

Abstract
Scope:
The accumulation of misfolded α-synuclein in dopaminergic neurons is the leading cause of Parkinson’s disease (PD). Resveratrol (RV), a polyphenolic compound derived from grapes and red wine, exerts a wide range of beneficial effects via activation of sirtuin 1 (SIRT1) and induction of vitagenes. Here, we assessed the role of RV in a 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) induced mouse model of PD and explored its potential mechanisms.
Methods and results:
RV and EX527, a specific inhibitor of SIRT1, were administered before and after MPTP treatment. RV protected against MPTP-induced loss of dopaminergic neurons, and decreases in tyrosine hydroxylase and dopamine levels, as well as behavioral impairments. Meanwhile, RV administration activated SIRT1. Microtubule-associated protein 1 light chain 3 (LC3) was then deacetylated and redistributed from the nucleus to the cytoplasm, which provoked the autophagic degradation of α-synuclein in dopaminergic neurons. Furthermore, EX527 antagonized the neuroprotective effects of RV by reducing LC3 deacetylation and subsequent autophagic degradation of α-synuclein.
Conclusion:
We showed that RV ameliorated both motor deficits and pathological changes in MPTP-treated mice via activation of SIRT1 and subsequent LC3 deacetylation-mediated autophagic degradation of α-synuclein. Our observations suggest that RV may be a potential prophylactic and/or therapeutic agent for PD.
Keywords: α-synuclein, LC3 deacetylation, Parkinson’s disease, Resveratrol, SIRT1
1. Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by the degeneration of dopaminergic neurons. The current therapies for PD are symptomatic and do not affect the disease course [1]. Degeneration of dopaminergic neurons is associated with the presence of intraneuronal proteinaceous cytoplasmic inclusions, termed Lewy bodies. Alpha-synuclein, an aggregation prone protein, has been identified as a major protein component of Lewy bodies and is the causative agent of PD [2–4]. Mounting evidence indicates that alterations in the autophagy lysosomal pathways of α-synuclein degradation might be preferentially involved in neuronal death and contribute to the pathogenesis of PD [2,5]. Therapies targeting these processes might provide preventive strategies and potential treatments, as they could delay, revert, or compensate for the neurodegeneration that leads to motor impairment [6–8]. In 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) or 6-hydroxydopamine (6-OHDA) induced PD models, resveratrol (RV), a natural polyphenolic compound, exerts neuroprotective effects against oxidative damage and neuronal injuries through its antioxidant and anti-inflammatory properties [9–11]. In addition to its antioxidant and anti-inflammatory actions, recent studies have suggested a neuroprotective role of RV through the activation of sirtuin 1 (SIRT1) and vitagenes, which could be important therapeutic targets for neurodegenerative disorders [12–14]. Several studies have indicated that RV induces autophagy by activating SIRT1 and thus protects neurons against neurodegenerative disorders [15–17]. Microtubule-associated protein 1 light chain 3 (LC3) is a key autophagy regulator that is distributed abundantly in the nucleus. Deacetylation of LC3 allows it to interact with the nuclear protein diabetes and obesity-regulated nuclear factor, which redistributes LC3 from the nucleus to the cytoplasm, where soluble LC3 (LC3-I) converts to the membrane-bound form (also known as LC3-II). LC3-II is then recruited to autophagosomal membranes, and plays a central role in autophagy [18]. Our previous study found that RV facilitated α-synuclein clearance and increased the levels of LC3-II in PC12 cells, indicating that neuroprotective effects of RV might be related to autophagic degradation of α-synuclein [6]. SIRT1 is an NAD+-dependent deacetylase enzyme [14]; therefore, deacetylation of LC3 by SIRT1 might be involved in neuroprotective effects of RV in PD. The present study aimed to clarify the importance of deacetylation of LC3 by SIRT1 and its downstream effects on autopahgic degradation of α-synuclein in the MPTP-induced mouse model of PD.
2. Materials and methods
2.1. Animal preparation
Male C57BL/6 mice (10–12 week, 24–28 g) were purchased from the Shanghai SLAC Laboratory Animal Company (Shanghai, China), with the permission number SCXK 2012–0002. All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publication no. 85–23, revised 1996). All animal care and experimental procedures were approved by the ethics committee for the use of experimental animals in Shanghai Jiao Tong University (Approval ID: SYXK2013–0052). Every attempt was made to limit animal numbers and their suffering. All mice were housed at 20–25°C and had free access to food and water during a 12h light/dark cycle in the Laboratory Animal Center of Shanghai Jiao Tong University.
2.2. Experimental design and treatment
Following acclimatization, the animals were randomized into four groups (Table 1), (n = 12). As shown in Fig. 1, MPTP (30 mg/kg/day free base) was injected intraperitoneally (i.p.) for 5 consecutive days. RV (100 mg/kg/day) was administrated by intragastric gavage (i.g.) for 33 days and was given 2 h before intraperitoneal injection of MPTP between the 8th and 12th day. EX527 (10 mg/kg/day), a specific SIRT1 inhibitor, was injected intraperitoneally once a day for 33 days at the same time as RV.
Table 1.
MPTP and drug treatments of mice groups
| Group | Treatment |
|---|---|
| CON | 1% DMSO/30% PEG-400/1% Tween 80 (i.p.) + 0.5% CMC (i.g.) + NS (i.p.) |
| MPTP | 1% DMSO/30% PEG-400/1% Tween 80 (i.p.) + 0.5% CMC (i.g.) + MPTP 30 mg/kg (i.p.) |
| RV + MPTP | 1% DMSO/30% PEG-400/1% Tween 80 (i.p.) + RV 100mg/kg (i.g.) + MPTP 30 mg/kg (i.p.) |
| EX527 + RV + MPTP | EX527 10 mg/kg (i.p.) + RV 100mg/kg (i.g.) + MPTP 30mg/kg (i.p.) |
CMC, carboxymethyl cellulose.
Figure 1.
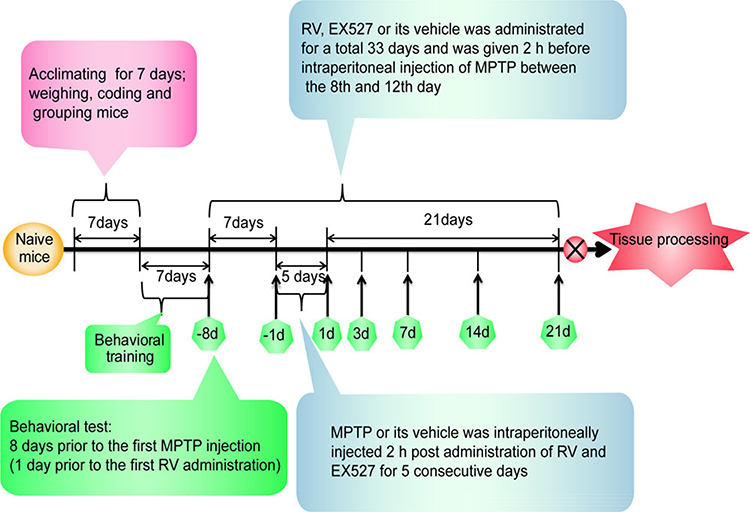
Experimental timeline. A timeline detailing the acclimatization, coding, grouping, behavioral training, and time points for behavioral tests as well as drug treatments and tissue processing.
All animals were pretrained for each behavioral test until a ceiling performance was reached. Behavioral tests were conducted 8 days prior to the first MPTP injection (1 day prior to the first RV administration), 1 day prior to the first MPTP injection and at 1st day, 3rd day, 7th day, 14th day, 21st day post the last MPTP injection. All the animals were sacrificed at the 22nd day post the last MPTP injection.
MPTP hydrochloride (M0896, Sigma) was dissolved in normal saline, RV (R5010, Sigma) was suspended in 0.5% carboxymethyl cellulose, and EX527 (2780, Tocris Bioscience) was reconstituted in 1% DMSO/30% PEG-400/1% Tween 80. The optimal dosage and timing of MPTP, RV and EX527 treatments employed in the current study were chosen on the basis of previous studies [19–23] and our pilot experiments.
2.3. Behavioral tests
2.3.1. Open field test
Each mouse was placed in a wooden locomotor activity chamber (open field: 80 × 80 × 28.5 cm). A camera mounted above the chamber connected to an automated video tracking system (Shanghai Mobile datum Information Technology Co. Ltd.) was used to monitor the activity of mice. Each session lasted for 5 min. Total distance traveled and the distance travelled in the central part were recorded. The number of rearing was manually counted by a blinded observer.
2.3.2. Stride length test
The stride length test was performed according to previously published methods [24], with a minor modification. The stride lengths of both limbs were measured manually as the distance between two paw prints. The three longest stride lengths were measured from each run. Runs in which the observer observed mice making stops or obvious decelerations were excluded from the analysis. The observer who monitored the behavior of the mice was blinded to the treatment groups.
2.3.3. Pole test
The pole test was performed according to previously published methods [25]. Briefly, the pole test required a vertical pole, 55 cm in height and 10 mm in diameter, with a rough surface that stood in the home cage. Mice were placed near the top of the pole, with their heads up, and the time to turn and climb down were recorded.
