

Cell-based immune therapies are treatment strategies based on the purposeful manipulation of cells to galvanize the immune system against specific antigens. Multiple types of immune cell have been used in this context, with cell-type selection depending on the cells’ intrinsic functions and therapeutic needs. Examples of immune-cell-based therapies include modified natural killer (NK) cells1 (such as those marketed by Dragonfly Therapeutics), chimeric antigen receptor (CAR)-T cells for the treatment of B-cell malignancies2 (Novartis and Kite Pharma) and dendritic cell (DC)-based therapies (Dendreon and DCPrime) against multiple cancers, such as prostate cancer and leukaemia3,4. The past decade has witnessed a flurry of advancements in such therapies, particularly in the use of DCs to co-opt the immune system for killing tumour cells in patients. A variety of DC vaccine formulations, delivery methods and antigen compositions are being explored in clinical trials, especially in combination with other potent therapies such as immune checkpoint-blockade inhibition. In this Comment, we discuss current DC-vaccine practices for treating cancer and speculate how far DCs can be pushed to achieve a ‘super-DC’ vaccine that drives strong antigen-specific immunity against cancer.
Dendritic cells
DCs are a heterogeneous population of leukocytes with an unmatched capacity for activating cellular and humoral immunity. Classically, DCs are characterized as (i) conventional DCs (cDCs), consisting of two subtypes, cDC1 and cDC2; (ii) plasmacytoid DCs (pDCs); (iii) Langerhan cells (LCs), a DC-like subset generally localized in the epidermis; and (iv) inflammatory DCs (iDCs), a subset of DCs that appears during inflammatory responses and originates mainly from monocyte precursors (Fig. 1). However, with advances in cell labelling and lineage-tracing technologies, the standard classification and lineage of DC subsets is being revised. For instance, LCs have been suggested to be more closely related to macrophages than to DCs (ref. 5). Moreover, new putative DC subsets branching from cDC2 and pDCs have been recently identified using single-cell RNA sequencing6. Additionally, a recent study using time-of-flight mass cytometry to phenotype human DCs indicated significant inter-individual heterogeneity within the cDC2 subset. The same study also found evidence of selective localization of certain DC subsets to skin or blood and to lymphoid organs7. However, the physiological relevance of these new subsets remains to be verified.
Fig. 1 |. Classical and newly discovered DC subsets.
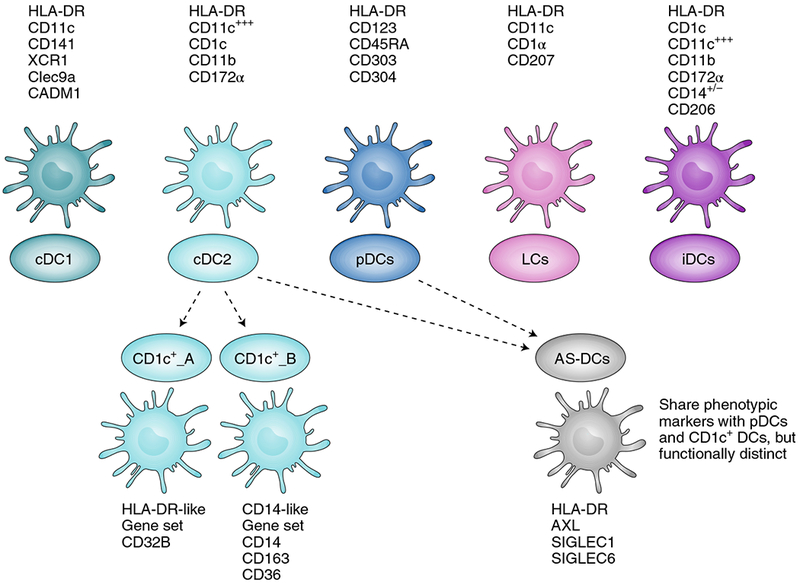
DCs have been divided into several subsets, namely conventional DCs (cDC1, cDC2), plasmacytoid DCs (pDCs), Langerhan cells (LCs) and inflammatory DCs (iDCs), according to their functional properties and phenotype. Cell surface markers generally used to identify these subsets from blood and tissue are listed with each cell type. cDC1 and cDC2 can present antigens on MHC-I and MHC-II, and can thus activate both CD8+ and CD4+ T cells. cDC1 are primarily lymph-node-resident DCs recognized for their exceptional capacity for antigen crosspresentation and for activating CD8+ T cells. They can secrete high levels of type-I and type-III IFNs and IL-12 when stimulated with double-stranded RNA (dsRNA) poly-IC:LC (poly-inosinic and