improvement noted at follow-up many months later. L, left. (Credit: unpublished data, S.K.J. The author is in compliance with Johns Hopkins School of Medicine institutional policy regarding single-case deidentified patient data.)
Conclusions
TBM continues to be a serious, life-threatening disease affecting vulnerable populations including HIV-infected individuals and young children. Key gaps exist in understanding the molecular basis and immunopathogenesis of TBM as well as the mechanisms of bacterial invasion into the CNS. New research in these areas is required to develop novel therapeutic strategies for TBM. Evidence that TBM-specific HDTs can improve survival is promising and should lead to proof-of-concept studies. Antimicrobial treatments, currently based on pulmonary TB regimens, need to be optimized for TBM, although a critical unanswered question is whether new regimens should be directed at decreasing inflammation, enhancing bacterial killing or both. To date, preclinical models have not been fully used to guide TBM clinical studies. Preclinical models could provide important mechanistic insights as well as a platform for the screening and initial evaluation of therapeutics. Live in vivo imaging techniques could provide vital information on the spatial and temporal kinetics of inflammation and intralesional antimicrobial exposures and the effects of bacterial killing at sites of infection, and some technologies such as MR spectroscopy and PET have the potential for clinical translation. Lastly, the field of TBM diagnostics suffers from the lack of a gold standard, and post-mortem studies could help determine the true sensitivity of current diagnostics. Substantially increased support for sustained basic and translational research to guide clinical studies is needed to advance the management of TBM and is an urgent priority.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Chiang SS, et al. Lancet Infect Dis. 2014;14:947–957. doi: 10.1016/S1473-3099(14)70852-7. [DOI] [PubMed] [Google Scholar]
- 2.Prasad K, Singh MB, Ryan H. Cochrane Database Syst Rev. 2016;(4):CD002244. doi: 10.1002/14651858.CD002244.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sánchez-Garibay C, Hernández-Campos ME, Tena-Suck ML, Salinas-Lara C. Tuberculosis. 2018;110:1–6. doi: 10.1016/j.tube.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 4.Rich AR, McCordock HA. Bull Johns Hopkins Hosp. 1933;52:5–37. [Google Scholar]
- 5.Rohlwink UK, et al. Pediatr Infect Dis J. 2016;35:e301–e310. doi: 10.1097/INF.0000000000001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matyszak MK, Perry VH. Neuroscience. 1995;64:967–977. doi: 10.1016/0306-4522(94)00448-e. [DOI] [PubMed] [Google Scholar]
- 7.Coureuil M, Lécuyer H, Bourdoulous S, Nassif X. Nat Rev Microbiol. 2017;15:149–159. doi: 10.1038/nrmicro.2016.178. [DOI] [PubMed] [Google Scholar]
- 8.Jain SK, Paul-Satyaseela M, Lamichhane G, Kim KS, Bishai WR. J Infect Dis. 2006;193:1287–1295. doi: 10.1086/502631. [DOI] [PubMed] [Google Scholar]
- 9.Skerry C, et al. PLoS One. 2013;8:e66310. doi: 10.1371/journal.pone.0066310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thuong NTT, et al. J Infect Dis. 2017;215:1020–1028. doi: 10.1093/infdis/jix050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Laarhoven A, et al. J Infect Dis. 2017;215:1029–1039. doi: 10.1093/infdis/jix051. [DOI] [PubMed] [Google Scholar]
- 12.van Laarhoven A, et al. Lancet Infect Dis. 2018 doi: 10.1016/S1473-3099(18)30053-7. [DOI]
- 13.Morris D, et al. Front Pharmacol. 2014;5:73. doi: 10.3389/fphar.2014.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsenova L, Sokol K, Freedman VH, Kaplan GJ. Infect Dis. 1998;177:1563–1572. doi: 10.1086/515327. [DOI] [PubMed] [Google Scholar]
- 15.Mai NT, et al. eLife. 2018;7:e33478. [Google Scholar]
- 16.Berger NA, et al. Br J Pharmacol. 2018;175:192–222. doi: 10.1111/bph.13748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeMarco VP, et al. Antimicrob Agents Chemother. 2015;59:5768–5774. doi: 10.1128/AAC.01146-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruslami R, et al. Lancet Infect Dis. 2013;13:27–35. doi: 10.1016/S1473-3099(12)70264-5. [DOI] [PubMed] [Google Scholar]
- 19.Heemskerk AD, et al. N Engl J Med. 2016;374:124–134. doi: 10.1056/NEJMoa1507062. [DOI] [PubMed] [Google Scholar]
- 20.Sun F, et al. Antimicrob Agents Chemother. 2014;58:6297–6301. doi: 10.1128/AAC.02784-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jullien S, Ryan H, Modi M, Bhatia R. Cochrane Database Syst Rev. 2016;9:CD012091. doi: 10.1002/14651858.CD012091.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tucker EW, et al. Dis Model Mech. 2016;9:1497–1506. doi: 10.1242/dmm.027326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tobin DM, et al. Cell. 2010;140:717–730. doi: 10.1016/j.cell.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weinstein EA, et al. Sci Transl Med. 2014;6:259ra146. doi: 10.1126/scitranslmed.3009815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bahr NC, et al. Lancet Infect Dis. 2018;18:68–75. doi: 10.1016/S1473-3099(17)30474-7. [DOI] [PMC free article] [PubMed] [Google Scholar]