2.4. Perfusion and tissue processing
Four animals in each group were anesthetized using chloral hydrate and perfused intracardially. Mouse brains were removed, postfixed in 4% paraformaldehyde at 4°C for 24 h and immersed sequentially in 20 and 30% sucrose solution at 4°C until sinking. The brains were then cut serially into coronal sections at a thickness of 20 μm on a Cryostat Microtome (Thermo Scientific Microm HM 525) and stored at –20°C in a cryoprotectant solution (30% glycerin and 30% ethylene glycol in 0.1 M PBS). Another eight animals in each group were sacrificed by cervical dislocation. The tissues of the striatum and substantia nigra (SN) were microdissected rapidly on ice [21], frozen in liquid nitrogen and stored at –80°C.
2.5. HPLC determination of dopamine and its metabolites in mice
The frozen mouse striatum was sonicated in ice-cold 0.2 M perchloric acid containing 10 mM EDTA. After being placed on ice for 15 min, the homogenates were centrifuged at 15 000 × g for 15 min at 4°C. The supernatants were used to determine monoamine concentrations. The levels of dopamine (DA), dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) were determined using HPLC with an electrochemical detector (Coulochem III, ESA, USA) equipped with a model 5041 analytical cell and a ESA 584 HPLC pump (ESA Biosciences Inc. USA). The column (Eclipse Plus C18, 3.5 μm, 2.1 × 150 mm, Agilent, USA) was kept at 35°C using a column heater. The mobile phase used comprised 100 mM NaH2PO4, 50 mM citric acid buffer, 200mg/Lsodium1-octanesulfonate, 8% methanol, and 50 μM EDTA, adjusted to pH 3.0 with sodium hydroxide. The flow rate of the mobile phase was 0.2 mL/min. The supernatant of the striatal tissue (20 μL) was injected directly into the column.
2.6. Immunoblotting and immunoprecipitation
Dissected mouse brain tissues were lysed in RIPA buffer and the protein homogenates were then separated by SDS-PAGE and transferred onto a polyvinylidene fluoride membrane. After blocking, the membranes were incubated overnight with primary antibodies, followed by washing and incubation with horseradish peroxidase conjugated secondary antibody for 1 h. Immunoreactive bands were then visualized using the ECL reagent (WBKLS0100, Merck Millipore). The densities of the immunoblotting bands were semiquantified using Image J software (National Institutes of Health). The primary antibodies used were as follows: anti-tyrosine hydroxylase (TH) (#T2928, Sigma), anti-α-synuclein (#ab1903, Abcam), anti-SIRT1 (#07–131, Millipore), anti-p62 (#18420–1-AP, Protein Tech Group), anti-LC3B (#18725–1-AP, Protein Tech Group), anti-acetyl-Lysine (#05–515, Millipore), anti-caspase-3 (#9662, CST), and anti-GAPDH (#10494–1-AP, Protein Tech Group).
For immunoprecipitation, extracted proteins were incubated with anti-LC3B (#3868, CST) at 4°C overnight with rotation, followed by precipitation with protein A agarose beads (P2012, Beyotime). Immunoprecipitates were then washed and subjected to SDS-PAGE separation. Subsequent immunoblotting procedures were the same as described above.
2.7. Immunofluorescence staining
Immunofluorescent analyses were performed based on the previous studies [26]. Brain sections were permeabilized and blocked for 1 h, and then incubated with the following primary antibodies overnight at 4°C: anti-TH, anti-α-synuclein, anti-SIRT1, anti-p62, and anti-LC3B. The sections were then incubated with anti-mouse IgG-Alexa Fluor 594 (A11032, Invitrogen) and anti-rabbit IgG-Alexa Fluor 488 (A11034, Invitrogen) for 1 h at 37°C. Thereafter, the sections were stained with 4’,6-diamidino-2-phenylindole (DAPI) for 5 min. Images were captured on an Olympus BX51 microscope with a DP-72 CCD camera. The number of TH positive cells in the SN was counted manually by a blinded investigator. The cell numbers in the SN were determined in three anatomically matched sections per animal (n = 4) according to previous studies [26–28]. For quantification of the immunoreactivities of SIRT1, p62, α-synuclein, LC3 in the cytoplasm, and LC3 in the nucleus, the mean intensity of fluorescence was measured using Image J software as described previously [29].
2.8. Statistical analysis
Data were analyzed using SPSS software (version 11.5, IBM Corp., New York, NY, USA). All values were expressed as means ± standard error. Statistical analysis of group differences was assessed by one-way analysis of variance followed by multiple comparisons using the least significant difference post hoc test. p < 0.05 was considered significant.
3. Results
3.1. RV improved the behavioral impairments of MPTP-treated mice
We studied the locomotor activity in MPTP-treated mice, and we found that mice in MPTP-treated group displayed a significant reduction in total distance, center distance, and the number of rearing at the 1st, 3rd, 7th, 14th, and 21st day post the last MPTP injection. Administration of RV significantly rescued these reductions in locomotor activities in MPTP-treated mice especially at 14th and 21st days. Treatment with EX527, a specific inhibitor of SIRT1, further reversed the beneficial effects of RV on locomotor activities (Fig. 2).
Figure 2.
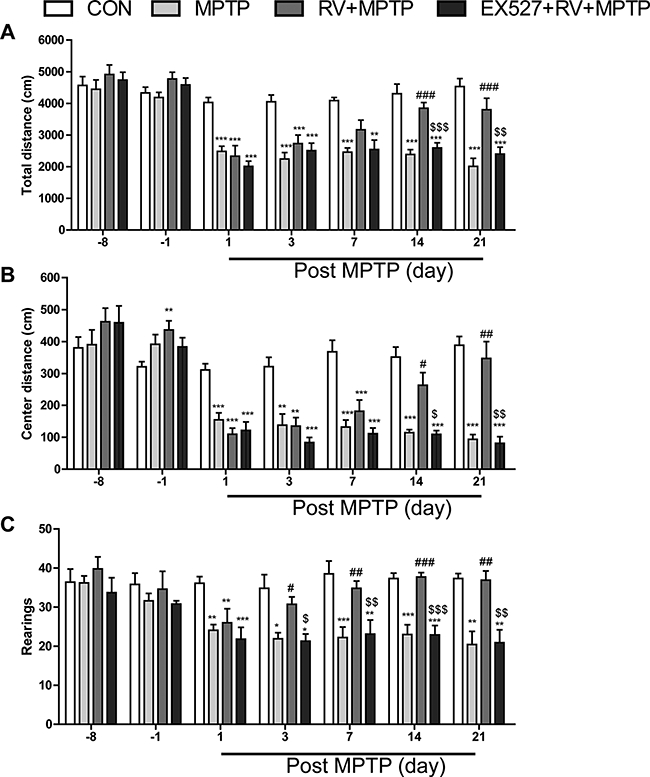
RV improved total distance (A), center distance (B), and rearings (C) of MPTP-treated mice in the open field test. Treatment with EX527, a specific inhibitor of SIRT1, significantly reversed the effect of RV. Data are presented as means ± standard error (n = 12). *p < 0.05, **p < 0.01, ***p < 0.001, compared with the CON group; #p < 0.05, ##p < 0.01, ###p < 0.001, compared with the MPTP group; $p < 0.05, $$p < 0.01, $$$p < 0.001, compared with the RV + MPTP group.
We studied gait abnormalities by measuring the stride length. As shown in Fig. 3, MPTP-treated mice had induced a significant decrease in forelimb and hindlimb stride lengths at the 1st, 3rd, 7th, 14th and 21st day post the last MPTP injection. After administration of RV, the stride lengths of the mice in the RV + MPTP group increased significantly compared with those in the MPTP group; furthermore, inhibition of SIRT1 with EX527 significantly abolished the effect of RV on the stride length in MPTP-treated mice.
Figure 3.
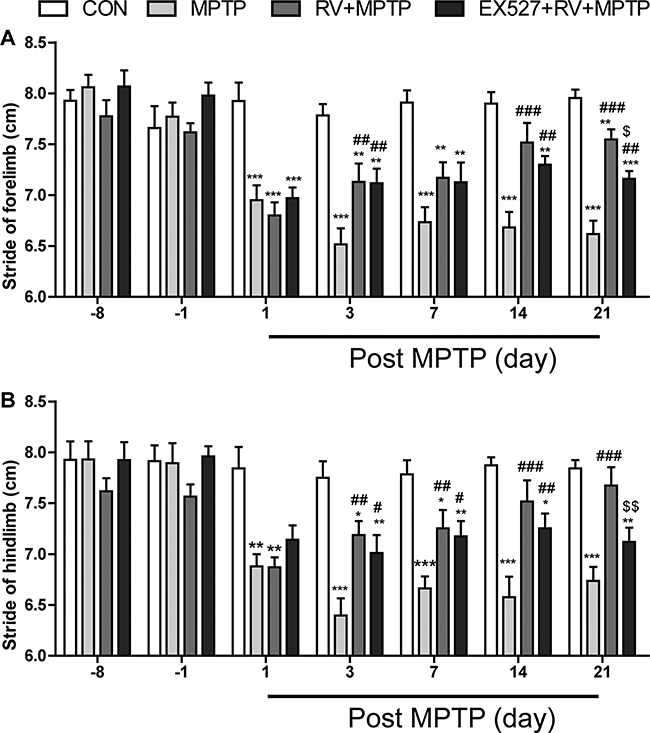
RV improved gait abnormalities of MPTP-treated mice in the stride length test. (A) Stride of forelimb; (B) stride of hindlimb. Inhibition of SIRT1 with EX527 significantly abolished the effect of RV. Data are presented as means ± standard error (n = 12). *p < 0.05, **p < 0.01, ***p < 0.001, compared with the CON group;#p < 0.05,##p < 0.01,###p < 0.001, compared with the MPTP group; $p < 0.05, $$p < 0.01, compared with the RV + MPTP group.