poly-cytidylic acids stabilized with poly-lysine). cDC2 are migratory DCs known for activating CD4+ T cells and for secreting high levels of IL-12 on activation. pDCs are primarily type-I IFN-secreting cells, and play a role in activating other DC subsets, T cells and B cells. Single-cell sequencing and unbiased genome classification of HLA-DR+Lin− cells isolated from blood suggests the existence of additional DC subsets (dotted lines), namely CD1c+_A, CD1c+_B and Axl+SIGLEC6+ DCs (AS-DCs). CD1c+_A and CD1c+_B are new subsets found within conventional cDC2 (CD1c+). The newly described AS-DC subset bears phenotypic markers similar to those found in both pDC and CD1c subsets, but deeper analysis revealed that AS-DCs were functionally distinct from pDCs despite their phenotypic similarity6.
Each DC subset is naturally and uniquely specialized to promote CD4+ and/or CD8+ T-cell activation, and their capabilities as natural adjuvants makes them particularly well suited for cell-based therapies8. With this knowledge in mind, multiple clinical trials over the past two decades have applied monocyte-derived DCs (MoDCs) or CD34+ stem-cell-derived DCs as vaccines in the context of cancer therapy. In 2010, Sipuleucel-T (marketed as PROVENGE; Dendreon), an autologous MoDC vaccine loaded with the tumour antigen PA2024 (a complex of prostatic acid phosphatase fused with the cytokine granulocyte\p=n-\macrophage colony-stimulating factor (GM-CSF)) received approval by the US Food and Drug Administration (FDA) as therapy against castration-resistant prostate cancer; but as a monotherapy its performance in the clinic was ultimately underwhelming. However, several DC vaccines have yielded favourable immune or clinical responses. Examples of such vaccines include a DC–tumour fusion vaccine, generated by fusing DCs with patient-derived tumour cells for treatment of acute myeloid leukaemia (AML)9; MoDCs transfected with mRNA encoding the shared antigen Wilms’ tumour protein (WT-1) for treating AML10; and MoDCs pulsed with the shared antigens tyrosinase and gp100 for treating melanoma11. Nevertheless, despite promising safety, tolerability and immunogenicity profiles, DC-vaccine trials, overall, have elicited only limited clinical responses. The apparent lack of success of MoDC vaccines may be attributed to several causes, including enhanced tumour burden and immune suppression (see Box 1; additional information on DC activation and mechanisms of tumour-induced suppression have been reviewed elsewhere12). Consequently, the main focus now is on designing improved DC-based vaccines that can override tumour-driven dysregulation of the immune system13,14. Increasing efforts are also being poured into testing new combinations of immunotherapies that can induce lasting antitumour immune responses.
Box 1 |. Possible reasons for the limited efficacy of DC vaccines in clinical trials.
Inherent differences between MoDCs and tissue-resident or lymph-node-resident bona fide DC subsets.
Possible need for engaging multiple DC subsets to prime and enhance immunity21,22.
The type of antigens, loading strategy and vaccine delivery approaches used in trials require refinement.
Inefficient DC migration from the site of injection to the draining lymph nodes.
Immune suppression brought on by the tumour.
Need for effective combination approaches to treat advanced disease.
