

Abstract
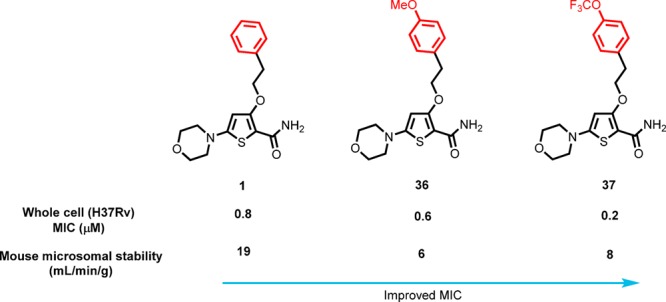
With the emergence of multidrug-resistant strains of Mycobacterium tuberculosis there is a pressing need for new oral drugs with novel mechanisms of action. Herein, we describe the identification of a novel morpholino–thiophenes (MOT) series following phenotypic screening of the Eli Lilly corporate library against M. tuberculosis strain H37Rv. The design, synthesis, and structure–activity relationships of a range of analogues around the confirmed actives are described. Optimized leads with potent whole cell activity against H37Rv, no cytotoxicity flags, and in vivo efficacy in an acute murine model of infection are described. Mode-of-action studies suggest that the novel scaffold targets QcrB, a subunit of the menaquinol cytochrome c oxidoreductase, part of the bc1-aa3-type cytochrome c oxidase complex that is responsible for driving oxygen-dependent respiration.
Introduction
Mycobacterium tuberculosis (M. tuberculosis) is the causative agent of tuberculosis (TB), an airborne transmitted disease that is fatal if not properly treated. TB disproportionately affects developing countries and now outranks HIV as the leading cause of death from an infection worldwide. In 2015, 1.8 million people died of TB (1.4 million HIV-negative and 0.4 million HIV positive).1 The current treatment regimen is considered antiquated and inadequate, taking a minimum of 6 months to cure patients, which contributes to high default rates leading to increased transmission, drug resistance, and even death.2−4 As a consequence, new TB drugs working through existing and novel mechanisms are urgently required.5,6 Although there has been increased investment in TB drug discovery over the past 10 years this has remained a significant challenge, and high-quality novel chemical starting points are still urgently required.7
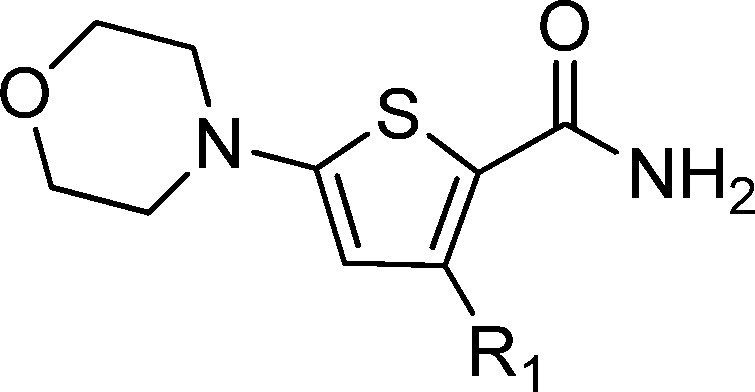
Identification of TB drug discovery starting points through in vitro target-directed screens has been largely unsuccessful.6 As a result, cell-based phenotypic screening has emerged as a preferred means to identify novel antitubercular scaffolds. In this study a cluster of novel morpholino–thiophenes (MOT) analogues was identified from the Lilly corporate screening deck using an aerobic whole cell phenotypic screen of M. tuberculosis strain H37Rv. The initial hit had good antitubercular activity with a minimum inhibitory concentration (MIC) of 0.72 ± 0.30 (n = 14). Resynthesis and profiling of hits from the primary screen confirmed antitubercular activity with good potency and selectivity over both VERO and HepG2 cytotoxicity counter screens (see Table 1 for an example). The representative confirmed hit 1 had good M. tuberculosis MIC-derived metrics such as lipophilic ligand efficiency,8 where LLEM. tuberculosis = pMIC – c log P or log D and Property Forecast Index9 where PFI = no. of aromatic rings + log P, suggesting that the series had potential to be optimized into a high-quality oral drug candidate.10,11 Physiochemical properties such as MW, log P, log D, and TPSA were in an acceptable range for oral drugs.11,12 Kinetic solubility suggested 1 was highly soluble, although later additional solubility profiling of analogues found thermodynamic solubility to be an issue. Compound 1 showed moderate CYP inhibition and no in vitro hERG liability. The perceived issue for 1 was metabolic stability as a result of high mouse microsomal clearance, albeit human microsomal clearance was moderate.
Table 1. In Vitro Profile of a Selected MOT Confirmed Hit.
| MIC (H37RvLP) | 0.72 ± 0.30 (n = 14) |
| Vero cytotoxicity | Vero > 100 μM |
| LLE | 4.2 |
| PFI | 4 |
| Measure CHI Log D | 2 |
| Kinetic solubility | >250 μM |
| MW | 332 |
| Clog P/D | 2.6/2.0 |
| TPSA | 65 |
| Microsomal clearance | Mouse = 19 mL·min−1·g−1 |
| Human = 3.5 mL·min−1·g−1 | |
| CYP inhibition (M) | 2D6 (5), 2C9 (5.1), 1A2 (<5), 2C19 (5.9) |
Related thiophene pharmacophores, where the morpholine is replaced by a benzimidazole group, have been investigated for other disease areas. Analogues with no ether chain and a benzimidazole functionality have been reported as inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase.13 Analogues with a benzimidazole functionality and an ether side chain have been reported as polo-like kinase (PLK) and Never in Mitosis Gene A-Related Kinase 2 (Nek2) inhibitors.14−17 The thiophene-2-carboxamide substructure has also been observed within antitubercular inhibitor agents (Figure 1). The structure of the thiophene 2 inhibitor of mycoyltransferase bound to M. tuberculosis FbpC has been solved.18 Recently, TCA1 3 has been reported to target both decaprenylphosphoryl-β-d-ribose oxidase (DprE1) and MoeW and was found to have in vitro bactericidal activity against both replicating and nonreplicating M. tuberculosis as well as being efficacious in both acute and chronic mouse models of TB infection.19
Figure 1.

Antitubercular agents with a thiophene-2-carboxamide substructure.
Herein, we report on the profiling of emerging leads from the MOT series that suggested that they likely target QcrB, a subunit of the menaquinol cytochrome c oxidoreductase (bc1 complex) that is part of the bc1-aa3-type cytochrome c oxidase complex, driving oxygen-dependent respiration. The imidazo[1,2-a]pyridine-3-carboxamides have recently been shown to target QcrB and are currently undergoing clinical development.20−24 The profile and novelty of representative confirmed hit 1 was considered promising, and a medicinal chemistry program was established to derive a better understanding of the pharmacophore based on structure–activity relationship (SAR) studies.
Chemistry
Synthetic Routes
Synthesis of the MOT scaffold was initially achieved using the route outlined in Scheme 1. Amide 5 was formed by reaction of acyl chloride 4 with morpholine and then converted to thioamide 6 following treatment with Lawesson’s reagent. Thioamide 6 was reacted with 2-aminoacetyl bromide to yield hydroxy-thiophene 7. Mitsunobu25 or alkylation reactions yielded the desired thiophene 8. However, a major drawback of this route was that 2-substituted-5-morpholino-3-oxo-2, 3-dihydrothiophene-2-carboxamide 9 was the major product, regardless of whether the Mitsunobu or alkylation reaction was employed to introduce the ether chain.
Scheme 1. General Synthesis of Morpholino–Thiophines.

Reagents and conditions: (i) Morpholine, DCM, 0 °C to rt; (ii) Lawesson’s reagent, 100 °C; (iii) 2-Bromoacetamide, NaH, THF, 0 °C to rt; (iv) DIAD, PPh3, phenylethylalcohol (R1OH), THF, rt.
To circumvent the issues with formation of 9, an alternative route was developed that relied on the early introduction of the ether side chain followed by introduction of the morpholine (Scheme 2). Using methyl 3-hydroxythiophene-2-carboxylate 10 in the Mitsunobu ether formation afforded phenyl ethyl derivative 11. Regioselective iodination with LDA and iodine afforded the versatile iodo-intermediate 12. The ester functional group of 12 was converted to the primary amide 13 by reaction with calcium chloride and ammonia in methanol.26 Suzuki27 or Buchwald28 reactions afforded a range of derivatives in this position (for example, 32, 34, and 35). Hydroxamic acid derivative 16 was prepared from intermediate 12 using a Buchwald-type reaction to form 14, followed by reaction of the ester with a tetrahydropyran protected hydroxylamine to afford 15, which was treated with p-toluenesulfonic acid to afford 16. Treatment of intermediate 11 with LDA and DMF afforded aldehyde 17, which was reacted with a range of amines, such as morpholine to give 18.
Scheme 2. Alternative Synthesis of Morpholino–Thiophines.

Reagents and conditions: (i) DIAD, PPh3, phenylethylalcohol, THF, rt; (ii) Iodine, LDA, THF, −78 °C; (iii) CaCl2, NH3, MeOH, 150 °C, μW; (iv) Suzuki: Boronic acid, Cs2CO3, Pd(PPh3)4, DMF, 80 °C or Buchwald: Amine, Proline, CuI, K3PO4, DMSO, 80 °C, μW; (v) LDA, DMF, THF, −78 °C; (vi) Amine, NaBH(OAc)3, CHCl3, rt; (vii) O-tetrahydropyran-2-yl hydroxylamine, LiHDMS, THF, −78 °C; (viii) p-toluenesulfonic acid monohydrate, MeOH.
Results and Discussion
The initial SAR investigation focused on systematically probing the pharmacophore to understand the impact of each functional group on antibacterial activity. Deletion analogues and nearest neighbors were examined as well as functionality on the phenyl ring with an overall aim of identifying key structural modifications that could be made to improve microsomal stability.
Primary Amide SAR
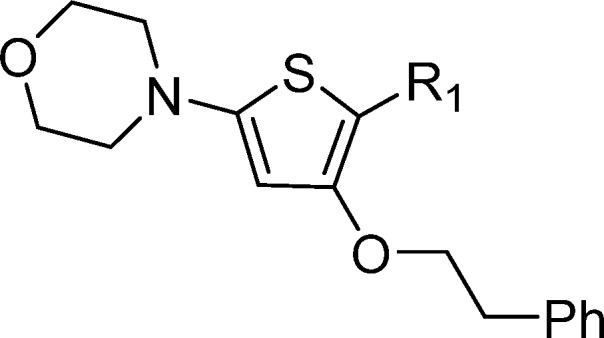
Modifications to the primary amide provided scope to improve MIC potency and mouse microsomal stability (Table 2). Removal of the primary amide 19 or replacement with an ester 14 resulted in a loss of activity, indicating it may be involved in the binding mode of the MOT scaffold with its target or uptake into M. tuberculosis. The methyl ketone 20 was found to be equipotent with 1 but had a negative impact on mouse microsomal clearance. The N-methyl amide 21 showed similar activity but also had a negative impact on the mouse microsomal stability. The ethyl 22 and trifluoroethyl 23 derivatives both led to a drop in potency. There were significant improvements in microsomal stability for the N-cyclobutyl amide 24; however, it came with a loss in antitubercular activity. Encouragingly, both the Weinreb amide 25 and the hydroxamic acid 16 showed improved microsomal stability together with a moderate MIC. A mix and match approach between TCA1 compound 3 was also explored; however, 26 was found to be inactive. Overall, the SAR for the primary amide replacement suggested that the carbonyl is important for antitubercular activity, and microsomal stability can be improved (Table 2).
Table 2. Primary Amide SAR.
| ID | log P/TPSA | R1 | MIC,a μM (n) | VEROb IC50, μM (n) | Cli mouse,c mL.min–1.g–1 |
|---|---|---|---|---|---|
| 1 | 2.6/65 | CONH2 | 0.72 ± 0.30 (14) | >100 (7) | 19 |
| 19 | 3.7/22 | H | >20 (4) | 68 ± 42 (2) | 37 |
| 14 | 3.7/48 | CO2Me | >20 (4) | >100 (4) | 50 |
| 20 | 3.4/39 | COMe | 0.42 ± 0.19 (5) | 84 ± 15 (2) | 41 |
| 21 | 2.8/51 | CONHMe | 0.12 ± 0.052 (5) | 91 ± 16 (3) | 36 |
| 22 | 3.1/51 | CONHEt | 1.7 ± 0.42 (2) | 56 (1) | 50 |
| 23 | 3.6/51 | CONHCH2CF3 | 3.4 ± 0.64 (2) | >100 (1) | 30 |
| 24 | 3.7/51 | CONHcyclobutyl | >20 (2) | 24 (1) | 0.5 |
| 25 | 2.6/51 | CONMe(OMe) | 1.8 ± 1.4 (10) | 78 ± 32 (5) | 0.5 |
| 16 | 2.0/71 | CONHOH | 3.5 ± 1.6 (2) | 58 (1) | 6 |
| 26 | 3.3/77 | CONHCO2Et | >20 (3) | >100 (2) | 1 |
Minimum inhibitory concentration (MIC90) is the minimum concentration required to inhibit the growth of M. tuberculosis in liquid culture. Data are the average ± standard deviation of a minimum of two independent experiments; numbers of experiments is given (n).
Inhibitory concentration (IC50) is the concentration required to inhibit growth of Vero cells by 50%. Data are the average ± standard deviation of a minimum of two independent experiments; numbers of experiments is given (n).
Intrinsic clearance (Cli) using CD1 mouse liver microsomes
Morpholine Side Chain SAR
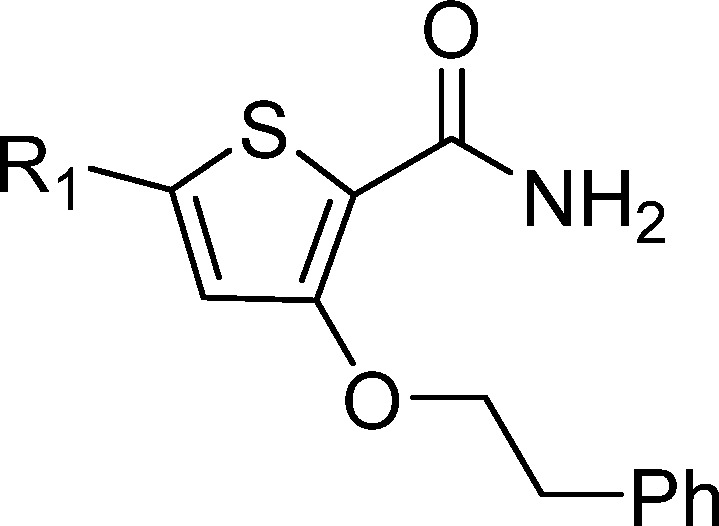
Replacement of the morpholine 1 with hydrogen 27 or piperidine 28 resulted in a loss of activity. The morpholin-3-one 29 showed improved microsomal stability; however, there was a >10-fold drop in potency. Bridged morpholine 30 showed a >5-fold drop in activity, whereas the methyl-substituted morpholine 31 lost activity, significant improvements in microsomal stability were not observed. Increasing the flexibility by opening up the morpholine ring to give 32 resulted in loss of activity. In light of the similarity to reported PLK inhibitors,14,15 heteroaromatic replacements were considered. The PLK-related benzimidazole derivative 33 was inactive; however, the triazine 34 and 4-pyridyl 35 displayed good potency. The microsomal stability was improved by extending the morpholine with a methylene linker 18; however, no antibacterial activity was observed for an array of amines (data not shown). Overall, the SAR for the morpholine substituent suggested that there was limited scope for improving potency and microsomal stability (Table 3).