We further evaluated MPTP-induced bradykinesia and coordination in mice by the pole test. We discovered that the return time and the total time were both significantly prolonged after MPTP treatment, especially at the 21st day post the last MPTP injection. Administration of RV significantly reversed the MPTP-induced prolongation of return time and total time in the pole test. Treatment with EX527 effectively counteracted the effect of RV, even though the inhibitory effect of EX527 was only significant at 7th day for the total time, probably because of the great individual differences between animals (Fig. 4). Taken together, our results suggested that RV improved the behavioral impairments induced by MPTP, and this effect of RV was SIRT1 dependent.
Figure 4.
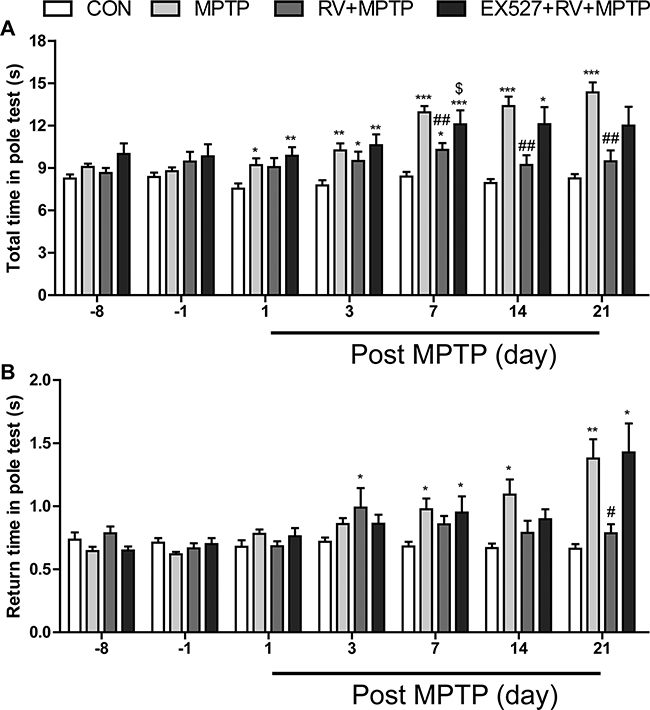
RV improved MPTP-induced bradykinesia and coordination in the pole test. (A) Total time; (B) return time. Inhibition of SIRT1 with EX527 effectively counteracted the effect of RV. Data are presented as means ± standard error (n = 12). *p < 0.05, **p < 0.01, ***p < 0.001, compared with the CON group; #p < 0.05, ##p < 0.01, compared with the MPTP group; $p < 0.05, compared with the RV + MPTP group.
3.2. RV attenuated MPTP-induced depletion of DA in the striatum
HPLC was used to investigate whether RV affected the levels of neurotransmitters. MPTP administration significantly reduced the levels of DA, DOPAC, and HVA (Fig. 5). The reduction of DA induced by MPTP was significantly attenuated by administration of RV. Treatment with EX527 counteracted the effect of RV-induced increase of DA significantly. There were no significant changes in DOPAC and HVA levels in mice treated with RV. These results suggested that RV might affect the synthesis of DA, but has no effect on the catabolism of DA.
Figure 5.
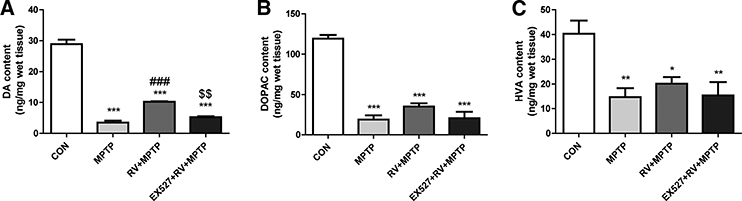
RV attenuated MPTP-induced depletion of DA in the striatum. The levels of (A) DA, (B) DOPAC, and (C) HVA were calculated and expressed as nanogram per milligram wet tissue weight. Treatment with EX527 significantly counteracted the effect of RV on the DA level. Data are presented as means ± standard error (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, compared with the CON group; ###p < 0.001, compared with the MPTP group; $$p < 0.01, compared with the RV + MPTP group.
3.3. RV rescued the loss of nigral TH positive neurons and the decrease of striatal TH protein induced by MPTP
First, we observed the loss of dopaminergic neurons in MPTP-treated mice using immunofluorescence staining for TH-positive neurons. As shown in Fig. 6A, MPTP treatment reduced the number of nigral TH positive neurons remarkably (Fig. 6B). Treatment with RV significantly increased the number of nigral TH positive neurons compared with the MPTP group. By contrast, EX527 treatment reversed the effect of RV in the EX527 + RV + MPTP group compared with the RV + MPTP group.
Figure 6.
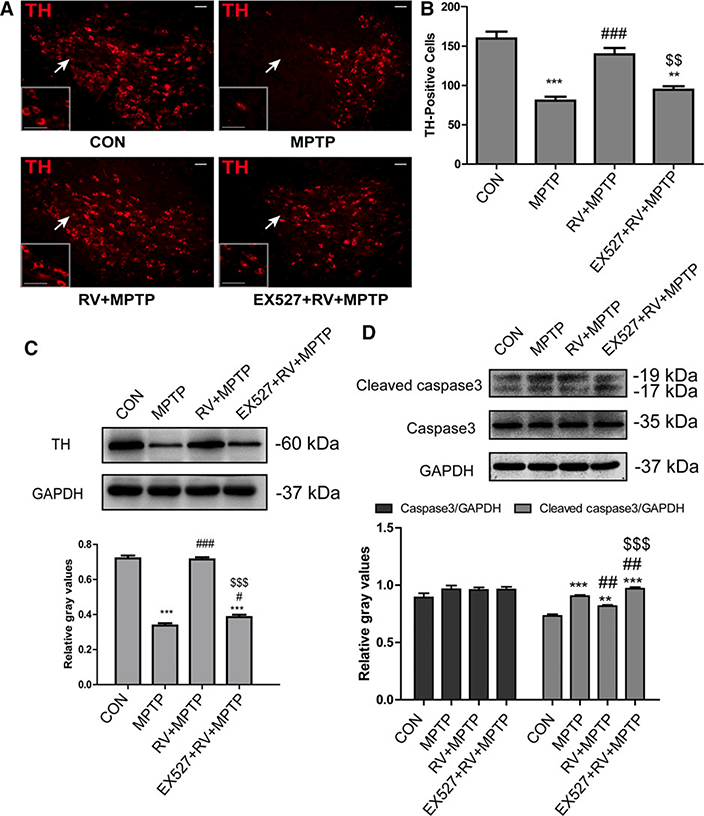
RV rescued the loss of nigral TH positive neurons and the decrease of striatal TH protein induced by MPTP Inhibition of SIRT1 with EX527 significantly reversed the effect of RV. (A) Representative microphotographs of dopaminergic neurons stained for TH in the SN. The white arrows indicate the areas showing the most differentiation and higher magnifications of the areas are shown in the rectangle. Scale bars: 50 pm. (B) Statistical results for the relative number of TH-positive neurons in the SN. Values were presented as means ± standard error (n = 4). (C) Western blotting analysis and quantification of relative TH protein abundance in the striatum. (D) Western blotting analysis and quantification of relative caspase 3 and cleaved caspase 3 protein abundance in the striatum. GAPDH protein served as the internal control. Values are presented as means ± standard error (n = 3). **p < 0.01, ***p < 0.001, compared with the CON group; #p < 0.05, ##p < 0.01, ###p < 0.001, compared with the MPTP group; $ $p < 0.01, $ $ $p < 0.001, compared with the RV + MPTP group.
Second, we examined the abundance of the striatal TH protein by Western blotting to determine whether DA terminals were also affected by RV (Fig. 6C). Similar to results above, after treatment with MPTP, the abundance of the striatal TH protein decreased significantly, while in the striatum of mice in the RV + MPTP group, the abundance of TH protein increased significantly compared with the MPTP-treated mice. EX527 treatment significantly reversed the effect of RV on the MPTP-induced decrease in the abundance of the striatal TH protein.