In vivo DC targeting
Techniques that target DCs in vivo focus on manipulating DCs within the host without requiring ex vivo DC modulation. These include in situ vaccines that target DCs within the tumour microenvironment through intratumoral injections of Toll-like receptor (TLR) ligands (such as poly-IC:LC, CpG or TLR7/8 agonists)15 or FMS-like tyrosine kinase 3 ligand (Flt3L), which trigger recruitment and local activation of DCs in the tumour. Furthermore, intratumoral administration of a combination of DC-activating factors, by way of injecting mRNA encoding CD40L, CD70 and constitutively active TLR4 (TriMix mRNA; eTheRNA immunotherapies), has been shown to reprogram suppressed DCs to induce functional antitumour T-cell immunity16,17. Systemically, DCs may be broadly targeted by vaccination with tumour-associated antigens (TAAs) conjugated with antibodies specific for cell-surface receptors expressed by all DCs, such as CDX-1401 (an anti-DEC-205 antibody conjugated with the NY-ESO1 tumour antigen from Celldex Therapeutics being tested in clinical trials NCT03358719 and NCT02166905), Clec12A (ref. 18) and potentially by CD40 agnostic antibodies (being evaluated in clinical trials NCT02376699, NCT02482168 and NCT01103635).
Several studies indicate that eliciting a strong and sustained cytotoxic T-cell response requires simultaneous or sequential activation of multiple subsets of antigen-presenting cells (APCs) such as DCs and macrophages19,20. Indeed, data from preclinical studies exploring nanoparticles encapsulating TAA-encoding RNA and lipid complexes demonstrate that activated pDCs drive the initial wave of immune activation by secreting type-I interferons (IFNs), which leads to cDC maturation, activation, lymph-node trafficking and T-cell activation. Then, a second wave of immune activation triggered by cytokine-secreting macrophages further promotes T-cell activation and antigen-specific functions21,22. However, broad APC-targeting vaccines run the inherent risk of inducing a deleterious cytokine storm. To avoid this adverse consequence, specific DC subsets may be engaged by using antibodies that are reactive against subset-specific factors. For example, the cDC1 subset could be targeted via XCR1, Clec9A or chemokines such as XCL1 (a ligand for XCR1)23. Similarly, immature DCs and iDCs may be targeted by antibodies specific for mannose receptor (as in the use of CDX-1307, tested in clinical trials NCT00648102 and NCT00709462). Another approach for DC subset-specific targeting employs vaccination with nanoparticles (encapsulating TAAs and TLR ligands) conjugated with the above-mentioned antibodies to avoid damaging systemic inflammation. This nanoparticle-based approach can potentially also be used to deliver cytokines, short or long peptides, total protein or RNA molecules to the target cells24.
Oncolytic virus therapy (virotherapy) is yet another mode of targeting DCs in vivo. Virotherapy involves vaccination with attenuated viral agents (including adenovirus, vaccinia virus, Maraba virus, measles virus, Coxsackie virus, New Castle Disease virus or influenza virus) that are genetically modified to boost tumour tropism and to express TAAs and cytokines for the activation and recruitment of DCs to the tumour microenvironment12. Furthermore, oncoviruses have been used to induce immunogenic cell death in tumour cells, enabling the TAA acquisition by recruited DCs and exposing the DCs to inflammatory maturation signals25. Talimogene Laherparepvec (IMLYGIC; Amgen), an attenuated GM-CSF-expressing herpes simplex virus, has been approved by the FDA for use as an intratumoral vaccine for the treatment of inoperable melanoma lesions26.
Other means of targeting DCs indirectly in vivo include the induction of immunogenic tumour cell death by chemotherapy, radiation and ultrasound27, and the use of improved RNA vaccines (such as those being developed by BioNTECH (LipoMerit; NCT02410733), Moderna, eTheRNA and Curevac (for detailed information on RNA vaccine formulation, see ref. 28)), DNA vaccines (such as Vaccibody’s VB10.6 and Innovio’s INO-9012), and irradiated tumour cells manipulated to secrete GM-CSF (the GVAX system)15.