Table 3. Morpholine Side Chain SAR.

Minimum inhibitory concentration (MIC90) is the minimum concentration required to inhibit the growth of M. tuberculosis in liquid culture. Data are the average ± standard deviation of a minimum of two independent experiments; numbers of experiments is given (n).
Inhibitory concentration (IC50) is the concentration required to inhibit growth of Vero cells by 50%. Data are the average ± standard deviation of a minimum of two independent experiments; numbers of experiments is given (n).
Intrinsic clearance (Cli) using CD1 mouse liver microsomes
Phenyl Ring and Ethyl Ether Linker SAR
The SAR around the phenyl ring and the ethyl linker was examined to see if the mouse microsomal stability could be improved by changing the substituents and physiochemical properties of the phenyl ethyl ether (Table 4). The 4-methoxy 36 was equipotent with 1 and also had improved microsomal stability, presumably as a result of a blocking metabolism on the phenyl ring. The 4-trifluoromethoxyphenyl 37 also afforded improved antibacterial activity and microsomal stability. The o-, m-, and p-substitution patterns of fluoro (38, 39, and 40) and o- and m-methoxy (41 and 42) groups were tolerated but did not significantly improve the overall properties of the molecule, suggesting that p-substitution of the phenyl group is favored. The 4-pyridyl 43 and p-cyanophenyl 44 derivatives lost activity, suggesting that polarity was not tolerated in this region. Saturated polar heterocyclic alkyl analogues were prepared, 45, 46, and 47, while they significantly improved microsomal stability, they lost whole cell activity, supporting the notion that polarity was not tolerated in this region. The phenethyl linker was found to be important for potency; shortening the chain to a methylene linker 48 resulted in loss of activity. Replacement of the O-linker to afford an n-propyl 49 was tolerated but with a drop off in potency. Introduction of a gem-difluoro group 50 as well as hydroxylation 51 on the benzylic position of the ethyl chain, designed in an effort to block benzylic oxidation, resulted in a loss of activity. Constraining the ethyl linker to afford 52 and 53 gave improved microsomal stability but had a detrimental effect on potency. The SAR for the phenyl ring and ethyl linker suggested that there is scope in this region for improving microsomal stability (Table 4).
Table 4. Phenyl Ring and Ethyl Linker SAR.

Minimum inhibitory concentration (MIC90) is the minimum concentration required to inhibit the growth of M. tuberculosis in liquid culture. Data are the average ± standard deviation of a minimum of two independent experiments; numbers of experiments is given (n).
Inhibitory concentration (IC50) is the concentration required to inhibit growth of Vero cells by 50%. Data are the average ± standard deviation of a minimum of two independent experiments; numbers of experiments is given (n).
Intrinsic clearance (Cli) using CD1 mouse liver microsomes
Core Replacement SAR
Phenyl was investigated as a bioisosteric thiophene replacement, employing two substitution patterns (Table 5). The 4-morpholino 54 showed low whole cell activity, whereas the 5-morpholino 55 had moderate whole cell activity as well as improved microsomal stability. The m-fluorophenyl derivative 56 showed similar activity to 55. Additional SAR for the phenyl bioisosteric replacement was explored and is similar to the reported thiophene SAR (data not shown). The pyridyl core bioisoster 57 showed significantly improved microsomal stability with similar potency to 55. Overall, the SAR for the thiophene core and its bioisoster replacements suggested that there is scope in this region for improving microsomal stability while maintaining whole cell activity (Table 5).
Table 5. Core Replacement SAR.

Minimum inhibitory concentration (MIC90) is the minimum concentration required to inhibit the growth of M. tuberculosis in liquid culture. Data are the average ± standard deviation of a minimum of two independent experiments; numbers of experiments is given (n).
Inhibitory concentration (IC50) is the concentration required to inhibit growth of Vero cells by 50%. Data are the average ± standard deviation of a minimum of two independent experiments; numbers of experiments is given (n).
Intrinsic clearance (Cli) using CD1 mouse liver microsomes
Summary of Pharmacokinetic (PK) Studies
Representative compounds 1, 55, 37, 46, and 25 were selected for mouse pharmacokinetic profiling to understand the correlation between in vitro microsomal turnover and in vivo clearance (Table 6). Compounds 55, 37, and 46, with improved in vitro microsomal stability, showed improved Cmax and AUC oral exposures versus 1. However, there was an in vitro microsomal versus in vivo clearance disconnect for 1, 55, 37, and 46 where there is an overprediction of clearance. The phenyl bioisosteric replacement 55 showed improved in vitro clearance as well as in vivo exposure versus 1. However, it had a reduced MIC potency. Similarly, the saturated polar heterocycle 46 showed significant improved bioavailability but no measurable MIC activity. Overall 37 had the best activity and in vivo PK profile with a moderate volume of distribution and bioavailability. Weinreb amide 25, despite having excellent in vitro microsomal stability, showed similar poor Cmax and AUC compared to 1. To better understand the optimization parameters required to improve the oral exposures for compounds with improved in vitro microsomal stability, a rat hepatic portal vein (HPV) PK study was undertaken on lead 37.
Table 6. Oral (10 mg/kg) and IV (3 mg/kg) Pharmacokinetic Profiles in Female C57 Mice for 1, 55, 37, 45, and 25 Based on Mouse Clearance and MIC Potency.
| ID | H37Rv MIC,a ng/mL | in vitro mouseb Clint, mL/min/g | route | Cmax ng/mL | T1/2, h | AUC ng-min/mL | scaled Clpre,c mL/min/kg | CLB, mL/min/kg | Vdss, L/kg | % F |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 130 | 19 | IV | 0.7 | 290 000 | 81 | 10 | 0.5 | 0.5 | |
| PO | 8 | 5300 | ||||||||
| 55 | 1900 | 4 | IV | 1.2 | 95 200 | 68 | 32 | 0.9 | 5 | |
| PO | 280 | 15 000 | ||||||||
| 37 | 83 | 8 | IV | 104 000 | 76 | 29 | 1.7 | 18 | ||
| PO | 430 | 64 000 | ||||||||
| 46 | >6200 | 2.1 | IV | 0.7 | 980 000 | 56 | 31 | 0.8 | 38 | |
| PO | 1700 | 123 000 | ||||||||
| 25 | 640 | <0.5 | PO | 160 | 5200 |
Minimum inhibitory concentration (MIC90) is the minimum concentration required to inhibit the growth of M. tuberculosis in liquid culture. Data are the average ± standard deviation of a minimum of two independent experiments; numbers of experiments is given (n).
Intrinsic clearance (Cli) using CD1 mouse liver microsomes.
Scaled predicted Clb (Clpre) using well stirred model.
Rat HPV PK Study for 37
The fraction of drug absorbed (Fa) from the gastrointestinal tract was low at 21% of the total administered drug, suggesting that the poor oral exposure was largely due to low solubility and/or metabolism in the gut wall during absorption since the permeability was observed to be rapid (see below). A 44% hepatic extraction ratio (ER), determined by measuring blood samples from the HPV (before first pass metabolism) versus the cardiac vein (systemic circulation), suggested that metabolic stability was also an issue, Table 7.
Table 7. HPV Study on 37.
| IV 1 mg/kg | PO HPV 5 mg/kg | PO Cardiac 5 mg/kg | |
|---|---|---|---|
| Cmax (ng/mL) | 200 | 140 | |
| Tmax (h) | 1 | 1 | |
| T1/2 (h) | 0.5 | ||
| AUC 0–24 (ng-min–1mL) | 33 000 | 35 000 | 19 000 |
| Clb | 30 | ||
| Vdss (L/kg) | 1.1 | ||
| F (%) | 12 | ||
| hepatic extraction ratio (ER) | 0.4 | ||
| fraction of dose absorbed (Fa) | 21 | ||
To better define the optimization parameters for improving oral exposure, the permeability and efflux of selected compounds were evaluated in a cell permeability assay. In addition, further solubility screens and metabolite identification studies were performed.
Madin Darby Canine Kidney (MDCK) Permeability Assay
Compounds 1, 37, 18, 46, and 56 were tested in an MDCK assay and found to show good permeability as well as no likely P-glycoprotein (P-gp) efflux issues (Table 8). This was unsurprising considering the favorable MW, log P, low hydrogen bond donor (HBD) count, and TPSA observed for the examples in Tables 1–5. The good permeability data would suggest that solubility-limited absorption was likely to be an underlying optimization issue for the series, not reflected in the kinetic solubility screen. It is known that the electron-deficient bivalent sulfur atoms in thiophenes have low-lying σ* orbitals available for interaction with electron donors such as oxygen. As such an intramolecular interaction between the thiophene sulfur σ* orbital and the carbonyl is likely stabilizing a planar conformation.29 Furthermore, primary amides, in particular aromatic ones, effectively generate two-dimensional intermolecular hydrogen-bonded networks30 that can also have a dramatic effect on the thermodynamic solubility.
Table 8. MDCK Profiling of Selected Compounds.
| ID | MW | log P | TPSA | HBD | A → B Papp 1 × 10e–6 cm/s | B → A Papp 1 × 10e–6 cm/s | P-gp Efflux B→ A/A → B |
|---|---|---|---|---|---|---|---|
| Quinidine | 324 | 2.6 | 45 | 1 | 4 | 89 | 20 |
| Verapamil | 454 | 3.8 | 64 | 0 | 14 | 29 | 2 |
| Atenolol | 266 | 0.2 | 85 | 4 | 1 | 1 | 1 |
| Metoprolol | 267 | 1.8 | 51 | 2 | 55 | 40 | 0.7 |
| 1 | 332 | 2.6 | 65 | 2 | 45 | 37 | 0.8 |
| 37 | 416 | 3.3 | 74 | 2 | 25 | 20 | 0.8 |
| 18 | 346 | 2.2 | 65 | 2 | 62 | 53 | 0.8 |
| 46 | 312 | 1.1 | 74 | 2 | 19 | 35 | 2 |
| 56 | 344 | 2.8 | 65 | 2 | 51 | 37 | 0.7 |
Solubility Screening
Considering the results of the HPV and permeability studies, we selected a number of compounds for additional solubility profiling. Compounds 37 showed poor solubility when tested in a high-throughput solubility assay (HTSA) at a range of pH values, pH 2/6/7.4. The ketone 20 showed improved solubility in the HTSA screens at different pH as well as in the FaSSIF and low pH HCl conditions. The improved solubility for the ketone versus the amide was likely to be as a result of the decreased potential for an intermolecular hydrogen-bonded network. While the methylene-homologated morpholine 18 was not active, it demonstrated improved solubility, in particular at low pH as a result of protonation of the morpholine. The phenyl bioisosteric replacement 55 also showed improved solubility versus 37, presumably as it is devoid of the intramolecular interaction between the thiophene sulfur and carbonyl, Table 9.
Table 9. Measured Solubility Data for Selected Compounds.
| pH 2.4, | pH 6 | pH 2 | pH 6 | pH 7.4 | |
|---|---|---|---|---|---|
| 0.01 N HCl | FaSSIF | HTSA | HTSA | HTSA | |
| ID | mg/mL | mg/mL | mg/mL | mg/mL | mg/mL |
| 20 | 0.4 | 0.2 | 0.6 | 0.03 | 0.03 |
| 18 | 1.7 | 0.04 | 0.7 | 0.1 | 0.3 |
| 55 | 1.3 | 0.3 | 0.6 | 0.3 | 0.3 |
| 37 | ND | ND | 0.002 | 0.001 | 0.001 |
Metabolite Identification
In order to be able to address and modify the metabolically labile moieties in the compound series, the in vivo metabolites of 37 were determined. The main metabolites observed were the products of oxidation of the morpholine, whereas de-ethylation was observed only in small quantities (Supporting Information Table 3).
Efficacy Studies
Compound 37 was profiled in an acute 4 day screening model of TB infection to determine if it had in vivo efficacy (infected with H37Rv).31 The compound was compared to moxifloxacin as the known control agent. The compounds were dosed orally at 100 mg/kg, and 37 showed a small but significant 0.8 log reduction of colony forming unit (CFU) counts in the lungs (Table 10). The in vivo efficacy, coupled with the understanding of the optimization parameters, led us to further investigate the mode of action for the series.
Table 10. Efficacy of 37 against M. tuberculosis in an Acute Murine Model of Intratracheal Infection.
| log
CFUs per mouse (lungs) |
|||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | mean | SD | |
| no treatment (day 9) | 6.8 | 6.8 | 6.5 | 6.9 | 7.0 | 6.8 | 0.2 |
| Moxifloxacin: 30 mg/kg 8 days | 3.8 | 4.4 | 4.0 | 3.9 | 3.8 | 4.0 | 0.3 |
| 37: 100 mg/kg 4 days | 6.1 | 5.8 | 6.0 | 0.2 | |||
Mode of Action Studies
Since the SAR suggested that the phenethyl linker and the amide carbonyl were critical aspects of the minimum pharmacophore, it appeared that the series shared a similar pharmacophore to a number of reported scaffolds targeting QcrB.24 QcrB is a subunit of the menaquinol cytochrome c oxidoreductase (bc1 complex) that is part of the bc1-aa3-type cytochrome c oxidase complex and is responsible for driving oxygen-dependent respiration.21,22,32 A number of inhibitors of QcrB have been identified recently through phenotypic screens,21,33−36 and Q203 is currently undergoing clinical development. Overlay of selected low local energy conformations of 37 with Q203(21) highlights the similarity in the pharmacophore (Figure 2).
Figure 2.

Overlay of known QcrB inhibitor Q203 (green) with 37 (yellow). Further 2D comparison of QcrB pharmacophores is shown in the Supporting Information (Table 1).
To test this hypothesis, a set of active compounds was tested against mutant strains of M. tuberculosis each with a single nucleotide polymorphism in QcrB; we used a QcrBT313I mutant since this was previously shown to confer resistance to multiple QcrB inhibitors including Q203 and the imidazopyridines, as well as QcrBM342T (Table 11).21,24 A homology model for the structure of QcrB has been reported previously based on multiple cytochrome b homologues,24 and the mutated residues map to the stigmatellin binding site within the bc1 complex modifying the shape of the binding pocket.24,37 The mutant strains were resistant to the panel of compounds chosen, with an increase in MIC of over 10-fold versus the wild-type (WT) strain. As for other QcrB inhibitors,21,24,35 the resistant mutant data suggests that the MOT series targets QcrB directly acting like stigmatellin and disrupting electron transport.
Table 11. Screening of Selected Compounds against M. tuberculosis QcrB Mutant Strains.
| MICa (μM) |
|||||
|---|---|---|---|---|---|
| ID | WTb | T313Ic | fold shift | M342Td | fold shift |
| 1 | 1.3 | 53 | 41 | 25 | 19 |
| 21 | 0.08 | >20 | >250 | 4.8 | 60 |
| 35 | 5.8 | >50 | >8.6 | >20 | >3 |
| 37 | 0.24 | 11 | 46 | ND | ND |
| 39 | 0.53 | 59 | 110 | 7 | 14 |
Minimum inhibitory concentration (MIC90) is the minimum concentration required to inhibit the growth of M. tuberculosis in liquid culture.
Wild-type strain.
QcrB T313I mutant strain.
QcrB M342T mutant strain. ND Not Done
To explore further the effect of the MOT compounds on QcrB, it was determined whether exposure resulted in a decrease in ATP levels, as would be expected for compounds which target QcrB.21,34 ATP and growth were monitored in the same experiment, and it was clear that ATP levels were depleted in a dose-dependent manner (Figure 3) as seen previously for compounds that also target QcrB.21,34,35,38 The depletion of ATP occurred at lower concentrations than required to inhibit growth, as for Q203 (a known QcrB inhibitor), suggesting that this is the mode of action. In contrast, depletion of ATP by rifampicin which targets RNA polymerase correlated with growth. These data support the conclusion that the MOT series targets QcrB.