In addition, Western blotting analysis of caspase 3 and cleaved caspase 3 in the striatum (Fig. 6D) showed that MPTP treatment increased the level of cleaved caspase 3 significantly, whereas RV significantly suppressed the increase in cleaved caspase 3. Inhibition of SIRT1 by EX527 abolished the suppressive effect of RV on the MPTP-induced increase in the level of cleaved caspase 3.
3.4. RV-mediated neuroprotection was SIRT1 dependent
To further determine RV-mediated neuroprotection is SIRT1 dependent, double immunofluorescence staining of TH (red) and SIRT1 (green) in the SN was performed. As shown in Fig. 7A, the loss of TH-positive neurons in SN and the decrease in SIRT1 in the SN appeared to occur simultaneously. RV activated SIRT1, and rescued the loss of TH-positive neurons induced by MPTP, whereas inhibition of SIRT1 by EX527 abolished the rescue effect of RV on MPTP-induced loss of TH-positive neurons. Further quantitative analysis of the immunoreactivity of SIRT1 in the SN showed a similar tendency to the relative number of TH-positive neurons in the SN (Figs. 6B and 7B).
Figure 7.
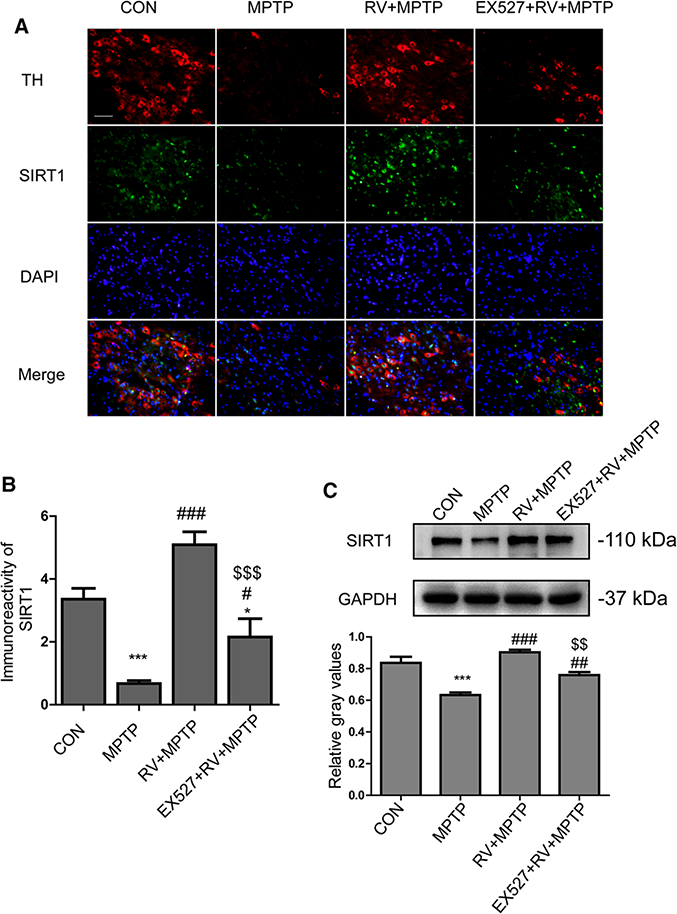
RV-mediated neuroprotection was SIRT1 dependent. As well as activating SIRT1, RV rescued the loss of TH-positive neurons induced by MPTP, whereas inhibition of SIRT1 with EX527 abolished the effect of RV. (A) Representative double immunofluorescence images of TH (red) and SIRT1 (green) in the SN. Nuclei were revealed by DAPI staining (blue), with the merged images depicted at the bottom. Scale bars: 100 μm. (B) Quantification of immunoreactivity of SIRT1 in SN. (C) Western blotting analysis and quantification of relative SIRT1 protein abundance in the striatum. GAPDH protein served as the internal control. Values are presented as means ± standard error (n = 3). *p < 0.05, ***p < 0.001, compared with the CON group; #p < 0.05, ##p < 0.01, ###p < 0.001, compared with the MPTP group; $ $p < 0.01, $ $ $p < 0.001, compared with the RV + MPTP group.
Meanwhile, we detected the protein level of SIRT1 in the striatum (Fig. 7C). Similar to the results shown for the SN, the level of striatal SIRT1 was reduced significantly in the mice of MPTP group. Administration of RV rescued the MPTP-induced decrease of striatal SIRT1 significantly. Inhibition of SIRT1 with EX527 counteracted the effect of RV. The results showed the similar tendency to the level of striatal TH protein (Fig. 6C); i.e., the decrease in the levels of TH and SIRT1 protein in the striatum happened simultaneously. These results suggested that RV-mediated neuroprotection was SIRT1 dependent.
3.5. Induction of autophagy by SIRT1 enhanced the degradation of a-synuclein
We investigated whether SIRT1 could induce autophagy, thereby enhancing the degradation of α-synuclein. As shown in Fig. 8A, activation of SIRT1 by RV significantly increased the level of LC3-II compared with the MPTP group whereas inhibition of SIRT1 by EX527 reversed the effect of RV on LC3-II abundance. Meanwhile, activation of SIRT1 by RV decreased the level of p62 and α-synuclein significantly compared with the MPTP group. Inhibition of SIRT1 by EX527 abolished the suppressive effect of RV on p62 and α-synuclein expression.
Figure 8.
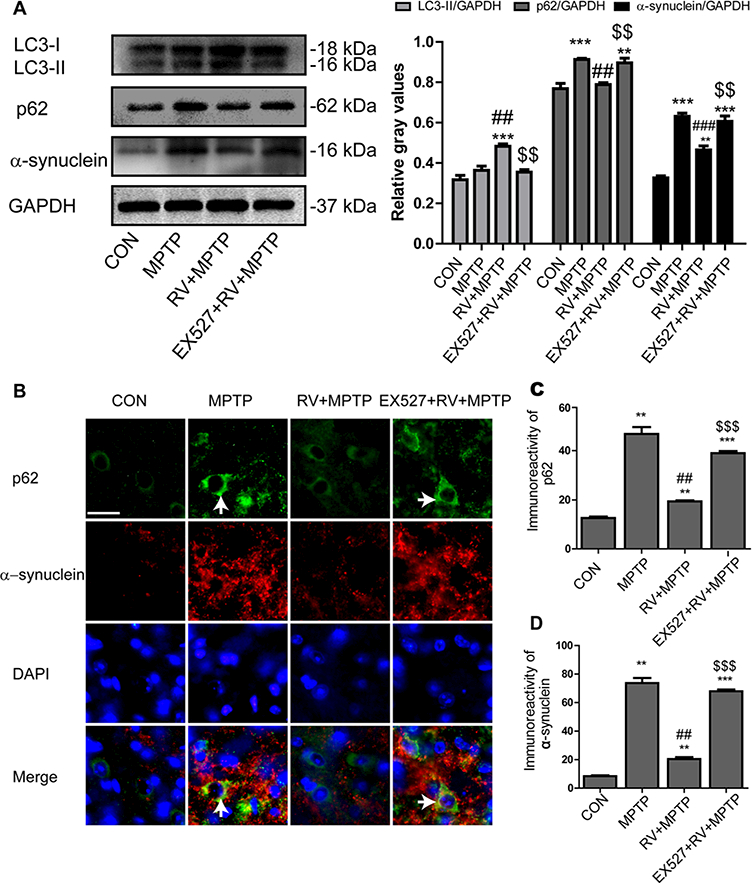
Induction of autophagy by SIRT1 enhanced degradation of α-synuclein. (A) Western blotting analysis and quantification of relative LC3, p62, and α-synuclein protein abundance in the striatum. GAPDH protein served as the internal control. (B) Representative double immunofluorescence images of α-synuclein (red) and p62 (green) in the SN. Nuclei were revealed by DAPI staining (blue), with the merged images depicted at the bottom. Arrows indicate α-synuclein and p62 coaccumulation (yellow) in the cytoplasm of degenerating dopaminergic neurons. Scale bars: 20 μm. (C) Quantification of the immunoreactivity of p62 in dopaminergic neurons. (D) Quantification of the immunoreactivity of α-synuclein in dopaminergic neurons. Values are presented as means ± standard error (n = 3). **p < 0.01, ***p < 0.001, compared with the CON group; ##p < 0.01, ###p < 0.001, compared with the MPTP group; $$p < 0.01, $$$p < 0.001, compared with the RV + MPTP group.
To further investigate the effect of SIRT1 on autophagy and the degradation of α-synuclein, double immunofluorescence staining of α-synuclein (red) and p62 (green) in the SN was conducted. Merged images showed α-synuclein and p62 coaccumulation (yellow) in the cytoplasm of degenerating dopaminergic neurons in the mice of the MPTP group. Together with SIRT1 activation, RV reduced the coaccumulation of α-synuclein and p62 induced by MPTP obviously. Treatment with EX527 caused a resumption of α-synuclein and p62 coaccumulation (yellow) in the cytoplasm of degenerating dopaminergic neurons again (Fig. 8B). Quantitative analysis of the immunoreactivity of p62 and α-synuclein in dopaminergic neurons showed a similar tendency to the Western blotting analysis of p62 and α-synuclein in the striatum (Fig. 8C and D). These results suggested that SIRT1 could induce autophagy, as shown by the increases in the LC3-II level and in p62 degradation, accompanied by enhanced degradation of α-synuclein.