Ex vivo preparation of DC vaccines
Currently, the most common DC-vaccine approach entails isolating monocytes from patient-derived peripheral blood mononuclear cells (PBMCs) and differentiating them into mature MoDCs under the influence of exogenous interleukin-4 (IL-4) and GM-CSF together with maturation factors such as TLR ligands, other cytokines and CD40L. DCs loaded with desired antigens (delivered via peptides, proteins, RNA or viral vectors) are administered to patients through multiple approaches such as intranodal, intravenous or intratumoral injections. However, MoDCs, despite being immunogenic, inherently lack the superior adjuvant capacity and immune-activating properties of natural DCs, such as cross-presentation and optimal production of IL-12 and type-I interferon (IFN) (ref. 29). Moreover, the generation of MoDC vaccines is a multistep and time-consuming process. Using natural DCs would provide an elegant solution for avoiding MoDC differentiation and their lack of potent physiological characteristics. However, given the low frequency of circulating DCs in blood (<1.0% of circulating PBMCs), isolating a sufficient number of cells for vaccinating in multiple rounds is difficult and requires large-scale PBMC isolation via leukapheresis. Additionally, there are no commercially available kits for the isolation of specialized DC subsets such as the cross-presenting CD141+ DCs. Flt3L-administration-based mobilization is a promising strategy for in vivo amplification of all DC subsets (making it possible to isolate rare DC subsets such as pDCs and CD1c+ DCs using commercially available kits) for use in vaccines30,31. This strategy may also provide an opportunity to isolate the DC subsets functionally specialized for antigen cross-presentation, such as CD141+ DCs, using cell-sorting technology.
Antigen selection.
The choice of TAA to be loaded onto DCs is critical for the optimization of ex vivo DC vaccines (for a detailed discussion regarding the selection of antigens, see refs 15,32). Neoantigens — unique, cancer-specific antigens that arise due to tumorigenic mutations — have shown particular promise as candidates for cancer vaccines. Notably, the intravenous immunization of three melanoma patients with autologous MoDCs loaded with personalized neoantigen peptides elicited clear evidence of CD8+ T-cell activation, which was specific to several neoantigens in the vaccine. This study set the standard for future clinical trials testing ex vivo DC vaccines loaded with personalized neoantigens33.
However, not all neoantigens are immunogenic, nor do all tumours express high levels of neoantigens. Pulsing DCs with tumour lysates34 or fusing tumour cells with MoDCs (ref. 9) can also yield significant cytotoxic T-lymphocytic (CTL) responses and even lead to clinical activity, especially when administered with checkpoint-blockade inhibitors and strong adjuvants. The mode of antigen loading, for example by RNA transfection (in the case of TriMix DCs)35 or lentiviral transduction (in the case of self-differentiated myeloid-derived antigen-presenting-cells reactive against tumours; SMART-DCs)36, can also substantially impact the final clinical outcome of DC vaccines. DCs may be modulated (through the lentivirus-driven expression of extracellular vesicle-internalizing receptors) to derive specific antigens from tumour-released extracellular vesicles, and present these antigens on their major histocompatibility complex (MHC)-I molecules (by a process known as ‘cross-dressing’) to activate CD8+ T cells37. Furthermore, induction of tumour-cell necrosis with oncoviruses, chemicals, radiation or high-intensity ultrasound leads to the release of alarmins and tumour antigens and to the recruitment of DCs to the tumour microenvironment, thereby enhancing antitumour CTL responses25,27. Therefore, there are many ways in which non-personalized neoantigen vaccines may be improved. In this regard, several questions need to be raised: are the massive resources required for identifying epitopes and then generating personalized neoantigen vaccines justified? Should resources also be directed towards improving DC–tumour fusion vaccines, whole tumour lysate vaccines and shared TAA vaccines?
Envisioning a super-DC vaccine
In what follows, we speculate how recent developments and technological advances in cell-based vaccines can be taken a step further towards the development of a super-DC vaccine. Ideally, a super-DC vaccine would be able to induce and restore functional antitumour immunity with little or negligible off-target toxicity, particularly when delivered in combination with other modalities such as checkpoint-blockade inhibition.