Figure 3.

ATP levels were measured in M. tuberculosis exposed to compounds for 24 h. Growth was monitored by OD590: (A–D) MOT compounds, (E) Q203, and (F) Rifampicin. Results are representative of 2 independent experiments.
Conclusion
We describe the identification of a novel morpholino–thiophene series from whole cell screening against M. tuberculosis. The confirmed hit 1 was extensively profiled, and the profile suggested that microsomal stability was a key optimization parameter. Structure–activity relationships led to an understanding of the minimal pharmacophore as well as emerging leads such as 37 with improved whole cell activity against M. tuberculosis and improved microsomal stability. Selected compounds were profiled in an acute in vivo efficacy model of TB, and 37 was found to have a small but significant effect. Mode of action profiling suggested the series targets QcrB, a subunit of the menaquinol cytochrome c oxidoreductase (bc1 complex) involved in bacterial respiration. In contrast to Q203, the novel lead series reported herein provides starting points with scope for optimization to reduce the high log P associated with Q203. Further lead optimization efforts will be reported elsewhere.
Experimental Section
Determination of Minimum Inhibitory Concentration (MIC90)
MICs were determined against M. tuberculosis H37Rv (ATCC 25618) and mutant strains grown in Middlebrook 7H9 medium containing 10% v/v OADC (oleic acid, albumin, dextrose, catalase) supplement (Becton Dickinson) and 0.05% w/v Tween 80 (7H9-Tw-OADC) under aerobic conditions as previously described.39 Bacterial growth was measured after 5 days of incubation at 37 °C. MIC90 was defined as the concentration of compound required to inhibit growth of M. tuberculosis by 90% and was determined by plotting growth and curve fitting using the Levenberg–Marquardt least-squares plot.
Cytotoxicity Assay
The Vero cell line (ATCC CRL-1587) was grown in DMEM, high glucose, GlutaMAX (Invitrogen), 10% fetal bovine serum (FBS), and 1× penicillin–streptomycin solution (100 U/mL). Compounds were solubilized in DMSO and assayed as a 10-point 3-fold serial dilution. Compounds were incubated with cells for 2 days at 37 °C, 5% CO2. CellTiter-Glo Reagent (Promega) was added and relative luminescent units (RLU) measured. Inhibition curves were fitted using the Levenberg–Marquardt algorithm; TC50 was calculated as the compound concentration giving 50% inhibition of growth.
Measurement of Intrabacterial ATP Levels
M. tuberculosis H37Rv was exposed to compounds for 24 h. ATP levels were measured using the BacTiter-Glo assay kit (Promega) and expressed as RLU (relative luminescence units). Growth was monitored by OD590.
Intrinsic Clearance (Cli) Experiments
Test compound (0.5 μM) was incubated with female CD1 mouse liver microsomes (Xenotech LLC; 0.5 mg/mL 50 mM potassium phosphate buffer, pH 7.4) and the reaction started with addition of excess NADPH (8 mg/mL 50 mM potassium phosphate buffer, pH 7.4). Immediately, at time zero, and then at 3, 6, 9, 15, and 30 min an aliquot (50 μL) of the incubation mixture was removed and mixed with acetonitrile (100 μL) to stop the reaction. Internal standard was added to all samples, the samples centrifuged to sediment precipitated protein, and the plates then sealed prior to UPLCMSMS analysis using a Quattro Premier XE (Waters Corp., USA). XLfit (IDBS, UK) was used to calculate the exponential decay and consequently the rate constant (k) from the ratio of peak area of test compound to internal standard at each time point. The rate of intrinsic clearance (CLi) of each test compound was then calculated using the following calculation
where V (mL/mg protein) is the incubation volume/mg protein added and microsomal protein yield is taken as 52.5 mg protein/g liver. Verapamil (0.5 μM) was used as a positive control to confirm acceptable assay performance. The human biological samples were sourced ethically, and their research use was in accord with the terms of the informed consents.
Aqueous Solubility
The aqueous solubility of the test compounds was measured using laser nephelometry. Compounds were subject to serial dilution from 10 to 0.5 mM in DMSO. An aliquot was then mixed with Milli-Q water to obtain an aqueous dilution plate with a final concentration range of 250–12 μM, with a final DMSO concentration of 2.5%. Triplicate aliquots were transferred to a flat-bottomed polystyrene plate which was immediately read on the NEPHELOstar (BMG Lab Technologies). The amount of laser scatter caused by insoluble particulates (relative nephelometry units, RNU) was plotted against compound concentration using a segmental regression fit, with the point of inflection being quoted as the compounds aqueous solubility (μM).
Thermodynamic Solubility via High-Throughput Method, HTSA
Samples prepared in DMSO were dried for 12 h. The powder or film was redissolved in the solvent at various pH (2, 6, and 7.4) and DMSO control at 2 mM target concentration. Samples are stirred for 20 h and filtered through a 0.7 μm GF filter. The filtrate was analyzed by HPLC assay for concentration against DMSO standard curve.40
Mouse Pharmacokinetics
Test compound was dosed as a bolus solution intravenously at 3 mg of free base/kilogram (dose volume 5 mL/kg; dose vehicle Saline or 10% DMSO; 40% PEG400; 50% saline) to female C57 Black (n = 3) or dosed orally by gavage as a solution at 10 mg of free base/kilogram (dose volume 10 mL/kg; dose vehicle 10% DMSO; 40% PEG400; 50% distilled water) to female C57 Black (n = 3/dose level). Blood samples were taken from each mouse tail vein at predetermined time points postdose, mixed with two volumes of distilled water, and stored frozen until UPLC/MS/MS analysis. Pharmacokinetic parameters were derived from the blood concentration time curve using PK Solutions software v 2.0 (Summit Research Services, USA).
Therapeutic Efficacy of 37 against M. tuberculosis in an Acute Murine Model of Intratracheal Infection
Specific pathogen-free, 8–10 week old female C57BL/6 mice were purchased from Harlan Laboratories and allowed to acclimate for 1 week. Mice were intrathecal infected with 100.000 CFU/mouse (M. tuberculosis H37Rv strain). Compound 39 was administered for 4 consecutive days starting 5 days after the infection. Moxifloxacin was used as an interassay control and administered for 8 consecutive days starting 1 day after the infection. Lungs were harvested on day 9, 24 h after the last administration. All lung lobes were aseptically removed, homogenized, and frozen. Homogenates were plated in 10% OADC-7H11 medium supplemented with activated charcoal 0.4% for 18 days at 37 °C. All animal studies were ethically reviewed and carried out in accordance with European Directive 2010/63/EEC and the GSK Policy on the Care, Welfare and Treatment of Animals. The number of CFU/mouse measured for each mouse is shown in Table 10. The differences in the lung microorganism burden (log 10 CFUs/lungs) obtained in the treated mice with respect to untreated controls (day 9 after infection) are shown in Table 10. No adverse clinical signs were observed in any animal. Moxifloxacin (30 mg/kg) reduced 2.8 logs the CFU lung number with respect to untreated mice. This quality control value is included in the accepted interval. CFU number in lungs of untreated mice: 6.8 log CFU. This value is inside the interval mean ±2 SD of the values of the last experiments. In these experimental conditions, 37 was able to inhibit the growth of the bacteria in the lungs of the mice compared to untreated mice. This difference was statistically significant.
Rat Hepatic Portal Vein (HPV) Study
Test compound was dosed orally by gavage at 5 mg of free base/kilogram (dose volume 5 mL/kg; dose vehicle 1.0% carboxy methyl cellulose, CMC) to male Sprague–Dawley Rats (n = 1 per time point). At predetermined time points, blood samples (100 μL) were taken from the hepatic portal vein into a tube containing EDTA. Immediately afterward, the remaining blood was taken by cardiac puncture into a separate tube containing EDTA to determine the concentration of compound reaching the systemic circulation. The blood was mixed with two volumes of distilled water and stored frozen until UPLC/MS/MS analysis. Pharmacokinetic parameters were derived from the blood concentration time curve using PK Solutions software v 2.0 (Summit Research Services, USA).
General Chemistry Methods
Chemicals and solvents were purchased from commercial vendors and were used as received, unless otherwise stated. Dry solvents were purchased in Sure Seal bottles stored over molecular sieves. Unless otherwise stated herein reactions have not been optimized. Analytical thin-layer chromatography (TLC) was performed on precoated TLC plates (Kieselgel 60 F254, BDH). Developed plates were air dried and analyzed under a UV lamp (UV 254/365 nm) and/or KMnO4 was used for visualization. Flash chromatography was performed using Combiflash Companion Rf (Teledyne ISCO) and prepacked silica gel columns purchased from Grace Davison Discovery Science or SiliCycle. Mass-directed preparative HPLC separations were performed using a Waters HPLC (2545 binary gradient pumps, 515 HPLC makeup pump, 2767 sample manager) connected to a Waters 2998 photodiode array and a Waters 3100 mass detector. Preparative HPLC separations were performed with a Gilson HPLC (321 pumps, 819 injection module, 215 liquid handler/injector) connected to a Gilson 155 UV/vis detector. On both instruments, HPLC chromatographic separations were conducted using Waters XBridge C18 columns, 19 mm × 100 mm, 5 μm particle size, using 0.1% ammonia in water (solvent A) and acetonitrile (solvent B) as mobile phase.1H NMR spectra were recorded on a Bruker Advance II 500, 400, or 300 spectrometer operating at 500, 400, or 300 MHz (unless otherwise stated) using CDCl3 or DMSO-d6 solutions. Chemical shifts (δ) are expressed in ppm recorded using the residual solvent as the internal reference in all cases. Signal splitting patterns are described as singlet (s), doublet (d), triplet (t), multiplet (m), broadened (b), or a combination thereof. Coupling constants (J) are quoted to the nearest 0.1 Hz (Hz). Low-resolution electrospray (ES) mass spectra were recorded on a Bruker Daltonics MicrOTOF mass spectrometer run in positive mode. High-resolution mass spectroscopy (HRMS) was performed using a Bruker Daltonics MicrOTOF mass spectrometer. LC-MS analysis and chromatographic separation were conducted with a Bruker Daltonics MicrOTOF mass spectrometer or an Agilent Technologies 1200 series HPLC connected to an Agilent Technologies 6130 quadrupole LC/MS where both instruments were connected to an Agilent diode array detector. The column used was a Waters XBridge column (50 mm × 2.1 mm, 3.5 μm particle size), and the compounds were eluted with a gradient of 5–95% acetonitrile/water + 0.1% ammonia. All final compounds showed chemical purity of ≥95% as determined by the UV chromatogram (190–450 nm) obtained by LC-MS analysis. High-resolution electrospray measurements were performed on a Bruker MicroTof mass spectrometer. Microwave-assisted chemistry was performed using a CEM microwave synthesizer.
5-Morpholino-3-phenethoxythiophene-2-carboxamide (1)
7 (200 mg, 0.88 mmol), 2-iodoethylbenzene (224 mg, 0.96 mmol), and K2CO3 (121 mg, 0.88 mmol) were dissolved in MeCN and heated for 15 min at 120 °C in μW. Excess MeCN was removed in vacuo and the crude residue partitioned between EtOAc/H2O, the organic solvent was removed in vacuo, and the residue was purified by column chromatography eluting with 0–100% EtOAc in heptanes to afford the desired product, colorless solid (60 mg, 21%). 1H NMR (500 MHz, CDCl3) δ 7.36 (dd, J = 7.3, 7.3 Hz, 2H), 7.27–7.26 (m, 1H), 5.83 (s, 1H), 4.36 (dd, J = 6.7, 6.7 Hz, 2H),3.85–3.82 (m, 4H), 3.21–3.12 (m, 6H). HRMS (ES + ve), C17H20N2O3S [M + H]+: calcd 333.1267, found 333.1261.
Methyl 3-[2-ethoxyethyl(methyl)amino]-3-oxo-propanoate (5)
Morpholine (5.0 g, 57 mmol) and dimethylpropanedioate (11.4 g, 86 mmol) were heated at 60 °C for 16 h. The reaction was stopped, and the excess reagent was removed in vacuo. The crude mixture was taken on to the next step without further purification (9.7 g, 90%). 1H NMR (300 MHz, CDCl3) δ 3.69–3.68 (s, 3H), 3.65–3.56 (m, 6H), 3.41 (s, 2H), 3.38 (d, J = 4.5 Hz, 2H). LC-MS m/z 204 [M + H]+.
Methyl 3-morpholino-3-thioxopropanoate (6)
5 (28.1 g, 150 mmol) was dissolved in 1,4-dioxane (72 mL), and Lawesson’s Reagent (34.0 g, 84 mmol) was added. The reaction was heated to 100 °C for 2 h, stopped, and cooled to 0 °C. The resulting precipitate was filtered washing with EtOAc, the filtrate was washed with NaHCO3 (100 mL), and the organic layer was dried though a hydrophobic frit before being concentrated in vacuo to afford an orange oil. Trituration with Et2O afforded the desired product as a pale orange solid (22.5 g, 74%). 1H NMR (500 MHz, CDCl3) δ 4.38 (t, J = 5.0 Hz, 2H), 4.09 (s, 2H), 3.84 (t, J = 5.0 Hz, 2H), 3.82–3.79 (m, 2H), 3.79 (s, 3H), 3.77–3.75 (m, 2H). LC-MS m/z 204 [M + H].
3-Hydroxy-5-morpholino-thiophene-2-carboxamide (7)
Sodium hydride (1.2 g, 29.5 mmol) was dissolved in THF (18 mL) at rt, and 6 (6.0 g, 29.5 mmol) was added. The reaction was stirred at rt for 30 min and then cooled to 0 °C. 2-Bromoacetamide (4.1 g, 29.5 mmol) was added. The reaction was maintained at 0 °C for a further 1 h, then warmed to rt, and stirred for a further 2 h. A second equivalent of NaH (1.2 g, 29.5 mmol) was added, and the reaction was stirred at rt for 2 h. LCMS indicated that the desired product had formed. The reaction was quenched with water, and excess THF was removed in vacuo. The aqueous layer was acidified with 1 M HCl and partitioned with EtOAc. The desired product precipitated from the organic extracts (6.7 g, 65%). 1H NMR (500 MHz, CDCl3) δ 11.91 (bs, 1H), 7.00 (bs, 2H), 5.83 (s, 1H), 4.37 (J = 5.1 Hz, 4H), 5.11 (J = 5.1 Hz, 4H). LC-MS m/z 479 [2 M + Na].
General Procedure A: LDA
Thiophene derivative (1 equiv) was added to a flame-dried flask under N2 and dry THF (4 mL/mmol) added. The solution was cooled to −78 °C, and LDA (1.1 equiv) was added dropwise. The reaction mixture was stirred for 5 min, and iodine (1.1 equiv) was added. The reaction was further stirred for 10 min at −78 °C, then warmed to rt for 3 h, quenched with water, and 1 N HCl (1 mL/mmol) added. The organic phase was separated and washed with an aqueous solution of sodium bisulphite and dried over MgSO4, and the solvent was removed in vacuo. The crude material was triturated with hexane to afford the desired product.
Methyl 5-Iodo-3-(2-phenylethoxy)thiophene-2-carboxylate (12)
Prepared using general procedure A starting from methyl 3-(2-phenylethoxy)thiophene-2-carboxylate (2.5 g, 9.5 mmol), pale yellow solid (2.2 g, 60%). 1H NMR (300 MHz, CDCl3) δ 7.35–7.28 (m, 5H), 6.98 (s, 1H), 4.30 (2H, t, J = 7.1 Hz), 3.85 (3H, s), 3.16 (2H, t, J = 7.0 Hz). LC-MS m/z 389 [M + H].