3.6. Deacetylation of LC3 by SIRT1 was involved in autophagic degradation of α-synuclein
We performed coimmunoprecipitation assays for the analysis of the acetylation levels of LC3. The results showed that acetylation of LC3 was markedly increased in the MPTP group. RV reversed LC3 acetylation significantly. When mice were treated with the specific SIRT1 inhibitor EX527, the level of acetylated LC3 in the mice of the EX527 + RV + MPTP group showed no obvious difference compared with the MPTP group, indicating that SIRT1 is required for deacetylation of LC3 (Fig. 9A).
Figure 9.
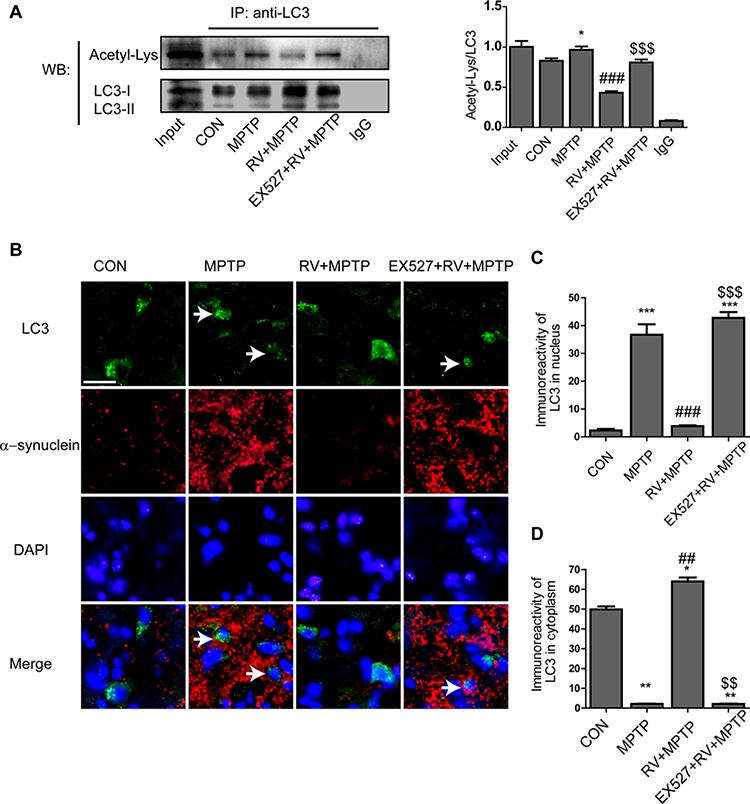
Deacetylation of LC3 by SIRT1 was involved in the autophagic degradation of α-synuclein. (A) The acetylation level of LC3 in the striatum. (B) Representative double immunofluorescence images of α-synuclein (red) and LC3 (green) in the SN. Nuclei were revealed by DAPI staining (blue), with the merged images depicted at the bottom. Arrows indicate that the nuclear to cytoplasmic redistribution of LC3 was prevented, with α-synuclein accumulation in the cytoplasm of degenerating dopaminergic neurons. Scale bars: 20 μm. (C) Quantification of the immunoreactivity of LC3 in the nuclei of dopaminergic neurons. (D) Quantification of the immunoreactivity of LC3 in the cytoplasm of dopaminergic neurons. Values are presented as means ± standard error (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, compared with the CON group; ##p < 0.01, ###p < 0.001, compared with the MPTP group; $ $p < 0.01, $ $ $p < 0.001, compared with the RV + MPTP group.
To further explore whether deacetylation of LC3 by SIRT1 is involved in the autophagic degradation of α-synuclein, double immunofluorescence staining of α-synuclein (red) and LC3 (green) in the SN was conducted. The results showed that treatment with RV caused LC3 to redistribute from the nucleus to the cytoplasm, which promoted the autophagic degradation of α-synuclein in dopaminergic neurons. By contrast, treatment with EX527, a specific SIRT1 inhibitor, blocked the decrease of acetylated LC3, prevented the nuclear to cytoplasmic redistribution of LC3, resulting in α-synuclein accumulation in the cytoplasm of dopaminergic neurons (Fig. 9B). The quantitative analysis of the immunoreactivity of LC3 in the nucleus and cytoplasm of dopaminergic neurons was shown in Fig. 9C andD. These results together with the results shown in Fig. 8 suggested that deacetylation of LC3 by SIRT1 promotes the autophagic degradation of α-synuclein.
4. Discussion
In this study, we demonstrated that RV attenuated MPTP-induced dopaminergic neurodegeneration and behavioral impairments. In addition, we showed that the protective effects of RV were mediated by activation of SIRT1. The subsequent deacetylation of LC3 by SIRT1 mediated autophagic degradation of α-synuclein. Inhibition of SIRT1 with EX527 antagonized the neuroprotective effects of RV on MPTP-induced motor deficits and pathological changes by blocking LC3 deacetylation and the subsequent autophagic degradation of α-synuclein.
RV, a natural polyphenolic compound, is present in a wide variety of plants including vegetables, fruits, and grains. It has been shown to have protective effects against a number of cardiovascular and neurodegenerative diseases [9,19,20,30–33]. In the PD model, RV administration protects mice from MPTP-induced motor coordination impairment, hydroxyl radical overloading, and neuronal loss [9]. Similarly, RV has also been demonstrated to be beneficial to the 6-OHDA-induced PD rat models [10,11,34]. Notably, in a study using gerbils, RV reached a concentration peak in the brain 4 h after intraperitoneal administration [35]. The capacity of RV to cross the blood-brain barrier in animal models suggested the neuroprotective potential of systemically administered RV in the brain. Despite its poor solubility, our study demonstrated that intragastric administration of RV, before and after MPTP-induced neurotoxicity, ameliorated the behavioral impairments, reduced the loss of dopaminergic neurons, and increased the levels of TH and DA. These findings suggest that dietary consumption rich in polyphenols like RV could offer benefits in PD.
The in vivo bioavailability of RV has been studied extensively in humans, rats, gerbils, and mice because of the “RV paradox”: i.e., the high bioactivity and the low bioavailability of RV [35–48]. RV is rapidly absorbed in the intestine and metabolized to glucuronide- and sulfate-conjugates in the liver after oral consumption [44]. The enterohepatic cycle of RV exists in human and animals. This rapid metabolism and enterohepatic cycle reduces the concentration of RV in blood [36,37,49]. Johnson et al. [39] analyzed the serum bioavailability of RV and found that oral RV (100 mg/kg body weight) achieved a Cmax of 12 μM at 15 min, an area under the curve of 368 nm/mL min, and the t1/2 for RV was 5.40 h. In addition, although RV is absorbed satisfactorily from the rodent gastrointestinal tract and presents undeniable activity, the peak plasma levels of unmetabolized RV were very low (below 10 μM), even after a high oral dose of 50 mg/kg. Elimination of RV is rather rapid [41]. Being a lipophilic molecule, RV has been demonstrated to be able to cross the blood-brain barrier and is maintained at an effective level in the brain to exert its biological function. Radioactivity of 14C-trans-RV was found in mouse brain after oral administration (5 mg/kg), suggesting that 14C-trans-RV are able to enter brain tissues and go into effect [46]. Wang et al. analyzed the bioavailability of RV in the brain after i.p. injection (30 mg/kg) in gerbils [35]. They found that the brain concentration of RV reached a peak at 4 h (406.3 ± 21.2 ng/g tissue), followed by a steady decline over time. RV could be detected in the brain up to 4 days later [35]. The rate of RV clearance from the brain was substantially lower than in other organs, as demonstrated by RV concentration at 18 h falling to 43% of the initial measurement in the brain, which was much slower than the clearance from the liver, where only 10% of the initial measurement remained [43]. Moreover, recent pharmacokinetic studies in mice found that the RV was delivered to target tissues in a stable sulfate-conjugated form and that the parent compound was gradually regenerated after repeated ingestion of RV. Thus, these metabolites act as “depots” for RV [40]. The generation of RV via this route might be more important than the unmetabolized form, and could explain the relationship between the low bioavailability and the high bioactivity of RV [40,42]. After daily administration for one week, RV (1.25–25 mg/kg, i.p.) decreased the brain malondialdehyde level dose dependently and increased brain superoxide dismutase, catalase, and peroxidase activities [47]. These findings suggested that the bioavailability of RV is well maintained in the brain and might be responsible for the beneficial effects of using different administration routes in vivo. In the present study, given the bioactivity and bioavailability of RV in the brain, RV was administrated at 100 mg/kg/day by intragastric gavage for 33 days,based on the previous studies [19,20,39,45,47,50,51] and our pilot experiments. We demonstrated that this treatment protocol could effectively protect mice from MPTP-induced behavioral impairments, neuronal loss, and decreased levels of TH and DA. The behavioral experiments also suggested that the neuroprotective effect of RV is time dependent, with more pronounced effects at 21 days. This time-dependent effect of RV might be related to its bioavailability properties; i.e. the accumulation and regeneration of RV in the brain after repeated ingestion.