Bulk production of DCs.
The first step towards this goal is to acquire bulk quantities of the desired DC subsets. To this end, we and others are successfully developing platforms to use CD34+ haematopoietic stem cells for the large-scale generation (on the order of millions of cells) of specific DC subsets such as CD141+Clec9a+ DCs, pDCs, LCs and CD1d+ DCs (ref. 38). The bulk production of DC subsets will enable further testing of the important observation that engagement of more than one distinct DC subset (cDCs and pDCs) is required for the optimal induction of immunity21,22. Once matured, these DC subsets would be loaded with the antigen of choice via a variety of methods (such as antigen pulsing, mRNA transfection and lentiviral transduction) and administered to patients29.
Off-the-shelf DC vaccine.
The ideal scenario for any DC vaccine is the bulk production of universal off-the-shelf vaccines. DCOne (DCPrime’s allogeneic DC vaccine) may be one such vaccine candidate. DCOne is a DC-like cell line, generated from an AML patient, expressing multiple co-stimulatory molecules and presenting multiple shared TAAs (such as WT-1). In a phase I/II clinical trial (NCT01373515) involving patients with advanced stage AML, the DCOne vaccine displayed no adverse events while inducing antigen-specific immunity. In vitro experiments have shown that DCOne cells can also induce protective immunity against multiple myeloma cells through the process of transfer of antigens (such as MUC-1 and survivin) to a patient’s endogenous APCs (ref. 4). These results are consistent with previous observations showing the importance of antigen transfer by exogenous DCs to host DCs in the generation and amplification of a potent CTL response39. Similarly, an allogeneic MoDC–tumour fusion vaccine tested in patients with renal cell carcinoma has proved to be safe and immunogenic40.
Importantly, allogeneic DC formulation, Ilixadencel (Immunicum; NCT02686944 and NCT02432846) has been used safely as an intratumoral adjuvant. Thus, desired super-DC subsets generated from CD34+ stem cells or monocyte precursors could potentially be used as an allogeneic vaccine without significant adverse events, using human leukocyte antigen (HLA) mismatch to provide adjuvant-like activation signals. Moreover, the allogeneic DCs could be employed using shared TAAs such as WT-1, MUC-1 or survivin, or loaded with patient-specific neoantigens, for personalized tumour therapies. Additional studies are still required to fully confirm the antigen-specific clinical benefits of this system.
Targeted inhibition of checkpoint genes on specific DC subsets.
Tumours can induce immune exhaustion, extending beyond T cells and affecting APCs. Therefore, DC vaccines are now being tested in tumour-free or early tumour stage settings, in which immunogenicity is higher owing to the lack of tumour-mediated immunosuppression and where different DC systems can be more readily compared. To overcome immune suppression in advanced cancers, DC vaccinations are being evaluated in combination with checkpoint-blockade inhibition at different stages of tumorigenesis (Table 1). So far, checkpoint-blockade interventions using antibodies against PD-1/PD-L1 (programmed cell death protein 1; programmed death-ligand 1) and CTLA-4 (cytotoxic T-lymphocyte-associated protein 4) have focused mechanistically on either the T cell or the tumour cell. Notably, all DC subsets express a myriad of immune inhibitory and stimulatory molecules that can dictate activation of both DCs and T cells. We analysed microarray datasets from published literature41–43 for the expression of select inhibitory checkpoint and stimulatory genes on DC subsets activated in vitro (Fig. 2). Our analysis revealed distinct checkpoint-blockade gene signatures for CD141+ DCs, MoDCs and pDCs. For example, both CD141+ DCs and MoDCs expressed high levels of PD-L1 and PD-L2 even before activation, whereas pDCs acquired PD-L1 and PD-L2 expression only on activation. By contrast, CTLA-4 was expressed at high levels on MoDCs (basal and post-activation), but not on CD141+ DCs and pDCs. Conversely, CD200 was found to be induced only on activated CD141+ DCs. Indoleamine 2,3-dioxygenase 1 (IDO1) was expressed at high levels on CD141+ DCs (basal and post-activation), on activated MoDCs and, to a lesser degree, on activated pDCs. Homeodomain-interacting protein kinase-1 (HPK-1), a kinase recently identified as a negative regulator of DC and T-cell function44, was found to be highly expressed at steady state on CD141+ DCs and was downregulated on activation. These observations suggest that the same checkpoint-blockade approach may not be optimal for all DC subsets. Hence, the targeted inhibition of checkpoint-blockade factors enriched in specific DC subsets should be used to enhance the efficacy of super-DC vaccines.