5-Iodo-3-(2-phenylethoxy)thiophene-2-carboxamide (13)
Prepared using general procedure B starting from 12 (600 mg, 1.55 mmol), pale yellow solid (432 mg, 75%). 1H NMR (300 MHz, CDCl3) δ 7.38–7.26 (m, 5H), 7.04 (s, 1H), 6.62 (br.s, 1H), 5.27 (br.s, 1H), 4.39 (t, J = 6.6 Hz, 2H), 3.14 (t, J = 6.6 Hz, 2H). LC-MS m/z 374 [M + H].
Methyl 5-Morpholino-3-(2-phenylethoxy)thiophene-2-carboxylate (14)
12 (1500 mg, 3.9 mmol), Cs2CO3 (1888 mg, 5.8 mmol), RuPhos (361 mg, 0.77 mmol), and Pd2(dba)3 (177 mg, 0.19 mmol) were placed in a vial. The vial was sealed, purged, and backfilled with N2 three times. Then dry THF (10 mL) was added followed by morpholine (438 mg, 5.0 mmol). The sealed vial was heated at 100 °C for 20 h. The reaction was filtered through a Celite pad, and the solvent was removed in vacuo. The residue was purified by flash chromatography eluting with 0–100% EtOAc in hexanes, yellow solid (943 mg, 70%).1H NMR (CDCl3; 300 MHz) δ 7.37–7.23 (5H, m), 5.78 (1H, s), 4.28 (2H, t, J = 7.2 Hz), 3.85–3.79 (4H, m), 3.81 (3H, s), 3.22–3.13 (6H, m). HRMS (ES + ve), C18H21NO4S [M + H]+: calcd 348.1264, found 348.1257.
5-Morpholino-3-(2-phenylethoxy)-N-tetrahydropyran-2-yloxy-thiophene-2-carboxamide (15)
To a solution of 14 (140 mg, 0.40 mmol) and O-tetrahydropyran-2-ylhydroxylamine (47 mg, 0.40 mmol) at −78 °C in dry THF (1.5 mL) was added LiHMDS (1 M in THF) (1.25 mL, 1.25 mmol) dropwise. The reaction was stirred for 5 min and then allowed to reach rt and stirred overnight. The reaction mixture was adsorbed onto silica and purified by column chromatography eluting with 0–100% EtOAc in heptanes. The selected fractions were combined, and the solvent was removed in vacuo to afford the desired product (82 mg, 46%) as a pale brown solid. 1H NMR (500 MHz, CDCl3) δ 9.29 (s, 1H), 7.39–7.35 (m, 2H), 7.31–7.29 (m, 3H), 5.77 (s, 1H), 5.01 (t, J = 3.1 Hz, 1H), 4.32 (t, J = 6.9 Hz, 2H), 4.05–3.99 (m, 1H), 3.84–3.81 (m, 4H), 3.66–3.61 (m, 1H), 3.20–3.13 (m, 6H), 1.93–1.82 (m, 3H), 1.71–1.59 (m, 3H). LC-MS m/z 438.1 [M + H], 349 [M + H – THP].
5-Morpholino-3-(2-phenylethoxy)thiophene-2-carbohydroxamic Acid (16)
To a solution of 15 (26 mg, 0.06 mmol) in MeOH (1 mL) was added p-toluenesulfonic acid monohydrate (1 mg, 0.006 mmol). The reaction was stirred until no SM was observed by TLC. The reaction mixture was adsorbed onto silica and purified by flash chromatography eluting with 30–100% EtOAc in heptanes, pale brown solid (12.6 mg, 55%). 1H NMR (400 MHz, CDCl3) δ 8.86 (s, 1H), 7.40–7.38 (m, 2H), 7.32–7.26 (m, 3H), 5.79 (s, 1H), 4.33 (t, J = 6.5 Hz, 2H), 3.83–3.81 (m, 4H), 3.19–3.13 (m, 6H). LC-MS m/z 350 [M + H].
Methyl 5-Formyl-3-(2-phenylethoxy)thiophene-2-carboxylate (18a)
Prepared using general procedure A starting from methyl 3-(2-phenylethoxy)thiophene-2-carboxylate (400 mg, 1.5 mmol), colorless oil (440 mg, 99%). 1H NMR (300 MHz, CDCl3) δ 9.74 (s, 1H), 7.94 (s, 1H), 7.27–7.24 (m, 2H), 7.22–7.17 (m, 3H), 4.28 (t, J = 7.0 Hz, 2H), 3.82 (s, 3H), 3.09 (t, J = 7.0 Hz, 2H). LC-MS m/z 313 [M + Na].
Methyl 5-(Morpholinomethyl)-3-(2-phenylethoxy)thiophene-2-carboxylate (18b)
A mixture of morpholine (159 mg, 1.83 mmol) and 18a (443 mg, 1.53 mmol) was dissolved in chloroform (10 mL) and stirred at rt for 1 h. Then NaBH(OAc)3 (485 mg, 2.29 mmol) was added, and the mixture was stirred in a sealed tube for 20 h. The mixture was diluted with water (4 mL), shaken vigorously, and filtered through a phase separator, and the filtrate was concentrated in vacuo to afford a colorless oil (540 mg, 98%). 1H NMR (500 MHz, CDCl3) δ 7.24–7.23 (m, 4H), 7.19 (s, 1H), 7.17–7.16 (m, 1H), 6.60 (s, 1H), 4.21 (t, J = 7.2 Hz, 2H), 3.76 (s, 3H), 3.68–3.63 (m, 4H), 3.52 (s, 2H), 3.07 (t, J = 7.2 Hz, 2H), 2.44–2.40 (m, 4H). LC-MS m/z 362 [M + H].
General Procedure B: Using CaCl2 To Form the Primary Amide
Acid (1 equiv) and CaCl2 (1.2 equiv) were added to a solution of NH3 in MeOH (7 M) (20 equiv). The vessel was sealed, and the mixture was stirred at 150 °C for 3 h under microwave conditions. The reaction mixture was diluted with DCM and a saturated aqueous solution of NH4Cl, and the organic phase was separated through a hydrophobic frit and concentrated in vacuo. The crude material was purified by flash chromatography eluting with 20–100% EtOAc in heptanes.
5-(Morpholinomethyl)-3-(2-phenylethoxy)thiophene-2-carboxamide (18)
Prepared by general procedure B starting from 18b (551 mg, 1.53 mmol), pale yellow solid (172 mg, 31%). 1H NMR (300 MHz, CDCl3) δ 7.41–7.30 (m, 4H), 7.27–7.24 (s, 1H), 6.77–6.73 (m, 2H), 5.32 (br.s, 1H), 4.40 (t, J = 6.7 Hz, 2H), 3.76–3.71 (m, 4H), 3.62 (s, 2H), 3.15 (t, J = 6.7 Hz, 2H), 2.55–2.49 (m, 4H). HRMS (ES + ve), C18H23N2O3S [M + H]+: calcd 347.1424, found 347.1442.
4-[4-(2-Phenylethoxy)-2-thienyl]morpholine (19)
tButyl 5-morpholino-3-(2-phenylethoxy)thiophene-2-carboxylate (73 mg, 0.19 mmol) was dissolved in MeOH (7 mL), and KOH (316 mg, 5.62 mmol) was added. The reaction mixture was heated to reflux for 26 h and cooled, and the solvent was removed in vacuo. H2O was added to the residue that was acidified with 2 M HCl and extracted with DCM (2 × 10 mL). The combined organics were dried and purified by column chromatography eluting with 20% EtOAc in heptane, colorless oil (5 mg, 8%). 1H NMR (CDCl3; 500 MHz) δ 7.28–7.13 (5H, m), 5.76 (1H, s), 5.44 (1H, s), 4.03 (2H, t, J = 7.1 Hz), 3.77–3.70 (4H, m), 3.03–2.95 (6H, m). LC-MS m/z 289 [M + H].
3-Hydroxy-5-morpholinothiophene-2-carbonitrile (20a)
NaOEt (10.4 g, 32 mmol) was dissolved in EtOH (90 mL), and 6 (5.00 g, 24.6 mmol) was added. The mixture was then cooled to 0 °C, and 2-bromoacetonitrile (2.95 mg, 24.6 mmol) was added. The reaction was stirred at 0 °C for 1 h and then allowed to warm to rt for a further 1 h before addition of further NaOEt (2.18 g, 31.98 mmol). The reaction was stirred for 12 h at rt. The precipitate was filtered and dissolved in H2O before acidifying with 1 M HCl and extracting with DCM (3 × 50 mL). The combined organics were dried through a hydrophobic frit and concentrated in vacuo. The resulting solid was triturated with Et2O and filtered off to afford a beige solid (2.83 g, 55%). 1H NMR (CDCl3; 500 MHz) δ 5.25 (1H, s), 4.56 (1H, s), 3.82 (4H, t, J = 4.9 Hz), 3.49 (4H, t, J = 4.9 Hz). LC-MS m/z 211 [M + H].
5-Morpholino-3-(2-phenylethoxy)thiophene-2-carbonitrile (20b)
20a (1.50 g, 7.1 mmol), Cs2CO3 (3.49 g, 10.7 mmol), and 2-bromoethylbenzene (1.98 g, 10.7 mmol) were combined in MeCN (15 mL) and heated in the microwave at 120 °C for 30 min. The reaction mixture was extracted with DCM (2 × 10 mL), washing with H2O (15 mL). The organics were dried through a hydrophobic frit and concentrated in vacuo to afford an orange oil. The crude material was columned eluting with 20–100% EtOAc in hexane to afford an off-white solid (844 mg, 37%). 1H NMR (CDCl3; 500 MHz) δ 7.37–7.32 (2H, m), 7.31–7.25 (4H, m), 5.66 (1H, s), 4.39 (2H, t, J = 7.0 Hz), 3.85–3.81 (4H, m), 3.19–3.16 (4H, m), 3.11 (2H, t, J = 7.0 Hz). LC-MS m/z 315 [M + H].
1-[5-Morpholino-3-(2-phenylethoxy)-2-thienyl]ethanone (20)
MeMgBr (53 mg, 0.45 mmol) was dissolved in toluene (5 mL) under N2, and 20b (70 mg, 0.22 mmol) was added. The mixture was heated to reflux for 3 h. The reaction mixture was allowed to cool to rt before acidifying with 2 M HCl to pH 2. The mixture was then heated to reflux for 30 min. The mixture was allowed to cool to rt and basified with a saturated aqueous NaHCO3 solution and extracted with EtOAc (2 × 15 mL). The organics were dried through a phase separating frit and concentrated in vacuo to afford an orange oil which solidified on standing at rt. The crude material was columned eluting with 0–20% MeOH in DCM, pink solid (2 mg, 2%). 1H NMR (CDCl3; 500 MHz) δ 7.30–7.23 (2H, m), 7.21–7.17 (3H, m), 5.67 (1H, s), 4.22 (2H, t, J = 6.8 Hz), 3.75–3.71 (4H, m), 3.18–3.14 (4H, m), 3.05 (2H, t, J = 6.8 Hz), 2.29 (3H, s). HRMS (ES + ve), C18H21NO3S [M + H]+: calcd 332.1315, found 332.1314.
3-Hydroxy-N-methyl-5-morpholinothiophene-2-carboxamide (21a)
NaOEt (1.04 g, 3.20 mmol) was dissolved in EtOH (9 mL), and methyl 6 (500 mg, 2.5 mmol) was added. The mixture was then cooled to 0 °C, and 2-bromo-N-methyl-acetamide (374 mg, 2.46 mmol) was added. The reaction was stirred at 0 °C for 1 h and then allowed to warm to rt for a further 1 h before addition of further NaOEt (218 mg, 3.2 mmol). The reaction was then stirred at rt for 2 h. The reaction was quenched with H2O, and the volatiles were removed in vacuo. The mixture was acidified using 2 M HCl and extracted with DCM (2 × 20 mL). The combined organics were dried and concentrated in vacuo to afford an off-white solid which was purified by column chromatography eluting with 5% MeOH in DCM to afford the desired product as a white solid (127 mg, 21%). 1H NMR (CDCl3; 300 MHz) δ 7.90 (1H, br s), 5.21 (1H, s), 4.69 (1H, s), 3.83–3.75 (4H, m), 3.54–3.45 (4H, m), 2.86 (3H, t, J = 4.8). LC-MS m/z 243 [M + H].
N-Methyl-5-morpholino-3-(2-phenylethoxy)thiophene-2-carboxamide (21)
21a (127 mg, 0.52 mmol), Cs2CO3 (256 mg, 0.79 mmol), and 2-bromoethylbenzene (146 mg, 0.79 mmol) were combined in MeCN (2 mL) and heated in the microwave at 120 °C for 30 min. The reaction mixture was extracted with DCM (2 × 10 mL), washing with H2O (15 mL). The organics were dried through a phase-separating frit and concentrated in vacuo to afford a brown oil. The crude material was columned eluting with 3% MeOH in DCM. It was purified further using the Waters autoprep acidic (20–95%), colorless solid (45 mg, 25%). 1H NMR (CDCl3; 500 MHz) δ 7.39–7.33 (2H, m), 7.31–7.25 (3H, m), 6.52 (1H, br s), 5.83 (1H, s), 4.35 (2H, t, J = 6.4 Hz), 3.83–3.79 (4H, m), 3.17–3.13 (4H, m), 3.10 (2H, t, J = 6.4 Hz), 2.76 (3H, d, J = 4.9 Hz). HRMS (ES + ve), C18H22N2O3S [M + H]+: calcd 347.1424, found 347.1443.
General Procedure C: AlCl3-Catalyzed Amide Formation
Acid (1 equiv) and trimethylaluminum in toluene (4 equiv) were stirred at rt, and ethanamine (1 equiv) was added. The reaction was heated at reflux for 3 h, stopped, and cooled to rt. Excess solvent was removed in vacuo; 1 M NaOH was added (20 mL/mmol), and the mixturewas extracted with EtOAc. The organic layer was dried in vacuo, and the crude residue was purified by column chromatography eluting with DCM to 10% DCM.
N-Ethyl-5-morpholino-3-(2-phenylethoxy)thiophene-2-carboxamide (22)
Prepared using general procedure C starting from 14 (50 mg, 0.14 mmol), colorless solid (77 mg, 88%). 1H NMR (400 MHz, CDCl3) δ 7.38–7.35 (m, 2H), 7.30–7.27 (m, 3H), 6.63 (bs, 1H), 5.84 (s, 1H), 4.36 (dd, J = 6.5, 6.5 Hz, 2H), 3.83 (dd, J = 4.7, 4.7 Hz, 4H), 3.34–3.25 (m, 2H), 3.19–3.10 (m, 6H), 1.07 (dd, J = 7.2, 7.2 Hz, 3H). HRMS (ES + ve), C19H25N2O3S [M + H]+: calcd 361.1589, found 361.1580.
5-Morpholino-3-phenethoxy-N-(2,2,2-trifluoroethyl)thiophene-2-carboxamide (23)
Prepared using general procedure C starting from 14 (50 mg, 0.14 mmol), colorless solid (20 mg, 33%). 1H NMR (500 MHz, CDCl3) δ 7.37 (dd, J = 7.3, 7.3 Hz, 2H), 7.31–7.27 (m, 3H), 6.93 (dd, J = 6.3, 6.3 Hz, 1H), 5.82 (s, 1H), 4.39 (dd, J = 6.6, 6.6 Hz, 2H), 3.92–3.82 (m, 6H), 3.21–3.12 (m, 6H). HRMS (ES + ve), C19H22F2N2O3S [M + H]+: calcd 415.1313, found 415.1298.