Herein, we found the RV improved the behavioral impairments of MPTP-treated mice significantly, while the reduction of DA was attenuated but not to the normal levels, remaining much lower compared with the control level. Similar results were reported by Suo et al., who found that pretreatment of trichostatin A fully restored MPTP-induced motor deficits, but only partially improved the reduction in DA [52]. These results are consistent with studies of PD patients and related animal models, which showed that a variety of active compensatory processes serve to delay the onset of neurological symptoms [53]. The intracerebral administration of 6-OHDA induces gross behavioral impairments resembling PD, such as akinesia, decreased nutrient intake and sensory neglect, only in animals with very large depletions of DA. Moreover, these animals often recover from the initial deficits caused by the large DA depletions, as long as a minimal amount of striatal DA remains [54,55]. These results are in agreement with the clinical findings that patients with relatively mild PD symptoms have striatal DA depletions of at least 70–80% [52, 56–58]. Many clinical symptoms appear late in progress of PD: the emergence of neurological or psychiatric symptoms might represent the end stage of the neurodegenerative processes. However, to reverse the clinical symptoms one may not need to reverse the entire neurobiological deficit; instead, clinical recovery might be achieved by a relatively modest restoration of the injured faculties. The natural compensatory processes of the nervous system might be responsible for this recovery, even without restoring the connections that have been lost [53]. Under normal conditions, the field of influence of a dopaminergic terminal is relatively small because of rapid uptake by that terminal and its immediate neighbors. Compensatory events, such as the prolonged action of a transmitter, upregulation of DA receptors, and axonal growth, might occur after degeneration of the DA component of the nigrostriatal bundle to retain postsynaptic function under dopaminergic control. Neurological deficits develop after extreme lesions (>98% DA depletion) when the compensatory responses are inadequate to restore function immediately [53,59]. In the present study, RV treatment might have contributed to improving the capacity of the remaining DA neurons to continue to modulate DAsensitive targets in the striatum to restore neurobiological function.
Numerous studies have demonstrated the beneficial effects of RV function via its antioxidant, anti-inflammatory, and metal-chelating properties [33]. RV administration elicited neuroprotective effects on MPTP-induced Parkinsonian mice via antioxidation, anti-inflammation, and increasing the expression of the suppressor of cytokine signaling-1 and peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α) [9,13,20]. In the 6-OHDA-induced rat model of PD, the protective effects of RV were shown to be related to reducing oxidative stress and increasing the antioxidant defense system [11, 60], or reducing inflammatory reactions, accompanied by decreased mRNA levels of cyclooxygenase-2 and tumor necrosis factor-α [10]. Recently, increasing evidence has indicated that the beneficial effects of RV also associated with activation of SIRT1, a NAD+-dependent protein deacetylase with numerous substrates, including histones, transcription factors, co-factors, and various enzymes [14, 33, 61–63]. Multiple target molecules are involved in the neuroprotective effects of RV via activation of SIRT1 in PD. In the MPTP-induced mouse model of PD, RV restored the cellular antioxidant defense system via SIRT1-dependent activation of PGC-1α, which, in turn, augmented the expression of antioxidant enzymes [13]. RV was also shown to prevent toxicity triggered by 6-OHDA via its ability to activate SIRT1: the inhibition, silencing, or downregulation of SIRT1 diminished the neuroprotective effects of RV [60,64]. In the present study, we observed that systemic treatment with RV activated SIRT1 in the nigra-striatum and significantly improved MPTP-induced behavioral impairments, as well as the pathological changes. These beneficial effects were blocked by administration of the specific SIRT1 inhibitor EX527. The SIRT1-dependent neuroprotective effects were also illustrated in our previous studies in rotenone-treated SH-SY5Y cells [6,30]. Therefore, the ability of RV to activate SIRT1 led to alterations in neuronal transcription profiles and ameliorated the deleterious effects in PD.
Activation of SIRT1 affects the functions of transcription factors, cofactors, and various enzymes. Investigations of these molecules and their related pathways indicated that RV protects against MPTP-induced cell degeneration and mitochondrial-oxidative damage by activating the SIRT1/PGC-1α pathways [13] and inhibiting the p53-caspase-3-dependent apoptotic pathway in PC12 cells by activation of SIRT1 [65] and preserves mitochondrial function via activating adenosine monophosphate activated protein kinase (AMPK), SIRT1, and transcription of PGC-1α target genes [16]. Recent studies revealed that SIRT1 is a potent regulator of autophagic degradation because it interacted with and deacetylates proautophagic proteins such Atg5, Atg7, and Atg8 [66]. In addition, RV reduces the activity of mammalian target of rapamycin, an evolutionarily conserved protein kinase that modulates autophagy, in a SIRT1-dependent manner [67]. Our previous study found that RV increased the clearance of α-synuclein in PC12 cells, which was accompanied by an increase in the level of LC3-II [6]. [18] In the current study, we demonstrated that following the activation of SIRT1 by RV, the abundance of acetylated LC3 decreased, leading to LC3 redistributing from the nucleus into the cytoplasm, which was accompanied by an increase in LC3-II and the autophagic degradation of α-synuclein and p62 in dopaminergic neurons. Inhibition of SIRT1 with EX527 blocked the decrease in acetylated LC3, prevented its nuclear to cytoplasmic redistribution, decreased the level of LC3-II and led to the accumulation of α-synuclein and p62. Consistent with our results, RV was observed recently to promote heme oxygenase-1-dependent LC3-II accumulation in dopaminergic SH-SY5Y cells. In addition, RV affected the phagosome content relevant to LC3-II levels in two primary fibroblast cultures from patients with early onset PD linked to different Park2 mutations [16,17]. Taken together, these results showed that activation of SIRT1, LC3 deacetylation, and subsequent autophagic degradation of α-synuclein are involved in the neuroprotective effects of RV. This hypothesis represents one molecular pathway for RV’s capacity to attenuate tissue injury in PD (Fig. 10).
Figure 10.
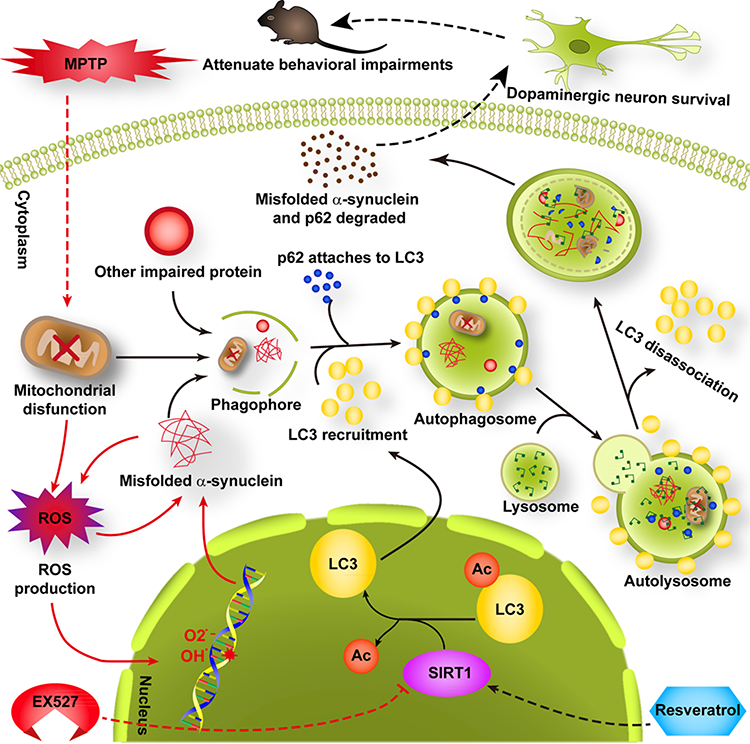
Activated SIRT1-induced LC3 deacetylation and subsequent autophagic degradation of α-synuclein was involved in the neuroprotective effects of RV in the MPTP mouse model of PD. Administration of RV activated SIRT1 (while EX527 inhibited SIRT1), which induced LC3 deacetylation, resulting in LC3 redistributing from the nucleus into the cytoplasm. This promoted the autophagic degradation of α-synuclein, which rescued the MPTP-induced loss of dopaminergic neurons, finally attenuating the behavioral impairments of MPTP-treated mice. Note: the red arrow indicates the action of MPTP or EX527; a black arrow indicates the action of RV.