Table 1 |.
Current clinical trials testing checkpoint-blockade inhibition in combination with DC vaccines for the treatment of cancer
| Condition | NCT number | Intervention | Phase | Status |
|---|---|---|---|---|
| Melanoma | NCT03092453 | Anti-PD-1 (Pembrolizumab) + autologous DC vaccine | I | Recruiting |
| Solid neoplasm | NCT02775292 | Anti-PD-1 (Nivolumab) + NY-ESO-1 reactive TCR retroviral vector transduced autologous peripheral blood lymphocytes + NY-ESO-1 peptide-pulsed autologous DC vaccine | I | Recruiting |
| Colorectal carcinoma | NCT03152565 | Anti-PDL1 (Avelumab) + autologous DC vaccine | I | Not yet recruiting |
| Advanced gastric cancer | NCT03393416 | Anti-PD-1 + multiple-antigen-specific cell therapy | I | Not yet recruiting |
| Non-Hodgkin lymphoma | NCT03035331 | Anti-PD-1 (Pembrolizumab) + autologous DC vaccine + pneumococcal 13-valent conjugate vaccine | I/II | Recruiting |
| Melanoma | NCT03325101 | Anti-PD-1 (Pembrolizumab) + autologous DC vaccine | I/II | Recruiting |
| Follicular lymphoma | NCT02677155 | Anti-PD-1 (Pembrolizumab) + anti-CD20 (Rituximab) + autologous DCs | II | Recruiting |
| Glioblastoma | NCT03014804 | Anti-PD-1 (Nivolumab) + autologous DCs pulsed with tumour lysate | II | Not yet recruiting |
| Prostate cancer | NCT02423928 | Anti-CTLA4 (Ipilimumab) + DC-based cryoimmunotherapy | I | Recruiting |
| Solid tumour | NCT02070406 | Anti-CTLA4 (Ipilimumab) + DC vaccine + gene-modified T cells | I | Recruiting |
| Metastatic melanoma | NCT02678741 | Standard-of-care checkpoint inhibitor + autologous DCs loaded with tumour lysate + yeast cell wall particle | I/II | Recruiting |
| Advanced non-small-cell lung cancer | NCT03360630 | Anti-PD-1 + autologous DC-CIK | I/II | Recruiting |
| Advanced solid tumour | NCT03190811 | Anti-PD-1 + autologous DCCIK | I/II | Recruiting |
| Small cell lung cancer | NCT03406715 | Anti-PD-1 (Nivolumab) + anti-CTLA4 (Ipilimumab) + autologous DC-based p53 vaccine | II | Not yet recruiting |
| Acute myelogenous leukemia | NCT03059485 | Anti-PD-L1 (Durvalumab) + autologous DC/AML fusion vaccine | II | Recruiting |
Source: clinicaltrials.gov. Data as current as May 2018. DC-CIK, dendritic cell and cytokine-induced killer cells.
Fig. 2 |. Expression of selected immune checkpoint factors on in vitro stimulated CD141+ DCs, MoDCs and pDCs.