N-Cyclobutyl-5-morpholino-3-phenethoxythiophene-2-carboxamide (24)
Prepared using general procedure C starting from 14 (50 mg, 0.14 mmol), colorless solid (17 mg, 30%). 1H NMR (400 MHz, CDCl3) δ 7.40–7.37 (m, 2H), 7.32–7.28 (m, 3H), 6.82 (bs, 1H), 5.84 (s, 1H), 4.52–4.43 (m, 1H), 4.39 (dd, J = 6.5, 6.5 Hz, 2H), 3.85–3.82 (m, 4H), 3.20–3.14 (m, 6H), 2.34–2.28 (m, 2H). HRMS (ES + ve), C21H27N2O3S (M+H)+: calcd 387.1737, found 387.1741.
Methyl 5-Morpholino-3-(2-phenylethoxy)thiophene-2-carboxylate (25a)
14 (1.5 g, 3.7 mmol), Cs2CO3 (1.9 g, 5.8 mmol), RuPhos (361 mg, 0.77 mmol), and Pd2(dba)3 (177 mg, 0.19 mmol) were placed in a vial. The vial was sealed, purged, and backfilled with nitrogen three times. Dry THF (10 mL) was added followed by morpholine (438 mg, 5.02 mmol). The reaction was heated in a sealed tube at 100 °C for 20 h. The reaction was filtered through a Celite pad, and the solvent was removed in vacuo. The crude residue was purified eluting with 0–10% EtOAc in heptane to afford the desired product as a yellow solid (1231 mg, 92% yield). 1H NMR (400 MHz, CDCl3) δ 7.34 (d, J = 4.5 Hz, 4H), 7.28–7.26 (m, 1H), 5.77 (s, 1H), 4.27 (dd, J = 7.2, 7.2 Hz, 2H), 3.84–3.81 (m, 6H), 3.22–3.14 (m, 6H), 1.28 (dd, J = 7.2, 7.2 Hz, 1H). LC-MS m/z 348 [M + H].
N-Methoxy-N-methyl-5-morpholino-3-phenethoxythiophene-2-carboxamide (25)
25a (500 mg, 1.4 mmol) and N,O-dimethylhydroxylamine hydochloride (211 mg, 2.15 mmol) were cooled to approximately −10 °C in THF (3 mL) under nitrogen. Isopropylmagnesium chloride (488 mg, 4.75 mmol) was then added dropwise. The reaction was warmed to rt and stirred for 3 h.The reaction was quenched with water, diluted in DCM, filtered through a hydrophobic frit, and concentrated in vacuo to afford a yellow oil. Recrystallization in EtOAc afforded an off-white solid (420 mg, 74%). 1H NMR (400 MHz, CDCl3) δ 7.34–7.31 (m, 4H), 7.28–7.23 (m, 1H), 5.81 (s, 1H), 4.26 (t, J = 7.6 Hz, 2H), 3.83 (t, J = 4.9 Hz, 4H), 3.72 (s, 3H), 3.27 (s, 3H), 3.21–3.16 (m, 6H). HRMS (ES + ve), C19H24N2O4S [M + H]+: calcd 377.1530, found 377.1526.
General Procedure D: Mitsunobu Reaction
Ethyl N-(3-hydroxy-5-morpholino-thiophene-2-carbonyl)carbamate (1.1 equiv), alcohol (1 equiv), and polymer-supported triphenylphosphine (1.1 equiv) were dissolved in DCM (15 mL/mmol). DIAD (1.1 equiv) was dissolved in DCM (12 mL/mmol) and added dropwise to the mixture. The reaction was stirred for 30 min, filtered, concentrated, and purified using prep LCMS to afford the desired product.
Ethyl N-[5-Morpholino-3-(2-phenylethoxy)thiophene-2-carbonyl]carbamate (26)
Prepared using general procedure D starting from ethyl N-(3-hydroxy-5-morpholino-thiophene-2-carbonyl)carbamate (100 mg, 0.33 mmol) and 2-phenylethanol (37 mg, 0.30 mmol), colorless solid (134 mg, 10%). 1H NMR (500 MHz, CDCl3) δ 8.93 (s, 1H), 7.34–7.28 (m, 5H), 5.73 (s, 1H), 4.36 (dd, J = 6.6, 6.6 Hz, 2H), 4.27 (q, J = 7.1 Hz, 2H), 3.82–3.79 (m, 4H), 3.22–3.13 (m, 6H), 1.34 (dd, J = 7.1, 7.1 Hz, 3H). LC-MS m/z 405 [M + H].
3-(2-Phenylethoxy)thiophene-2-carboxamide (27)
Methyl 3-(2-phenylethoxy)thiophene-2-carboxylate (90 mg, 0.34 mmol) was dissolved in 7 N NH3 in MeOH (1 mL) and heated to 80 °C for 16 h. The reaction was then heated to 100 °C for 4 h. The reaction mixture was transferred to a thick-walled vial, and ammonium hydroxide (33% aqueous, 0.5 mL) was added. The reaction was heated at 100 °C for 16 h. The volatiles were removed in vacuo to give a white solid. The residue was purified by flash chromatography eluting with 0–40% EtOAc in hexane, and the pure fractions were concentrated in vacuo to afford the desired product, colorless solid (27 mg, 30%). 1H NMR (DMSO, 500 MHz) δ 7.69 (d, 1H, J = 5.5 Hz), 7.42 (s, 1H), 7.33–7.32 (m, 4H), 7.26–7.22 (m, 1H), 7.14 (d, 1H, J = 5.5 Hz), 6.69 (s, 1H), 4.42 (t, 2H, J = 6.7 Hz), 3.10 (t, 2H, J = 6.6 Hz). HRMS (ES + ve), C13H14NO2S [M + H]+: calcd 248.075226, found 248.073976.
3-(2-Phenylethoxy)-5-(1-piperidyl)thiophene-2-carboxamide (28)
Ethyl 3-(2-phenylethoxy)-5-(1-piperidyl)thiophene-2-carboxylate (60 mg, 0.17 mmol) was dissolved in anhydrous THF (2 mL) and placed under N2. LiNH2 (38 mg, 1.67 mmol) was added in one portion, and the reaction was stirred for 48 h. The solvent was evaporated and acidified (pH 2) with the minimum amount of 2 M HCl. This was then extracted 3× with DCM and passed through a hydrophobic frit, and the organics were concentrated in vacuo. Purification by column chromatography eluting with 0–100% EtOAc in hexane afforded a white solid (55 mg, 51%).1H NMR (400 MHz, CDCl3) δ 7.37–7.33 (m, 2H), 7.30–7.26 (m, 3H), 5.75 (s, 1H), 4.34 (t, J = 7.03 Hz, 2H), 3.22 (m, 4H), 3.13 (t, J = 7.2 Hz, 2H), 1.73–1.67 (m, 4H), 1.65–1.60 (m, 2H). HRMS (ES + ve), C18H23N2O2S [M + H]+: calcd 331.1475, found 331.1466.
General Procedure E
l-Proline carboxamide (1 equiv), K2CO3 (4 equiv), l-Proline (0.38 equiv), and CuI (0.16 equiv) were added to a microwave vial and sealed, purged with nitrogen (×3), and then morpholin-3-one (1.7 equiv) and then anhydrous DMSO (2 mL) were added. The vial was purged again with nitrogen (×3) and then heated in the microwave reactor at 100 °C for 4 h. The reaction mixture was partitioned between water (8 mL/mmol) and DCM (16 mL/mmol), and the aqueous layer was extracted with further DCM (16 mL/mmol). The combined DCM extracts were evaporated in vacuo, and the residue was purified by mass-directed HPLC 5–95% MeCN acidic to afford the desired product.
5-(3-Oxomorpholino)-3-phenethoxythiophene-2-carboxamide (29)
Prepared using general procedure E starting from 13 (90 mg, 0.24 mmol) and morpholin-3-one (41 mg, 0.41 mmol), colorless solid (8 mg, 8%). 1H NMR (CDCl3, 400 MHz) δ 7.37 (dd, 2H, J = 7.2, 7.2 Hz), 7.28 (s, 3H), 6.58 (s, 1H), 4.42–4.38 (m, 4H), 4.09 (t, 2H, J = 5.1 Hz), 3.84 (t, 2H, J = 5.2 Hz), 3.16 (t, 2H, J = 6.8 Hz). HRMS (ES + ve), C17H18N2O4S [M + H]+: calcd 347.1060, found 347.1044
5-(2-Oxa-5-azabicyclo[2.2.1]heptan-5-yl)-N-methyl-3-phenethoxythiophene-2-carboxamide (30)
Prepared using general procedure E starting from 13 (70 mg, 0.18 mmol) and 2-oxa-5-azabicyclo[2.2.1]heptane hydrochloride (41 mg, 0.30 mmol), colorless solid (6 mg, 7%). 1H NMR (MeOD, 500 MHz,) δ 7.36–7.28 (m, 4H), 7.26–7.22 (m, 1H), 5.86 (s, 1H), 4.66 (s, 1H), 4.43–4.39 (m, 2H), 4.34 (s, 1H), 3.87 (d, 1H, J = 7.8 Hz), 3.79 (dd, 1H, J = 1.2, 7.6 Hz), 3.46 (dd, 1H, J = 1.4, 9.5 Hz), 3.13–3.07 (m, 3H), 2.67 (d, 3H, J = 4.4 Hz), 2.03–1.93 (m, 2H). HRMS (ES + ve), C19H22N2O3S [M + H]+: calcd 359.1424, found 359.1435.
5-(3-Methylmorpholino)-3-phenethoxythiophene-2-carboxamide (31)
Prepared using general procedure E starting from 13 (100 mg, 0.26 mmol) and 3-methylmorpholine (165 mg, 0.45 mmol), colorless solid (10 mg, 14%). 1HNMR (CDCl3, 500 MHz,) δ 7.38–7.34 (m, 3H), 7.29 (s, 2H), 5.74 (s, 1H), 4.35 (t, 2H, J = 6.9 Hz), 3.98 (dd, 1H, J = 3.7, 11.4 Hz), 3.80–3.74 (m, 2H), 3.69–3.60 (m, 2H), 3.29–3.23 (m, 1H), 3.16–3.12 (m, 3H), 1.29 (d, 3H, J = 6.6 Hz). HRMS (ES + ve), C18H22N2O3S [M + H]+: calcd 347.1424, found 347.1431.
5-[2-Methoxyethyl(methyl)amino]-3-(2-phenylethoxy)thiophene-2-carboxamide (32)
Prepared using general procedure E starting from 13 (80 mg, 0.21 mmol) and 2-methoxy-N-methyl-ethanamine (29 mg, 0.32 mmol), pale orange solid (15 mg, 19%). 1H NMR (500 MHz, CDCl3) δ 7.35–7.31 (m, 2H), 7.25–7.24 (m, 3H), 6.42 (br.s, 1H), 5.55 (s, 1H), 5.09 (br.s, 1H), 4.33 (t, J = 6.8 Hz, 2H), 3.57 (t, J = 5.5 Hz, 2H), 3.45 (t, J = 5.4 Hz, 2H), 3.34 (s, 3H), 3.11 (t, J = 6.8 Hz, 2H), 3.01 (s, 3H). HRMS (ES + ve), C17H23N2O3S [M + H]+: calcd 335.1424, found 335.1423.
Methyl 5-(Benzimidazol-1-yl)-3-hydroxy-thiophene-2-carboxylate (33a)
Benzimidazole (67 mg, 0.57 mmol) was dissolved in DCM (5 mL), methyl 2-chloro-3-oxo-thiophene-2-carboxylate (100 mg, 0.52 mmol) was added followed by potassium hydrogen carbonate (52 mg, 0.52 mmol), and the mixture was stirred for 4 h, partitioned between DCM/H2O and the organic layer passed through a hydrophobic frit, and concentrated in vacuo. The precipitate that formed was recrystallized from MeOH to afford a colorless solid (125 mg, 88%). 1H NMR (300 MHz, MeOD) δ 8.46 (s, 1H), 7.86–7.76 (m, 2H), 7.48–7.38 (m, 2H), 6.82 (s, 1H), 3.83 (s, 3H). LC-MS m/z 275 [M + H].
5-(Benzimidazol-1-yl)-3-(2-phenylethoxy)thiophene-2-carboxamide (33)
To a cooled solution of 33a (75 mg, 0.20 mmol) in DCM (1 mL) was added oxalyl chloride (0.019 mL, 0.22 mmol) and 1 drop of DMF. The reaction was allowed to warm to rt, and after 1 h ammonium hydroxide (33% aqueous, 0.5 mL) was added. The reaction was left at rt for 36 h. The volatiles were removed in vacuo, and the residue was partitioned between brine and EtOAc. The organics were separated, dried (Na2SO4), filtered, and concentrated in vacuo. The residue was purified by prep HPLC (Gilson, ammonia, 5–95% MeCN) to afford a white solid (20 mg, 25%). 1H NMR (DMSO, 500 MHz) δ 8.59 (s, 1H), 7.79 (d, 1H, J = 8.0 Hz), 7.75 (d, 1H, J = 8.0 Hz), 7.58 (s, 1H), 7.54 (s, 1H), 7.39–7.36 (m, 1H), 7.34–7.27 (m, 6H), 7.23–7.18 (m, 1H), 6.71 (s, 1H), 4.48 (t, 2H, J = 6.7 Hz), 3.11 (t, 2H, J = 6.7 Hz). HRMS (ES + ve), C20H17N3O2S [M + H]+: calcd 364.1114, found 364.1098.
3-(2-Phenylethoxy)-5-(triazol-2-yl)thiophene-2-carboxamide (34)
13 (250 mg, 0.67 mmol), CuI (13 mg, 0.067 mmol), and K2CO3 (204 mg, 1.47 mmol) were added to a vial, and the vial was sealed and purged with nitrogen. Then DMF (3 mL), 1H-triazole (69 mg, 1.0 mmol), and N,N′-dimethyl-1,2-cyclohexanediamine (29 mg, 0.20 mmol) were added, and the reaction was heated at 110 °C for 16 h and diluted with DCM. H2O was added, and the organic solvent was removed in vacuo. Purification by prep HPLC, white solid (12 mg, 6%) . 1H NMR (300 MHz, acetone) δ 8.03 (s, 2H), 7.48 (s, 1H), 7.45–7.32 (m, 4H), 7.31–7.24 (m, 1H), 6.80–6.77 (m, 1H), 6.66–6.65 (m, 1H), 4.66 (dd, J = 6.6, 6.6 Hz, 2H), 3.25 (dd, J = 6.6, 6.6 Hz, 2H),HRMS (ES + ve), C15H14N4O2S [M + H]+: calcd 315.0910, found 315.0906.
3-(2-Phenylethoxy)-5-(4-pyridyl)thiophene-2-carboxamide (35)
To a sealed vial containing 13 (130 mg, 0.35 mmol), Cs2CO3 (340 mg, 1.05 mmol), Pd(PPh3)4 (40 mg, 0.035 mmol), and 4-pyridinylboronic acid (66 mg, 0.49 mmol) under N2 was added DMF (3 mL). The reaction was then stirred at 80 °C for 16 h, diluted with EtOAc, and filtered through a silica pad, and the filtrate was concentrated in vacuo. Purification by flash chromatography eluting with 0–100% EtOAc in heptanes afforded a yellow solid (53 mg, 45%). 1H NMR (400 MHz, CDCl3) δ 8.68–8.65 (m, 2H), 7.48–7.46 (m, 2H), 7.46–7.35 (m, 2H), 7.33–7.29 (m, 3H), 7.23 (s, 1H), 6.78 (br.s, 1H), 5.36 (br.s, 1H), 4.51 (t, J = 6.6 Hz, 2H), 3.20 (t, J = 6.6 Hz, 2H). HRMS (ES + ve), C18H17N2O2S [M + H]+: calcd 325.1005, found 325.1008.