However, other mechanisms could be involved in the observed RV-mediated neuroprotection, including its antioxidant properties. Environmental toxins, such as MPTP, can induce oxidative stress and generate reactive oxygen species in dopaminergic neurons. Increased reactive oxygen species damages both proteins and mitochondria, leading to α-synuclein aggregates in dopaminergic neurons. The aggregated α-synuclein can cause more mitochondrial damage and worsen the oxidative stress, thus resulting in neurodegeneration [68]. Activation of SIRT1 and subsequent LC3 deacetylation mediated autophagy could remove the initial damaged mitochondria and the aggregated α-synuclein. Meanwhile, damaged mitochondria and aggregated α-synuclein-induced oxidative stress could also be blocked. Thus, activation of SIRT1 and the antioxidant effect are not mutually exclusive in the neuroprotective action of RV.
In conclusion, RV, as a multitarget compound with multiple neuroprotective roles, represents an intriguing potential candidate to treat neurodegenerative diseases including PD. Given the pleiotropic actions and benefits of RV and the identification of multiple related targets of action, it seems likely that RV acts via multiple pathways. In the present study, we showed that RV ameliorated both the motor deficits and pathological changes of PD mice by activating SIRT1, which led to LC3 deacetylation mediated autophagic degradation of α-synuclein. Our study also proposed a mechanism to support RV as a prophylactic or therapeutic agent for PD.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81171205, 81371410, 81471232) and the Biomedical Multidisciplinary Program of Shanghai Jiao Tong University (YG2014MS31).
Abbreviations:
- DA
dopamine
- DOPAC
dihydroxyphenylacetic acid
- HVA
homovanillic acid
- LC3
microtubule-associated protein 1 light chain 3
- MPTP
1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine
- 6-OHDA
6-hydroxydopamine
- PD
Parkinson’s disease
- PGC-1a
peroxisome proliferator activated receptor gamma coactivator 1a
- RV
resveratrol
- SIRT1
sirtuin1
- SN
substantia nigra
- TH
tyrosine hydroxylase
Footnotes
Y.-J. G., W.-J. Z., and Y.-C. W. designed the study; Y.-J. G., S.-Y. D.,X.-X. C, and Y. F. researched and interpreted data; Y.-J. G., W.-J. Z, and Y.-C. W. wrote the manuscript; Y.-J. G., W.-J. Z., S.-H. K, E.-K. T, T. L, M. Y, and Y.-C. W. discussed/edited the manuscript. All authors read and approved the final manuscript.
The authors have declared no conflict of interest.
5 References
- [1].Valera E, Masliah E, Therapeutic approaches in Parkinson’s disease and related disorders. J. Neurochem 2016. doi: 10.1111/jnc.13529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bourdenx M, Bezard E, Dehay B, Lysosomes and alphasynuclein form a dangerous duet leading to neuronal cell death. Front. Neuroanat 2014, 8, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Goedert M, Spillantini MG, Del Tredici K, Braak H, 100 years of Lewy pathology. Nat. Rev. Neurol 2013, 9, 13–24. [DOI] [PubMed] [Google Scholar]
- [4].Lashuel HA, Overk CR, Oueslati A, Masliah E, The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci 2013, 14, 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dehay B, Martinez-Vicente M, Caldwell GA, Caldwell KA et al. , Lysosomal impairment in Parkinson’s disease. Mov Disord. 2013, 28, 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wu Y, Li X, Zhu JX, Xie W et al. , Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neurosignals 2011, 19, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].He Q, Koprich JB, Wang Y, Yu WB et al. , Treatment with trehalose prevents behavioral and neurochemical deficits produced in an AAV alpha-synuclein rat model of Parkinson’s disease. Mol. Neurobiol 2016, 53, 2258–2268. [DOI] [PubMed] [Google Scholar]
- [8].Ghavami S, Shojaei S, Yeganeh B, Ande SR et al. , Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol 2014, 112, 24–49. [DOI] [PubMed] [Google Scholar]
- [9].Lu KT, Ko MC, Chen BY, Huang JC et al. , Neuroprotective effects of resveratrol on MPTP-induced neuron loss mediated by free radical scavenging. J. Agric. Food Chem 2008, 56, 6910–6913. [DOI] [PubMed] [Google Scholar]
- [10].Jin F, Wu Q, Lu YF, Gong QH et al. , Neuroprotective effect of resveratrol on 6-OHDA-induced Parkinson’s disease in rats. Eur. J. Pharmacol 2008, 600, 78–82. [DOI] [PubMed] [Google Scholar]
- [11].Khan MM, Ahmad A, Ishrat T, Khan MB et al. , Resveratrol attenuates 6-hydroxydopamine-induced oxidative damage and dopamine depletion in rat model of Parkinson’s disease. Brain Res. 2010, 1328, 139–151. [DOI] [PubMed] [Google Scholar]
- [12].Jeong JK, Moon MH, Lee YJ, Seol JW et al. , Autophagy induced by the class III histone deacetylase Sirt1 prevents prion peptide neurotoxicity. Neurobiol. Aging 2013, 34, 146–156. [DOI] [PubMed] [Google Scholar]
- [13].Mudo G, Makela J, Di Liberto V, Tselykh TV et al. , Transgenic expression and activation of PGC-1alpha protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cell. Mol. Life Sci 2012, 69, 1153–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Howitz KT, Bitterman KJ, Cohen HY, Lamming DW et al. , Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [DOI] [PubMed] [Google Scholar]
- [15].Vingtdeux V, Giliberto L, Zhao H, Chandakkar P et al. , AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem 2010, 285, 9100–9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ferretta A, Gaballo A, Tanzarella P, Piccoli C et al. , Effect of resveratrol on mitochondrial function: implications in parkin-associated familiar Parkinson’s disease. Biochim. Biophys. Acta 2014, 1842, 902–915. [DOI] [PubMed] [Google Scholar]
- [17].Lin TK, Chen SD, Chuang YC, Lin HY et al. , Resveratrol partially prevents rotenone-induced neurotoxicity in dopaminergic SH-SY5Y cells through induction of heme oxygenase-1 dependent autophagy. Int. J. Mol. Sci 2014, 15, 1625–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Huang R, Xu Y, Wan W, Shou X et al. , Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol. Cell 2015, 57, 456–466. [DOI] [PubMed] [Google Scholar]
- [19].Blanchet J, Longpre F, Bureau G, Morissette M et al. , Resveratrol, a red wine polyphenol, protects dopaminergic neurons in MPTP-treated mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1243–1250. [DOI] [PubMed] [Google Scholar]
- [20].Lofrumento DD, Nicolardi G, Cianciulli A, De Nuccio F et al. , Neuroprotective effects of resveratrol in an MPTP mouse model of Parkinson’s-like disease: possible role of SOCS-1 in reducing pro-inflammatory responses. Innate Immun. 2014, 20, 249–260. [DOI] [PubMed] [Google Scholar]
- [21].Jackson-Lewis V, Przedborski S, Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc 2007, 2, 141–151. [DOI] [PubMed] [Google Scholar]
- [22].Dietrich MO, Antunes C, Geliang G, Liu ZW et al. , Agrp neurons mediate Sirt1’s action on the melanocortin system and energy balance: roles for Sirt1 in neuronal firing and synaptic plasticity. J. Neurosci 2010, 30, 11815–11825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Smith MR, Syed A, Lukacsovich T, Purcell J et al. , A potent and selective Sirtuin 1 inhibitor alleviates pathology in multiple animal and cell models of Huntington’s disease. Hum. Mol. Genet 2014, 23, 2995–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fernagut PO, Diguet E, Labattu B, Tison FA, A simple method to measure stride length as an index of nigrostriatal dysfunction in mice. J. Neurosci. Methods 2002, 113, 123–130. [DOI] [PubMed] [Google Scholar]
- [25].Drucker-Colin R, Garcia-Hernandez F, A new motor test sensitive to aging and dopaminergic function. J. Neurosci. Methods 1991, 39, 153–161. [DOI] [PubMed] [Google Scholar]
- [26].Zhao W, Dumanis SB, Tamboli IY, Rodriguez GA et al. , Human APOE genotype affects intraneuronal Abeta1–42 accumulation in a lentiviral gene transfer model. Hum. Mol. Genet 2014, 23, 1365–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ho YJ, Ho SC, Pawlak CR, Yeh KY, Effects of D-cycloserine on MPTP-induced behavioral and neurological changes: potential for treatment of Parkinson’s disease dementia. Behav. Brain Res 2011, 219, 280–290. [DOI] [PubMed] [Google Scholar]
- [28].Geng X, Tian X, Tu P, Pu X, Neuroprotective effects of echinacoside in the mouse MPTP model of Parkinsonńs disease. Eur. J. Pharmacol 2007, 564, 66–74. [DOI] [PubMed] [Google Scholar]