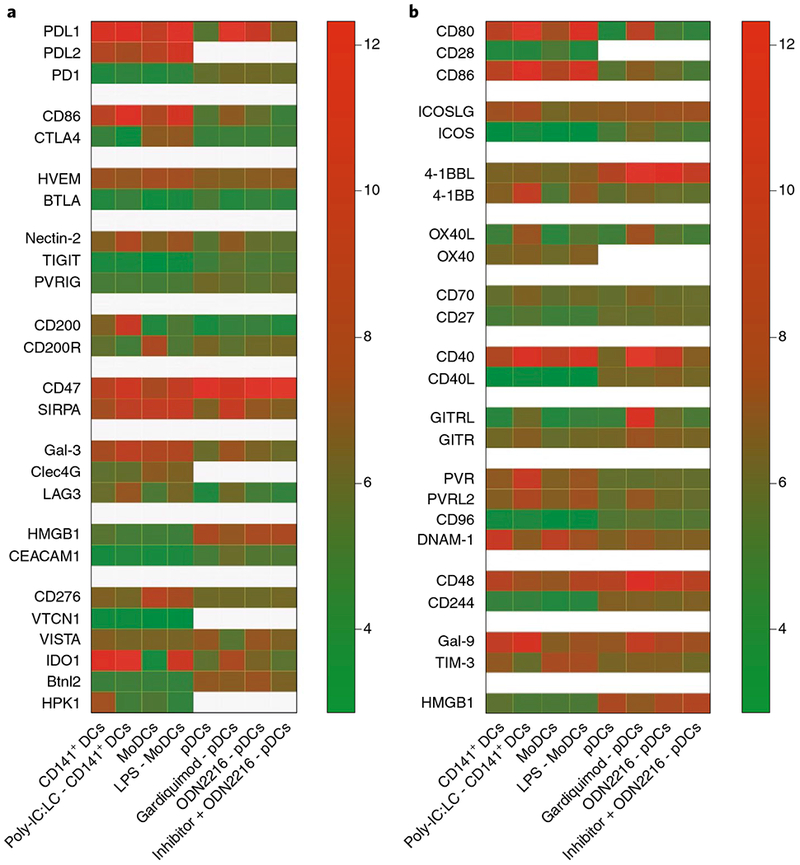
Different DC subsets display a unique pattern of expression for inhibitory and activating checkpoints both at basal levels and on activation. CD141+ DCs and MoDCs were differentiated from CD34+ precursors and monocytes, respectively41. Natural pDCs were sorted from blood of healthy donors42. The original microarray datasets were analysed using the statistical package limma in R. Expression values are normalized by housekeeping gene expression43, and were log2 transformed; mean values are shown on the heatmap. a, Inhibitory immune-checkpoint interactions. b, Stimulatory immune-checkpoint interactions. LPS, lipopolysaccharide-TLR4 agonist; Gardiquimod, TLR7 agonist; ODN2216, Class A CpG oligonucleotide-human TLR9 ligand.
To create a super-DC vaccine, CD34+ stem cells could be genetically engineered using small interfering RNA (siRNA), lentiviral transduction or, eventually, CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats–CRISPR-associated protein-9 nuclease) technology to permanently silence one or several of these inhibitory molecules, such as PD-L1, CD200, HPK-1 and IDO1, before differentiation into mature DCs. A similar approach is used for the generation of SMART-DCs, wherein DCs derived from monocytes are genetically reprogrammed by lentiviral co-transfection to express GM-CSF, IL-4 and TAAs to expedite DC differentiation36. Interestingly, siRNA-mediated knockdown of PD-L1 and PD-L2 on immature MoDCs was shown to improve the capacity of DCs to expand and activate both CD4 and CD8 T cells45, and a clinical trial is underway to investigate the clinical benefits of PD-L1-silenced MoDC vaccines in haematological malignancies (NCT02528682). Also, mRNA-modified MoDCs transiently expressing inhibitory soluble extracellular parts of PD-1 and PD-L1 (sPD-1 and sPD-L1) were reported to induce T-cell activation while avoiding T-cell exhaustion46.