3-[2-(4-Methoxyphenyl)ethoxy]-5-morpholinothiophene-2-carboxamide (36)
Prepared using general procedure D starting from 7 (250 mg, 1.09 mmol) and 2-(4-methoxyphenyl)ethanol (183 mg, 1.20 mmol), off-white solid (15 mg, 3%). 1H NMR (DMSO, 500 MHz) δ 7.24–7.21 (m, 2H), 6.95 (br s, 1H), 6.89–6.86 (m, 2H), 6.38 (br s, 1H), 6.22 (s, 1H), 4.29 (t, 2H, J = 6.8 Hz), 3.74 (s, 3H), 3.71 (t, 4H, J = 4.9 Hz), 3.15–3.12 (m, 4H), 3.00 (t, 2H, J = 6.8 Hz). HRMS (ES + ve), C18H22N2O4S [M + H]+: calcd 363.1373, found 363.1376.
5-Morpholino-3-[2-[4-(trifluoromethoxy)phenyl]ethoxy]thiophene-2-carboxamide (37)
Prepared using general procedure D starting from 7 (300 mg, 1.31 mmol) and 2-[4-(trifluoromethoxy)phenyl]ethanol (298 mg, 1.44 mmol), colorless solid (96 mg, 17%). 1H NMR (DMSO, 500 MHz) δ 7.46 (d, 2H, J = 8.7 Hz), 7.31 (d, 2H, J = 7.9 Hz), 6.95 (s, 1H), 6.39 (s, 1H), 6.24 (s, 1H), 4.35 (t, 2H, J = 6.8 Hz), 3.71 (m, 4H), 3.16–3.10 (m, 6H). HRMS (ES + ve), C18H19N2O4F3S [M + H]+: calcd 417.1094, found 417.1091.
3-[2-(2-Methoxyphenyl)ethoxy]-5-morpholinothiophene-2-carboxamide (38)
Prepared using general procedure D starting from 7 (300 mg, 1.31 mmol) and 2-(2-fluorophenyl)ethanol (202 mg, 1.44 mmol), colorless solid (105 mg, 22%). 1H NMR (DMSO, 500 MHz) δ 7.44–7.39 (m, 1H), 7.34–7.28 (m, 1H), 7.20–7.15 (m, 2H), 6.98 (br s, 1H), 6.40 (br s, 1H), 6.23 (s, 1H), 4.33 (t, 2H, J = 6.5 Hz), 3.71 (m, 4H), 3.15–3.10 (m, 6H). HRMS (ES + ve), C17H20FN2O3S [M + H]+: calcd 351.1173, found 351.1204.
3-[2-(3-Fluorophenyl)ethoxy]-5-morpholinothiophene-2-carboxamide (39)
Prepared using general procedure D starting from 2-(3-fluorophenyl)ethanol (337 mg, 2.40 mmol) and 7 (500 mg, 2.19 mmol), colorless solid (49 mg, 6%). 1H NMR (DMSO, 500 MHz) 7.36 (m, 1H), 7.21–7.15 (m, 2H), 7.09–7.04 (m, 1H), 6.96 (s, 1H), 6.37 (s, 1H), 6.25 (s, 1H), 4.36 (t, 2H, J = 6.8 Hz), 3.71(m, 4H), 3.16–3.08 (m, 6H). HRMS (ES + ve), C17H20FN2O3S [M + H]+: calcd 351.1173, found 351.1177.
3-(3-(4-Fluorophenyl)propyl)-5-morpholinothiophene-2-carboxamide (40)
Prepared using general procedure D starting from 7 (100 mg, 0.44 mmol)and 2-(4-fluorophenyl)ethanol (67.5 mg, 0.48 mmol), colorless solid (20 mg, 13%). 1H NMR (500 MHz, CDCl3) δ 7.13 (dd, J = 5.4, 8.5 Hz, 2H), 6.95 (dd, J = 8.6, 8.5 Hz, 2H), 5.72 (s, 1H), 4.24 (dd, J = 6.7, 6.7 Hz, 2H), 3.76–3.73 (m, 4H), 3.12–3.09 (m, 4H), 3.01 (dd, J = 6.6, 6.6 Hz, 2H). LCMS (acidic, 20–90% MeCN) m/z 351 (100) rt 2.5. HRMS (ES + ve), C17H20FN2O3S [M + H]+: calcd 351.1173, found 351.1180.
3-[2-(3-Methoxyphenyl)ethoxy]-5-morpholinothiophene-2-carboxamide (41)
Prepared using general procedure D starting from 7 (250 mg, 1.09 mmol) and 2-(3-methoxyphenyl)ethanol (183 mg, 1.20 mmol), colorless solid (5.8 mg, 1%). 1H NMR (CDCl3, 500 MHz) δ 7.18–7.16 (s, 1H), 6.78–6.71 (m, 3H), 5.73 (s, 1H), 4.25 (t, 2H, J = 6.7 Hz), 3.76–3.73 (m, 4H), 3.73 (s, 3H), 3.12–3.09 (m, 4H), 3.01 (t, 2H, J = 6.7 Hz). HRMS (ES + ve), C18H23N2O4S [M + H]+: calcd 363.1373, found 363.1373.
3-[2-(2-Methoxyphenyl)ethoxy]-5-morpholinothiophene-2-carboxamide (42)
Prepared using general procedure D starting from 7 (250 mg, 1.09 mmol) and 2-(2-methoxyphenyl)ethanol (183 mg, 1.20 mmol), colorless solid (1.8 mg, 0.4%). 1H NMR (CDCl3, 500 MHz) δ 7.27 (dd, 1H, J = 1.8, 8.1 Hz), 7.20 (dd, 1H, J = 1.7, 7.5 Hz), 6.96–6.90 (m, 2H), 5.86 (br s, 1H), 4.33 (t, 2H, J = 6.8 Hz), 3.86 (s, 3H), 3.83 (m, 4H), 3.22–3.19 (m, 4H), 3.17 (t, 2H, J = 6.6 Hz). LC-MS m/z 363 [M + H].
5-Morpholino-3-[2-(4-pyridyl)ethoxy]thiophene-2-carboxamide (43)
Prepared using general procedure D starting from 7 (100 mg, 0.43 mmol) and 2-(4-pyridyl)ethanol (59 mg, 0.48 mmol), off-white solid (4.3 mg, 3%). 1H NMR (CDCl3, 500 MHz) δ 8.47–8.45 (m, 2H), 7.09–7.07 (m, 2H), 4.27 (t, 2H, J = 6.5 Hz), 3.73–3.70 (m, 4H), 3.09–3.07 (m, 4H), 3.01 (t, 2H, J = 6.6 Hz). HRMS (ES + ve), C16H20N3O3S [M + H]+: calcd 334.1220, found 334.1233.
3-[2-(4-Cyanophenyl)ethoxy]-5-morpholino-thiophene-2-carboxamid (44)
Prepared using general procedure D starting from 4-(2-hydroxyethyl)benzonitrile (212 mg, 1.4 mmol) 7 (300 mg, 1.3 mmol), colorless solid (82 mg, 17%). 1H NMR (DMSO, 500 MHz) δ 7.79–7.78 (m, 2H), 7.55–7.53 (m, 2H), 6.94 (s, 1H), 6.28 (s, 1H) 6.24 (s, 1H), 4.39 (t, 2H, J = 6.6 Hz), 3.71 (m, 4H), 3.18 (t, 2H, J = 6.6 Hz), 3.15–3.12 (m, 4H). HRMS (ES + ve), C18H19N3O3S [M + H]+: calcd 358.1220, found 358.1218.
5-Morpholino-3-((tetrahydrofuran-2-yl)methoxy)thiophene-2-carboxamide (45)
Prepared using general procedure D starting from 7 (250 mg, 1.1 mmol) and tetrahydrofuran-2-ylmethanol (123 mg, 1.2 mmol), colorless solid (83 mg, 24%). 1H NMR (500 MHz, CDCl3) δ 6.82–6.82 (m, 1H), 5.82 (s, 1H), 5.43–5.43 (m, 1H), 4.09–4.08 (m, 2H), 4.00–3.95 (m, 1H), 3.88–3.76 (m, 6H), 3.23–3.20 (m, 5H), 2.79–2.72 (m, 1H), 2.21–2.13 (m, 1H), 1.83–1.76 (m, 1H). HRMS (ES + ve), C14H20N2O4S [M + H]+: calcd 313.1217, found 313.1218.
3-((3-Methyloxetan-3-yl)methoxy)-5-morpholinothiophene-2-carboxamide (46)
Prepared using general procedure D starting from 7 (250 mg, 1.1 mmol) and (3-methyloxetan-3-yl)methanol (123 mg, 1.2 mmol), colorless solid (129 mg, 37%). 1H NMR (500 MHz, CDCl3) δ 7.08 (s, 1H), 5.87 (s, 1H), 5.28 (s, 1H), 4.68 (d, J = 6.1 Hz, 2H), 4.59 (d, J = 6.1 Hz, 2H), 4.12 (s, 2H), 3.87–3.85 (m, 4H), 3.25–3.22 (m, 4H), 1.39 (s, 3H). HRMS (ES + ve), C14H21N2O4S (M+H)+: calcd 313.1217, found 313.1215.
5-Morpholino-3-(2-morpholinoethoxy)thiophene-2-carboxamide (47)
Prepared using general procedure D starting from 7 (250 mg, 1.1 mmol) and 2-morpholinoethanol (158 mg, 1.2 mmol), light pink solid (38 mg, 10%). 1H NMR (CDCl3, 500 MHz) δ 5.74 (s, 1H), 4.11 (t, 2H, J = 5.6 Hz), 3.74 (m, 4H), 3.62 (m, 4H), 3.11 (m, 4H), 2.67 (t, 2H, J = 5.5 Hz), 2.45 (m, 4H). HRMS (ES + ve), C15H23N3O4S [M + H]+: calcd 342.1482, found 342.1497.
3-Benzyloxy-5-morpholinothiophene-2-carboxamide (48)
Prepared using general procedure D starting from 7 (250 mg, 1.1 mmol) and phenylmethanol (130 mg, 1.2 mmol), off-white solid (3.4 mg, 0.9%). 1H NMR (CDCl3, 500 MHz,) δ 7.45–7.40 (m, 5H), 5.90 (s, 1H), 5.18 (s, 2H), 3.86–3.84 (m, 4H), 3.23–3.20 (m, 4H). HRMS (ES + ve), C16H18N2O3S [M + H]+: calcd 319.1111, found 319.1118.
Methyl 3-(3-Phenylpropyl)thiophene-2-carboxylate (49a)
Methyl 3-bromothiophene-2-carboxylate (600 mg, 2.7 mmol), 4,4,5,5-tetramethyl-2-[(E)-3-phenylprop-1-enyl]-1,3,2-dioxaborolane (928 mg, 3.8 mmol), Cs2CO3 (2652 mg, 8.1 mmol), Pd(OAc)2 (61 mg, 0.27 mmol), triphenylphosphine (285 mg, 1.1 mmol), and THF under N2 were heated at 80 °C for 16 h. The reaction was diluted with DCM, and H2O was added. The organic layer was separated with a hydrophobic frit, and the solvent was evaporated to afford a pale brown crude product. The crude product was dissolved in EtOH (10 mL), and then Pd/C (289 mg, 0.27 mmol) was added. The reaction mixture was stirred at rt under H2 until complete reduction of the double bond was observed by LCMS. The reaction was filtered through a Celite pad and the solvent evaporated. The resulting crude material was further purified by flash chromatography eluting with 0–30% EtOAc in heptanes to afford a colorless oil (640 mg, 91%). 1H NMR (400 MHz, CDCl3) δ 7.32 (d, J = 4.6 Hz, 1H), 7.24–7.17 (m, 2H), 7.14–7.09 (m, 3H), 6.88 (d, J = 4.8 Hz, 1H), 3.77 (s, 3H), 3.00 (t, J = 7.7 Hz, 2H), 2.61 (t, J = 7.7 Hz, 2H), 1.94–1.85 (m, 2H). LC-MS m/z 261 [M + H].
Methyl 5-Iodo-3-(3-phenylpropyl)thiophene-2-carboxylate (49b)
Prepared using general procedure A starting from 49a (640 mg, 2.5 mmol), pale yellow solid (791 mg, 66%). 1H NMR (500 MHz, CDCl3) δ 7.33–7.28 (m, 3H), 7.22–7.18 (m, 2H), 7.12 (s, 1H), 3.84 (s, 3H), 3.04 (t, J = 7.9 Hz, 2H), 2.70 (t, J = 7.8 Hz, 2H), 1.99–1.91 (m, 2H). LCMS m/z 387 [M+H, low intensity].
5-Iodo-3-(3-phenylpropyl)thiophene-2-carboxamide (49c)
Prepared using general procedure B starting from 49b (300 mg, 0.78 mmol), off-white solid (115 mg, 35%). 1H NMR (400 MHz, CDCl3) δ 7.34–7.28 (m, 2H), 7.22–7.19 (m, 3H), 7.12 (s, 1H), 5.41–5.41 (br.s, 2H), 2.95 (t, J = 7.7 Hz, 2H), 2.70 (t, J = 7.4 Hz, 2H), 2.01–1.94 (m, 2H). LC-MS m/z 372 [M + H].
5-Morpholino-3-(3-phenylpropyl)thiophene-2-carboxamide (49)
Prepared using general procedure E starting from 49c (120 mg, 0.32 mmol) and morpholine (48 mg, 0.55 mmol), off-white solid (39 mg, 36%). 1H NMR (500 MHz, CDCl3) δ 7.33–7.29 (m, 2H), 7.24–7.21 (m, 3H), 5.95 (s, 1H), 5.25 (br.s, 2H), 3.86–3.83 (m, 4H), 3.21–3.18 (m, 4H), 2.90 (t, J = 7.9 Hz, 2H), 2.73 (t, J = 7.6 Hz, 2H), 2.03–1.96 (m, 2H). HRMS (ES + ve), C18H23N2O2S [M + H]+: calcd 331.1475, found 331.1476.
Methyl 3-Phenacyloxythiophene-2-carboxylate (50a)
To a solution of methyl 3-hydroxythiophene-2-carboxylate (1000 mg, 6.3 mmol) in DMF (15 mL) were added 2-bromo-1-phenyl-ethanone (1510 mg, 7.6 mmol) and K2CO3 (1311 mg, 9.5 mmol). The resulting mixture was heated at 60 °C for 16 h. Then the reaction mixture was cooled to rt, diluted with DCM, and filtered to remove the base. Then the solvent was evaporated to dryness. The resulting crude was further purified by flash chromatography eluting with 0–100% EtOAc in heptanes to afford the desired product (1265 mg, 72%) as an orange solid. 1H NMR (400 MHz, CDCl3) δ 8.05–8.01 (m, 2H), 7.64 (t, J = 7.3 Hz, 1H), 7.52 (t, J = 7.4 Hz, 2H), 7.38 (d, J = 5.0 Hz, 1H), 6.76 (d, J = 5.0 Hz, 1H), 5.47 (s, 2H), 3.88 (s, 3H). LC-MS m/z 299 [M + Na].