- [29].Yu Y, Wu Y, Szabo A, Wang S et al. , Teasaponin improves leptin sensitivity in the prefrontal cortex of obese mice. Mol. Nutr. Food Res 2015, 59, 2371–2382. [DOI] [PubMed] [Google Scholar]
- [30].Feng Y, Liu T, Dong SY, Guo YJ et al. , Rotenone affects p53 transcriptional activity and apoptosis via targeting SIRT1 and H3K9 acetylation in SH-SY5Y cells. J. Neurochem 2015, 134, 668–676. [DOI] [PubMed] [Google Scholar]
- [31].Richard T, Pawlus AD, Iglesias ML, Pedrot E et al. , Neuroprotective properties of resveratrol and derivatives. Ann. N. Y.Acad. Sci 2011, 1215, 103–108. [DOI] [PubMed] [Google Scholar]
- [32].Sun AY, Wang Q, Simonyi A, Sun GY, Resveratrol as a therapeutic agent for neurodegenerative diseases. Mol. Neurobiol 2010, 41, 375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Diaz-Gerevini GT, Repossi G, Dain A, Tarres MC et al. , Beneficial action of resveratrol: How and why? Nutrition 2016, 32, 174–178. [DOI] [PubMed] [Google Scholar]
- [34].Wang Y, Xu H, Fu Q, Ma R, Xiang J, Protective effect of resveratrol derived from Polygonum cuspidatum and its liposomal form on nigral cells in parkinsonian rats. J. Neurol. Sci 2011,304, 29–34. [DOI] [PubMed] [Google Scholar]
- [35].Wang Q, Xu J, Rottinghaus GE, Simonyi A et al. , Resveratrol protects against global cerebral ischemic injury in gerbils. Brain. Res 2002, 958, 439–447. [DOI] [PubMed] [Google Scholar]
- [36].Marier JF, Vachon P, Gritsas A, Zhang J et al. , Metabolism and disposition of resveratrol in rats: extent of absorption, glucuronidation, and enterohepatic recirculation evidenced by a linked-rat model. J. Pharmacol. Exp. Ther 2002, 302, 369–373. [DOI] [PubMed] [Google Scholar]
- [37].Walle T, Hsieh F, DeLegge MH, Oatis JE Jr. et al. , High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab. Dispos 2004, 32, 1377–1382. [DOI] [PubMed] [Google Scholar]
- [38].Meng X, Maliakal P, Lu H, Lee MJ et al. , Urinary and plasma levels of resveratrol and quercetin in humans, mice, and rats after ingestion of pure compounds and grape juice. J. Agric. Food Chem 2004, 52, 935–942. [DOI] [PubMed] [Google Scholar]
- [39].Johnson JJ, Nihal M, Siddiqui IA, Scarlett CO et al. , Enhancing the bioavailability of resveratrol by combining it with piperine. Mol. Nutr. Food Res 2011, 55, 1169–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Patel KR, Andreadi C, Britton RG, Horner-Glister E et al. , Sulfate metabolites provide an intracellular pool for resveratrol generation and induce autophagy with senescence. Sci. Transl. Med 2013, 5, 205ra133. [DOI] [PubMed] [Google Scholar]
- [41].Gescher AJ, Steward WP, Relationship between mechanisms, bioavailibility, and preclinical chemopreventive efficacy of resveratrol: a conundrum. Cancer Epidemiol. Biomarkers Prev 2003, 12, 953–957. [PubMed] [Google Scholar]
- [42].Iwuchukwu OF, Sharan S, Canney DJ, Nagar S, Analytical method development for synthesized conjugated metabolites of trans-resveratrol, and application to pharmacokinetic studies. J. Pharm. Biomed. Anal 2012, 63, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Robb EL, Stuart JA, trans-Resveratrol as a neuroprotectant. Molecules 2010, 75, 1196–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Calamini B, Ratia K, Malkowski MG, Cuendet M et al. , Pleiotropic mechanisms facilitated by resveratrol and its metabolites. Biochem. J 2010, 429, 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shu XH, Wang LL, Li H, Song X et al. , Diffusion Efficiency and Bioavailability of Resveratrol Administered to Rat Brain by Different Routes: Therapeutic Implications. Neurotherapeutics 2015, 72, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Vitrac X, Desmouliere A, Brouillaud B, Krisa S et al. , Distribution of [14C]-trans-resveratrol, a cancer chemopreventive polyphenol, in mouse tissues after oral administration. Life Sci. 2003, 72, 2219–2233. [DOI] [PubMed] [Google Scholar]
- [47].Mokni M, Elkahoui S, Limam F, Amri M et al. , Effect of resveratrol on antioxidant enzyme activities in the brain of healthy rat. Neurochem. Res 2007, 32, 981–987. [DOI] [PubMed] [Google Scholar]
- [48].Juan ME, Maijo M, Planas JM, Quantification of transresveratrol and its metabolites in rat plasma and tissues by HPLC. J. Pharm. Biomed. Anal 2010, 57, 391–398. [DOI] [PubMed] [Google Scholar]
- [49].Pangeni R, Sahni JK, Ali J, Sharma S et al. , Resveratrol: review on therapeutic potential and recent advances in drug delivery. Expert Opin. Drug Deliv 2014, 7 7, 1285–1298. [DOI] [PubMed] [Google Scholar]
- [50].Singh N, Agrawal M, Dore S, Neuroprotective properties and mechanisms of resveratrol in in vitro and in vivo experimental cerebral stroke models. ACS Chem. Neurosci 2013, 4, 1151–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Anandhan A, Tamilselvam K, Vijayraja D, Ashokkumar N et al. , Resveratrol attenuates oxidative stress and improves behaviour in 1 -methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) challenged mice. Ann. Neurosci 2010, 77, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Suo H, Wang P, Tong J, Cai L et al. , NRSF is an essential mediator for the neuroprotection of trichostatin A in the MPTP mouse model of Parkinson’s disease. Neuropharmacology 2015, 99, 67–78. [DOI] [PubMed] [Google Scholar]
- [53].Zigmond MJ, Do compensatory processes underlie the preclinical phase of neurodegenerative disease? Insights from an animal model of parkinsonism. Neurobiol. Dis 1997, 4, 247–253. [DOI] [PubMed] [Google Scholar]
- [54].Ungerstedt U, Adipsia and aphagia after 6-hydroxydopamine induced degeneration of the nigrostriatal dopamine system. Acta Physiol. Scand Suppl 1971, 367, 95–122. [DOI] [PubMed] [Google Scholar]
- [55].Zigmond MJ, Stricker EM, Recovery of feeding and drinking by rats after intraventricular 6-hydroxydopamine or lateral hypothalamic lesions. Science 1973, 782, 717–720. [DOI] [PubMed] [Google Scholar]
- [56].Zigmond MJ, Hastings TG, Perez RG, Increased dopamine turnover after partial loss of dopaminergic neurons: compensation or toxicity? Parkinsonism Relat. Disord 2002, 8, 389–393. [DOI] [PubMed] [Google Scholar]
- [57].Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K et al. , Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J. Neurol. Sci 1973, 20, 415–455. [DOI] [PubMed] [Google Scholar]
- [58].Wu Y, Le W, Jankovic J, Preclinical biomarkers of Parkinson disease. Arch. Neurol 2011, 68, 22–30. [DOI] [PubMed] [Google Scholar]
- [59].Zigmond MJ, Berger TW, Grace AA, Stricker EM, Compensatory responses to nigrostriatal bundle injury. Studies with 6-hydroxydopamine in an animal model of parkinsonism. Mol. Chem. Neuropathol 1989, 70, 185–200. [DOI] [PubMed] [Google Scholar]
- [60].Albani D, Polito L, Batelli S, De Mauro S et al. , The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by alpha-synuclein or amyloid-beta (1–42) peptide. J. Neurochem 2009, 770, 1445–1456. [DOI] [PubMed] [Google Scholar]
- [61].Baur JA, Sinclair DA, Therapeutic potential of resveratrol: the in vivo evidence. Nat. Rev. Drug Discov 2006, 5, 493–506. [DOI] [PubMed] [Google Scholar]
- [62].Kaeberlein M, McDonagh T, Heltweg B, Hixon J et al. , Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem 2005, 280, 17038–17045. [DOI] [PubMed] [Google Scholar]
- [63].Zhang Y, Chen ML, Zhou Y, Yi L et al. , Resveratrol improves hepatic steatosis by inducing autophagy through the cAMP signaling pathway. Mol. Nutr. Food Res 2015, 59, 1443–1457. [DOI] [PubMed] [Google Scholar]
- [64].Chao J, Yu MS, Ho YS, Wang M et al. , Dietary oxyresveratrol prevents parkinsonian mimetic 6-hydroxydopamine neurotoxicity. Free. Radic. Biol. Med 2008, 45, 1019–1026. [DOI] [PubMed] [Google Scholar]
- [65].Ye J, Liu Z, Wei J, Lu L et al. , Protective effect of SIRT1 on toxicity of microglial-derived factors induced by LPS to PC12 cells via the p53-caspase-3-dependent apoptotic pathway. Neurosci. Lett 2013, 553, 72–77. [DOI] [PubMed] [Google Scholar]
- [66].Lee IH, Cao L, Mostoslavsky R, Lombard DB et al. , A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. U S A 2008, 705, 3374–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ghosh HS, McBurney M, Robbins PD, SIRT1 negatively regulates the mammalian target of rapamycin. PLoS One 2010, 5, e9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Giordano S, Darley-Usmar V, Zhang J, Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. 2014, 2, 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]