Methyl 3-(2,2-Difluoro-2-phenylethoxy)thiophene-2-carboxylate (50b)
To a solution of 50a (400 mg, 1.5 mmol) in DCM (10 mL) was added DAST (817 mg, 5.1 mmol). The reaction mixture was stirred at 60 °C for 16 h. The solvent was removed under vacuum, and the crude was purified by flash chromatography eluting with 0–70% EtOAc in heptanes to afford the desired product (376 mg, 81%) as a yellow oil. 1H NMR (500 MHz, CDCl3) 7.68–7.65 (2H, m), 7.50–7.47 (3H, m), 7.40 (1H, d, J = 5.5 Hz), 6.78 (1H, d, J = 5.5 Hz), 4.54 (2H, t, J = 11.9 Hz), 3.85 (3H, s). LC-MS m/z 321 [M + Na].
Methyl 3-(2,2-Difluoro-2-phenylethoxy)-5-iodo-thiophene-2-carboxylate (50c)
Prepared using general procedure A starting from 50b (376 mg, 1.3 mmol), pale yellow solid (468 mg, 77%). 1H NMR (400 MHz, CDCl3) δ 7.65–7.62 (m, 2H), 7.51–7.47 (m, 3H), 6.94 (s, 1H), 4.50 (t, J = 11.9 Hz, 2H), 3.83 (s, 3H). LC-MS m/z 425 [M + H].
3-(2,2-Difluoro-2-phenylethoxy)-5-iodothiophene-2-carboxamide (50d)
Prepared using general procedure starting from 50c (467 mg, 1.1 mmol), off-white solid (233 mg, 46%). 1H NMR (500 MHz, CDCl3) δ 7.58–7.50 (m, 5H), 6.98 (s, 1H), 6.60 (br.s, 1H), 5.35 (br.s, 1H), 4.51 (t, J = 12.4 Hz, 2H). LC-MS m/z 410 [M + H].
3-(2,2-Difluoro-2-phenylethoxy)-5-morpholinothiophene-2-carboxamide (50)
To a microwave vial under a N2 atmosphere and precharged with 50d (233 mg, 0.57 mmol), CuI (22 mg, 0.11 mmol), l-Proline (26 mg, 0.23 mmol), and K2CO3 (314 mg, 2.27 mmol) was added morpholine (84 mg, 0.97 mmol) followed by DMSO (3.5 mL) and water (0.10 mL). The vial was sealed and stirred at 60 °C under microwave conditions for 3 h. The reaction was diluted with DCM, and H2O was added. The organics were separated with a hydrophobic frit and concentrated to dryness, and the crude was purified by flash chromatography 0–100% EtOAc in heptanes to afford an off-white solid (69 mg, 32%). 1H NMR (500 MHz, CDCl3) δ 7.59–7.56 (m, 2H), 7.54–7.49 (m, 3H), 6.42 (br.s, 1H), 5.75 (s, 1H), 5.17 (br.s, 1H), 4.49 (t, J = 12.3 Hz, 2H), 3.85–3.82 (m, 4H), 3.20–3.18 (m, 4H). HRMS (ES + ve), C17H18N2O3F2S [M + H]+: calcd 367.1076, found 369.1079.
3-(2-Hydroxy-2-phenylethoxy)-5-morpholinothiophene-2-carboxamine (51)
A solution of 7 (200 mg, 0.88 mmol) in DMF (2.5 mL) and K2CO3 (242 mg, 1.75 mmol) was stirred for 5 min; then 2-phenyloxirane (158 mg, 1.31 mmol) was added. The vial was sealed, and the reaction was heated up to 70 °C overnight. LCMS showed SM consumed and formation of desired product as the main product. The reaction was diluted with DCM and filtered to remove the inorganic base, the solvent removed, and the crude purified by column chromatography eluting with 0–5% MeOH in DCM. The mixture was then purified by prep HPLC (basic) to an orange solid (12 mg, 3.9% yield). 1H NMR (500 MHz, CDCl3) δ 7.47–7.38 (m, 5H), 5.82 (s, 1H), 5.16–5.12 (m, 1H), 4.27–4.25 (m, 2H), 3.83 (t, J = 5.0 Hz, 4H), 3.19 (t, J = 5.0 Hz, 4H), 2.62 (d, J = 3.2 Hz, 1H). LC-MS m/z 349 [M + H].
5-Morpholino-3-tetralin-1-yloxy-thiophene-2-carboxamide (52)
Prepared using general procedure D starting from 7 (250 mg, 1.1 mmol) and tetralin-1-ol (178 mg, 1.2 mmol), off-white solid (14 mg, 3%). 1H NMR (DMSO, 500 MHz) δ 7.35–7.33 (m, 1H), 7.29–7.26 (m, 1H), 7.24–7.17 (m, 2H), 6.93 (s, 1H), 6.49 (s, 1H), 6.36 (s, 1H) 5.56 (dd, 1H, J = 4.5, 4.5 Hz), 3.74 (m, 4H), 3.19 (m, 4H), 2.88–2.82 (m, 1H), 2.78–2.70 (m, 1H), 2.08–1.98 (m, 2H), 1.87–1.79 (m, 2H). HRMS (ES + ve), C19H22N2O3S [M + H]+: calcd 359.1424, found 359.1419.
3-Indan-1-yloxy-5-morpholinothiophene-2-carboxamide (53)
Prepared using general procedure D starting from indan-1-ol (176 mg, 1.31 mmol) and 7 (300 mg, 1.31 mmol), light yellow solid (37 mg, 8%). 1H NMR (500 MHz, DMSO) δ 7.52 (m, 2H), 7.21–7.08 (m, 4H), 7.17–7.13 (m, 2H), 5.27 (s, 1H), 4.29 (t, 1H, J = 8.0 Hz), 3.67–3.62 (m, 4H), 3.44 (m, 4H), 2.92–2.85 (m, 1H), 2.81–2.74 (m, 1H), 2.06–1.99 (m, 1H), 1.61 (m, 1H). HRMS (ES + ve), C18H21N2O3S [M + H]+: calcd 345.1268, found 345.1267.
4-Morpholino-2-phenethoxybenzamide (54)
Methyl 2-hydroxy-4-morpholino-benzoate (400 mg, 1.69 mmol) was dissolved in MeCN (2 mL), Cs2CO3 (549 mg, 1.69 mmol) and 2-bromoethylbenzene (343 mg, 1.8 mmol) were added, and the reaction was stirred at rt for 16 h. The inorganics were filtered off, and the excess solvent was removed in vacuo. The crude residue was columned, eluting with 0–100% EtOAC in heptane. The desired product was isolated and reacted on without further purification with LiNH2 (37 mg, 1.7 mmol) in THF (2 mL) at rt for 16 h. The reaction mixture was poured over ice and extracted into EtOAC, excess solvent was removed in vacuo, and the crude residue was purified by column chromatography eluting with 0–10% DCM/MeOH in DCM to afford the desired product as a colorless solid (6 mg, 1%). 1H NMR (500 MHz, CDCl3) δ 8.09 (d, J = 8.8 Hz, 1H), 7.39–7.35 (m, 2H), 7.32–7.29 (m, 3H), 6.58 (dd, J = 2.3, 8.9 Hz, 1H), 6.38 (d, J = 2.2 Hz, 1H), 5.33 (s, 2H), 4.39 (t, J = 6.8 Hz, 2H), 3.87 (t, J = 4.9 Hz, 4H), 3.28–3.20 (m, 6H). HRMS (ES + ve), C19H23N2O3 [M + H]+: calcd 327.1700, found 327.1703.
Methyl 5-Bromo-2-(2-phenylethoxy)benzoate (55a)
Prepared using general procedure D starting from methyl 5-bromo-2-hydroxy-benzoate (0.50 g, 2.2 mmol) and 2-phenylethanol (0.29 g, 2.4 mmol), colorless solid (0.46 g, 63%). 1H NMR (300 MHz, CDCl3) δ 7.91 (d, J = 2.4 Hz, 1H), 7.53 (dd, J = 2.4, 8.9 Hz, 1H), 7.34 (s, 5H), 6.84 (d, J = 8.9 Hz, 1H), 4.24 (t, J = 7.0 Hz, 2H), 3.91 (s, 3H), 3.17 (t, J = 7.0 Hz, 2H). LC-MS m/z 335, 337 [M+H, 1:1 Br characteristic isotopic abundance ratio].
Methyl 5-Morpholino-2-(2-phenylethoxy)benzoate (55b)
To a sealed vial precharged with 55a (457 mg, 1.36 mmol), Pd(OAc)2 (15.4 mg, 0.068 mmol), BINAP (85 mg, 0.14 mmol), and Cs2CO3 (666 mg, 2.04 mmol) under N2 was added toluene (4 mL) and then morpholine (154 mg, 1.77 mmol). The resulting mixture was then heated to 100 °C for 16 h. The reaction was then diluted with EtOAc, and the organics were washed with brine. The organics were then separated and dried (MgSO4) before concentration to dryness. The crude was purified by flash column chromatography eluting with 0–60% EtOAc in heptanes to afford the desired product (263 mg, 56%) as a yellow oil. 1H NMR (300 MHz, CDCl3) δ 7.39–7.32 (m, 6H), 7.04 (dd, J = 3.0, 9.0 Hz, 1H), 6.91 (d, J = 9.0 Hz, 1H), 4.21 (t, J = 7.1 Hz, 2H), 3.90–3.85 (m, 7H), 3.19–3.08 (m, 6H). LC-MS m/z 342.1 [M + H].
5-Morpholino-2-(2-phenylethoxy)benzamide (55)
Prepared using general procedure B starting from 55b (266 mg, 0.77 mmol), pale yellow solid (77 mg, 29%). 1H NMR (300 MHz, CDCl3) δ 7.78 (d, J = 3.0 Hz, 1H), 7.49 (br.s, 1H), 7.32–7.30 (m, 5H), 7.04 (dd, J = 3.1, 8.9 Hz, 1H), 6.96 (d, J = 9.0 Hz, 1H), 5.43 (br.s, 1H), 4.37 (t, J = 6.6 Hz, 2H), 3.90–3.85 (m, 4H), 3.22–3.11 (m, 6H). HRMS (ES + ve), C19H23N2O3 [M + H]+: calcd 327.1703, found 327.1697.
5-Bromo-2-[2-(3-fluorophenyl)ethoxy]benzoate (56a)
Prepared using general procedure D starting from methyl 5-bromo-2-hydroxy-benzoate (1.5 g, 6.5 mmol) and 2-(3-fluorophenyl)ethanol (1.0 g, 7.1 mmol), white solid (1.45 g, 63%). 1H NMR (500 MHz, CDCl3) δ 7.91 (d, J = 2.5 Hz, 1H), 7.53 (dd, J = 2.5, 8.8 Hz, 1H), 7.30–7.28 (m, 1H), 7.11–7.07 (m, 2H), 6.98–6.94 (m, 1H), 6.83 (d, J = 8.8 Hz, 1H), 4.23 (t, J = 6.6 Hz, 2H), 3.91 (s, 3H), 3.16 (t, J = 6.5 Hz, 2H). LC-MS m/z 353, 355 [M+H, 1:1 Br characteristic isotopic abundance ratio].
2-[2-(3-Fluorophenyl)ethoxy]-5-morpholino-benzoate (56b)
To a sealed vial precharged with 56a (400 mg, 1.1 mmol), Pd(OAc)2 (12.7 mg, 0.05 mmol), BINAP (71 mg, 0.1 mmol), and Cs2CO3 (554 mg, 1.7 mmol) under nitrogen atmosphere was added toluene (4 mL) and then morpholine (128 mg, 1.5 mmol). The resulting mixture was then heated to 100 °C for 16 h. The reaction was then diluted with EtOAc, and the organics were washed with brine. The organics were then separated and dried (MgSO4) before concentration in vacuo. The crude was purified by flash column chromatography eluting with 0–60% EtOAc in heptanes. The desired fractions were concentrated to dryness in vacuo to obtain a yellow solid (184 mg, 45%). 1H NMR (500 MHz, CDCl3) δ 7.37 (d, J = 3.0 Hz, 1H), 7.11–7.01 (m, 4H), 6.97–6.92 (m, 1H), 6.90 (d, J = 9.0 Hz, 1H), 4.21 (t, J = 6.6 Hz, 2H), 3.90 (s, 3H), 3.88–3.76 (m, 4H), 3.15–3.09 (m, 6H). LC-MS m/z 360.15 [M + H].
2-[2-(3-Fluorophenyl)ethoxy]-5-morpholino-benzamide (56)
Prepared using general procedure B starting from 56b (184 mg, 0.51 mmol), colorless solid (114 mg, 65%). 1H NMR (300 MHz, CDCl3) δ 7.78 (d, J = 3.2 Hz, 1H), 7.47 (br.s, 1H), 7.37–7.30 (m, 1H), 7.10–6.93 (m, 5H), 5.55 (br.s, 1H), 4.36 (t, J = 6.6 Hz, 2H), 3.87–3.78 (m, 4H), 3.22–3.12 (m, 6H). HRMS (ES + ve), C19H22FN2O3 [M + H]+: calcd 363.1373, found 363.1373.
6-Morpholino-3-(2-phenylethoxy)pyridine-2-carboxamide (57)
Prepared using general procedure B starting from methyl 6-morpholino-3-(2-phenylethoxy)pyridine-2-carboxylate colorless gum (100 mg, 15%). 1H NMR (400 MHz, CDCl3) δ 7.35- 7.12 (m, 6H), 6.71 (d, J = 10.2 Hz, 1H), 5.66 (s, 1H), 4.16 (t, J = 6.9 Hz, 2H), 3.75 (m, 4H), 3.36 (m, 4H), 3.08 (t, J = 7.02 Hz, 2H). HRMS (ES + ve), C18H23N2O2S [M + H]+: calcd 328.1656, found 328.1654.
Acknowledgments
We thank the following for technical assistance: James Ahn, Lena Anoshchenko, Shilah Bonnett, Alex Cookson, Julie Early, Lindsay Flint, Douglas Joerss, James Johnson, Susan Lawrie, Allison Morley, Alastair Pate, James Roberts, David Roberts, Bjorn Sunde, Dean Thompson, Anisa Tracy, and James Vela. This work was funded in part by Eli Lilly and Co. in support of the mission of the Lilly TB Drug Discovery Initiative and by the Bill and Melinda Gates Foundation, under grant OPP1024038. The work was also funded in part by the Bill and Melinda Gates Foundation & Wellcome Trust grant awards to University of Dundee - A Centre of Excellence for Lead Optimisation for Diseases of the Developing World (OPP1066891& 100195/Z/12/Z).
Glossary
Abbreviations Used
- MOT
morpholino thiophenes
- Qcrb
Cytochrome bc1 complex Cytochrome b subunit
- TB
tuberculosis
- HIV
human immunodeficiency virus
- MIC
minimum inhibitory concentration
- PFI
property forecast index
- CYP
cytochrome P450
- hERG
human Ether-à-go-go-Related Gene
- TPSA
total polar surface area
- DIAD
diisopropyl azodicarboxylate
- LDA
lithium diisopropylamide
- Cli
intrinsic clearance
- DCM
dichloromethane
- DMF
dimethylforamide
- AUC
area under curve
- HPV
hepatic portal vein
- PK
pharmacokinetic
- ER
extraction ration
- MDCK
madin darby canine kidney
- P-gp
P-glycoprotein
- HBD
hydrogen bond donor
- HTSA
high throughout solubility assay
- FaSSIF
fasted state gastric fluid
- CFU
colony forming unit
- WT
wild type
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b00172.
Author Contributions
‡ L.A.T.C. and P.C.R.: These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Global tuberculosis report 2015; World Health Organization: Geneva, Switzerland, 2015.
- Zumla A.; George A.; Sharma V.; Herbert R. H. N.; Oxley A.; Oliver M. The WHO 2014 Global tuberculosis report-further to go. Lancet Global Health 2015, 3 (1), e10–e12. 10.1016/S2214-109X(14)70361-4. [DOI] [PubMed] [Google Scholar]
- Dheda K.; Barry C. E. 3rd; Maartens G. Tuberculosis. Lancet 2016, 387 (10024), 1211–1226. 10.1016/S0140-6736(15)00151-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsburgh C. R. J.; Barry C. E. I.; Lange C. Treatment of Tuberculosis. N. Engl. J. Med. 2015, 373 (22), 2149–2160. 10.1056/NEJMra1413919. [DOI] [PubMed] [Google Scholar]
- Koul A.; Arnoult E.; Lounis N.; Guillemont J.; Andries K. The challenge of new drug discovery for tuberculosis. Nature 2011, 469 (7331), 483–490. 10.1038/nature09657. [DOI] [PubMed] [Google Scholar]
- Lechartier B.; Rybniker J.; Zumla A.; Cole S. T. Tuberculosis drug discovery in the post-post-genomic era. EMBO Molecular Medicine 2014, 6 (2), 158–168. 10.1002/emmm.201201772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunatha U. H.; Smith P. W. Perspective: Challenges and opportunities in TB drug discovery from phenotypic screening. Bioorg. Med. Chem. 2015, 23 (16), 5087–5097. 10.1016/j.bmc.2014.12.031. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6 (11), 881–890. 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Young R. J.; Green D. V. S.; Luscombe C. N.; Hill A. P. Getting physical in drug discovery II: the impact of chromatographic hydrophobicity measurements and aromaticity. Drug Discovery Today 2011, 16 (17–18), 822–830. 10.1016/j.drudis.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Bayliss M. K.; Butler J.; Feldman P. L.; Green D. V. S.; Leeson P. D.; Palovich M. R.; Taylor A. J. Quality guidelines for oral drug candidates: dose, solubility and lipophilicity. Drug Discovery Today 2016, 21 (10), 1719–1727. 10.1016/j.drudis.2016.07.007. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. Improving Drug Design: An Update on Recent Applications of Efficiency Metrics, Strategies for Replacing Problematic Elements, and Compounds in Nontraditional Drug Space. Chem. Res. Toxicol. 2016, 29 (4), 564–616. 10.1021/acs.chemrestox.6b00043. [DOI] [PubMed] [Google Scholar]
- Waring M. J. Lipophilicity in drug discovery. Expert Opin. Drug Discovery 2010, 5 (3), 235–248. 10.1517/17460441003605098. [DOI] [PubMed] [Google Scholar]
- Booker M. L.; Bastos C. M.; Kramer M. L.; Barker R. H.; Skerlj R.; Sidhu A. B.; Deng X.; Celatka C.; Cortese J. F.; Guerrero Bravo J. E.; Crespo Llado K. N.; Serrano A. E.; Angulo-Barturen I.; Jiménez-Díaz M. B.; Viera S.; Garuti H.; Wittlin S.; Papastogiannidis P.; Lin J.-w.; Janse C. J.; Khan S. M.; Duraisingh M.; Coleman B.; Goldsmith E. J.; Phillips M. A.; Munoz B.; Wirth D. F.; Klinger J. D.; Wiegand R.; Sybertz E. Novel Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase with Anti-malarial Activity in the Mouse Model. J. Biol. Chem. 2010, 285 (43), 33054–33064. 10.1074/jbc.M110.162081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmitte K. A.; Andrews C. W.; Badiang J. G.; Davis-Ward R. G.; Dickson H. D.; Drewry D. H.; Emerson H. K.; Epperly A. H.; Hassler D. F.; Knick V. B.; Kuntz K. W.; Lansing T. J.; Linn J. A.; Mook R. A. Jr; Nailor K. E.; Salovich J. M.; Spehar G. M.; Cheung M. Discovery of thiophene inhibitors of polo-like kinase. Bioorg. Med. Chem. Lett. 2009, 19 (3), 1018–1021. 10.1016/j.bmcl.2008.11.041. [DOI] [PubMed] [Google Scholar]
- Emmitte K. A.; Adjebang G. M.; Andrews C. W.; Alberti J. G. B.; Bambal R.; Chamberlain S. D.; Davis-Ward R. G.; Dickson H. D.; Hassler D. F.; Hornberger K. R.; Jackson J. R.; Kuntz K. W.; Lansing T. J.; Mook R. A. Jr; Nailor K. E.; Pobanz M. A.; Smith S. C.; Sung C.-M.; Cheung M. Design of potent thiophene inhibitors of polo-like kinase 1 with improved solubility and reduced protein binding. Bioorg. Med. Chem. Lett. 2009, 19 (6), 1694–1697. 10.1016/j.bmcl.2009.01.094. [DOI] [PubMed] [Google Scholar]
- Rheault T. R.; Donaldson K. H.; Badiang-Alberti J. G.; Davis-Ward R. G.; Andrews C. W.; Bambal R.; Jackson J. R.; Cheung M. Heteroaryl-linked 5-(1H-benzimidazol-1-yl)-2-thiophenecarboxamides: Potent inhibitors of polo-like kinase 1 (PLK1) with improved drug-like properties. Bioorg. Med. Chem. Lett. 2010, 20 (15), 4587–4592. 10.1016/j.bmcl.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Solanki S.; Innocenti P.; Mas-Droux C.; Boxall K.; Barillari C.; van Montfort R. L. M.; Aherne G. W.; Bayliss R.; Hoelder S. Benzimidazole Inhibitors Induce a DFG-Out Conformation of Never in Mitosis Gene A-Related Kinase 2 (Nek2) without Binding to the Back Pocket and Reveal a Nonlinear Structure–Activity Relationship. J. Med. Chem. 2011, 54 (6), 1626–1639. 10.1021/jm1011726. [DOI] [PubMed] [Google Scholar]
- Ibrahim D. A.; Boucau J.; Lajiness D. H.; Veleti S. K.; Trabbic K. R.; Adams S. S.; Ronning D. R.; Sucheck S. J. Design, Synthesis, and X-ray Analysis of a Glycoconjugate Bound to Mycobacterium tuberculosis Antigen 85C. Bioconjugate Chem. 2012, 23 (12), 2403–2416. 10.1021/bc3004342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Sambandan D.; Halder R.; Wang J.; Batt S. M.; Weinrick B.; Ahmad I.; Yang P.; Zhang Y.; Kim J.; Hassani M.; Huszar S.; Trefzer C.; Ma Z.; Kaneko T.; Mdluli K. E.; Franzblau S.; Chatterjee A. K.; Johnsson K.; Mikusova K.; Besra G. S.; Fütterer K.; Robbins S. H.; Barnes S. W.; Walker J. R.; Jacobs W. R.; Schultz P. G. Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (27), E2510–E2517. 10.1073/pnas.1309171110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S.; Kim R. Y.; Seo M. J.; Lee S.; Kim Y. M.; Seo M.; Seo J. J.; Ko Y.; Choi I.; Jang J.; Nam J.; Park S.; Kang H.; Kim H. J.; Kim J.; Ahn S.; Pethe K.; Nam K.; No Z.; Kim J. Lead Optimization of a Novel Series of Imidazo[1,2-a]pyridine Amides Leading to a Clinical Candidate (Q203) as a Multi- and Extensively-Drug-Resistant Anti-tuberculosis Agent. J. Med. Chem. 2014, 57 (12), 5293–5305. 10.1021/jm5003606. [DOI] [PubMed] [Google Scholar]
- Pethe K.; Bifani P.; Jang J.; Kang S.; Park S.; Ahn S.; Jiricek J.; Jung J.; Jeon H. K.; Cechetto J.; Christophe T.; Lee H.; Kempf M.; Jackson M.; Lenaerts A. J.; Pham H.; Jones V.; Seo M. J.; Kim Y. M.; Seo M.; Seo J. J.; Park D.; Ko Y.; Choi I.; Kim R.; Kim S. Y.; Lim S.; Yim S.-A.; Nam J.; Kang H.; Kwon H.; Oh C.-T.; Cho Y.; Jang Y.; Kim J.; Chua A.; Tan B. H.; Nanjundappa M. B.; Rao S. P. S.; Barnes W. S.; Wintjens R.; Walker J. R.; Alonso S.; Lee S.; Kim J.; Oh S.; Oh T.; Nehrbass U.; Han S.-J.; No Z.; Lee J.; Brodin P.; Cho S.-N.; Nam K.; Kim J. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013, 19 (9), 1157–1160. 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- Abrahams K. A.; Cox J. A. G.; Spivey V. L.; Loman N. J.; Pallen M. J.; Constantinidou C.; Fernández R.; Alemparte C.; Remuiñán M. J.; Barros D.; Ballell L.; Besra G. S. Identification of Novel Imidazo[1,2-a]pyridine Inhibitors Targeting M. tuberculosis QcrB. PLoS One 2013, 7 (12), e52951. 10.1371/journal.pone.0052951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.; Moraski G. C.; Cramer J.; Miller M. J.; Schorey J. S. Bactericidal Activity of an Imidazo[1, 2-a]pyridine Using a Mouse M. tuberculosis Infection Model. PLoS One 2014, 9 (1), e87483. 10.1371/journal.pone.0087483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora K.; Ochoa-Montaño B.; Tsang P. S.; Blundell T. L.; Dawes S. S.; Mizrahi V.; Bayliss T.; Mackenzie C. J.; Cleghorn L. A. T.; Ray P. C.; Wyatt P. G.; Uh E.; Lee J.; Barry C. E.; Boshoff H. I. Respiratory Flexibility in Response to Inhibition of Cytochrome c Oxidase in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58 (11), 6962–6965. 10.1128/AAC.03486-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsunobu O.; Yamada M. Preparation of Esters of Carboxylic and Phosphoric Acid via Quaternary Phosphonium Salts. Bull. Chem. Soc. Jpn. 1967, 40 (10), 2380–2382. 10.1246/bcsj.40.2380. [DOI] [Google Scholar]
- Bundesmann M. W.; Coffey S. B.; Wright S. W. Amidation of esters assisted by Mg(OCH3)2 or CaCl2. Tetrahedron Lett. 2010, 51 (30), 3879–3882. 10.1016/j.tetlet.2010.05.075. [DOI] [Google Scholar]
- Miyaura N.; Yamada K.; Suzuki A. A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett. 1979, 20 (36), 3437–3440. 10.1016/S0040-4039(01)95429-2. [DOI] [Google Scholar]
- Muci A. R.; Buchwald S. L.. Practical Palladium Catalysts for C-N and C-O Bond Formation. In Cross-Coupling Reactions: A Practical Guide; Miyaura N., Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2002; pp 131–209. [Google Scholar]
- Beno B. R.; Yeung K.-S.; Bartberger M. D.; Pennington L. D.; Meanwell N. A. A Survey of the Role of Noncovalent Sulfur Interactions in Drug Design. J. Med. Chem. 2015, 58 (11), 4383–4438. 10.1021/jm501853m. [DOI] [PubMed] [Google Scholar]
- Seo M.; Park J.; Kim S. Y. Self-assembly driven by an aromatic primary amide motif. Org. Biomol. Chem. 2012, 10 (28), 5332–5342. 10.1039/c2ob25117e. [DOI] [PubMed] [Google Scholar]
- Rullas J.; García J. I.; Beltrán M.; Cardona P.-J.; Cáceres N.; García-Bustos J. F.; Angulo-Barturen I. Fast Standardized Therapeutic-Efficacy Assay for Drug Discovery against Tuberculosis. Antimicrob. Agents Chemother. 2010, 54 (5), 2262–2264. 10.1128/AAC.01423-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak P. A.; Rao S. P. S.; Ping Tan M.; Lin X.; Chyba J.; Tay J.; Ng S. H.; Tan B. H.; Cherian J.; Duraiswamy J.; Bifani P.; Lim V.; Lee B. H.; Ling Ma N.; Beer D.; Thayalan P.; Kuhen K.; Chatterjee A.; Supek F.; Glynne R.; Zheng J.; Boshoff H. I.; Barry C. E.; Dick T.; Pethe K.; Camacho L. R. A High-Throughput Screen To Identify Inhibitors of ATP Homeostasis in Non-replicating Mycobacterium tuberculosis. ACS Chem. Biol. 2012, 7 (7), 1190–1197. 10.1021/cb2004884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman R. C. Why are membrane targets discovered by phenotypic screens and genome sequencing in Mycobacterium tuberculosis?. Tuberculosis 2013, 93 (6), 569–588. 10.1016/j.tube.2013.09.003. [DOI] [PubMed] [Google Scholar]
- Chandrasekera N. S.; Berube B. J.; Shetye G.; Chettiar S.; O’Malley T.; Manning A.; Flint L.; Awasthi D.; Ioerger T. R.; Sacchettini J.; Masquelin T.; Hipskind P. A.; Odingo J.; Parish T. Improved Phenoxyalkylbenzimidazoles with Activity against Mycobacterium tuberculosis Appear to Target QcrB. ACS Infect. Dis. 2017, 3 (12), 898–916. 10.1021/acsinfecdis.7b00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phummarin N.; Boshoff H. I.; Tsang P. S.; Dalton J.; Wiles S.; Barry C. E. III; Copp B. R. SAR and identification of 2-(quinolin-4-yloxy)acetamides as Mycobacterium tuberculosis cytochrome bc (1) inhibitors. MedChemComm 2016, 7 (11), 2122–2127. 10.1039/C6MD00236F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Westhuyzen R.; Winks S.; Wilson C. R.; Boyle G. A.; Gessner R. K.; Soares de Melo C.; Taylor D.; de Kock C.; Njoroge M.; Brunschwig C.; Lawrence N.; Rao S. P. S.; Sirgel F.; van Helden P.; Seldon R.; Moosa A.; Warner D. F.; Arista L.; Manjunatha U. H.; Smith P. W.; Street L. J.; Chibale K. Pyrrolo[3,4-c]pyridine-1,3(2H)-diones: A Novel Antimycobacterial Class Targeting Mycobacterial Respiration. J. Med. Chem. 2015, 58 (23), 9371–9381. 10.1021/acs.jmedchem.5b01542. [DOI] [PubMed] [Google Scholar]
- Esser L.; Quinn B.; Li Y.-F.; Zhang M.; Elberry M.; Yu L.; Yu C.-A.; Xia D. Crystallographic Studies of Quinol Oxidation Site Inhibitors: A Modified Classification of Inhibitors for the Cytochrome bc1 Complex. J. Mol. Biol. 2004, 341 (1), 281–302. 10.1016/j.jmb.2004.05.065. [DOI] [PubMed] [Google Scholar]
- O’Malley T.; Alling T.; Early J. V.; Wescott H. A.; Kumar A.; Moraski G. C.; Miller M. J.; Masquelin T.; Hipskind P. A.; Parish T. Imidazopyridine compounds inhibit mycobacterial growth by depleting ATP levels. Antimicrob. Agents Chemother. 2018, 62 (6), e02439-17. 10.1128/AAC.02439-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollinger J.; Bailey M. A.; Moraski G. C.; Casey A.; Florio S.; Alling T.; Miller M. J.; Parish T. A Dual Read-Out Assay to Evaluate the Potency of Compounds Active against Mycobacterium tuberculosis. PLoS One 2013, 8 (4), e60531. 10.1371/journal.pone.0060531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwochert J.; Lao Y.; Pye C. R.; Naylor M. R.; Desai P. V.; Gonzalez Valcarcel I. C.; Barrett J. A.; Sawada G.; Blanco M.-J.; Lokey R. S. Stereochemistry Balances Cell Permeability and Solubility in the Naturally Derived Phepropeptin Cyclic Peptides. ACS Med. Chem. Lett. 2016, 7 (8), 757–761. 10.1021/acsmedchemlett.6b00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.