There have been attempts at the genetic reprogramming of DCs to overexpress activation and maturation stimuli along with the antigens of choice. One such example is the TriMix-DC vaccine for cancer, in which mRNA transfection is used to overexpress CD40L, CD70, active TLR4 and TAA on MoDCs to arm these cells with activating co-receptors and activation stimuli47–49. Another example is the SmyleDCpp65 vaccine, in which cord-blood- and peripheral-blood-isolated monocytes are transduced with lentivirus to enhance expression of GM-CSF, IFNα and cytomegalovirus antigen pp65 so as to facilitate overnight vaccine preparation and de novo DC maturation post-vaccination50. Similarly, super-DCs could be engineered to overexpress co-stimulatory molecules or express IL-12 to aid T helper type 1 (TH1) and CD8 T-cell activation. However, caution must be exercised when manipulating DC activation signals, as the cells may become refractory or upregulate inhibitory signals following T-cell contact. In fact, many inhibitory or T helper type 2 (TH2) signals on DCs are required in modicum to initiate T-cell activation and proliferation.
Route of vaccine administration.
It is well recognized that the route of administration for a DC vaccination can substantially affect clinical outcomes. It would stand to reason that intranodal vaccine administration should instil the best immunity by providing vaccine components direct access to lymph-node-resident XCR1+ DCs and CD8+ T cells51. However, it seems that, in addition to being arguably less invasive and painful for the patient, intravenous vaccinations are equally suited (if not more successful) at garnering DC activation, as shown for neoantigens-pulsed ex vivo DC vaccines33 and for BioNTECH’s Lipo-MERIT vaccine (NCT02410733), an RNA-based in vivo DC targeting vaccine for patients with melanoma.
Outlook
Despite the varying degrees of success with cell-based therapies, checkpoint-blockade inhibitors and chemotherapy, a large fraction of cancer patients remain unresponsive to therapy, or are prone to relapse. It seems that lasting antitumour immunity, especially in the wake of massive tumour-induced immune suppression, will require a combination of strategies. Increasing numbers of small-molecule inhibitors of suppressive signalling pathways, such as tyrosine kinase, arginase and IDO1, are being developed and used in combination with DC vaccines to alleviate tumour-related immunosuppression and to improve the immunogenicity of DC vaccines. Among these targets, HPK-1 has been identified as a novel potential druggable kinase target that could significantly improve both DC and T-cell responses in a cancer setting44. Moreover, PX478, an inhibitor of hypoxia inducible factor 1 α (HIF1α; a central regulator of tumour-induced hypoxia-associated immune suppression), was demonstrated to enhance the efficacy of a DC vaccine in a mouse model of breast cancer52.
Successful DC-targeted immunotherapy will probably require a multiphase approach; yet it remains unclear whether DC vaccination should be administered along with adjuvant therapies (chemotherapy or radiation therapy, or checkpoint-blockade inhibitors) or in sequence. Interestingly, while Sipuleucel-T failed to impress as a monotherapy, early observations from recent trials investigating Sipuleucel-T in combination with other therapies, such as checkpoint-blockade inhibition (NCT03024216 and NCT01804465), show promising results. Arguably, a regimen starting with DC vaccination followed by a checkpoint-blockade inhibitor or chemotherapy may improve effector-T-cell generation enhanced by PD-1/PD-L1 blockade and reduced myeloid-derived suppressor cells. Inversely, checkpoint inhibition or immunogenic cell death induced by chemotherapy or radiation may release tumour antigens (including neoantigens), and the response to these antigens could be amplified by subsequent DC vaccination. Vaccination could be achieved not only through optimized DC subset selection and administration, as described above, but also through administration of Flt3L to mobilize DC populations from progenitors, and through tumour microenvironment in situ vaccination with TLR ligands, STING agonists or oncolytic viruses. Detailed mechanistic exploration and carefully controlled preclinical and clinical studies are however required before off-the-shelf DC vaccines could become approved for general treatment.
Acknowledgements
This work was supported by the National Institutes of Health grants RO1CA201189, R01CA180913 and R01AI081848, the Cancer Research Institute and the Melanoma Research Alliance. N.B. is a member of the Parker Institute for Cancer Immunotherapy, which supports the Icahn School of Medicine at Mount Sinai, NY, Cancer Immunotherapy Program.
Footnotes
Competing interests
N.B. is on the senior advisory board of Check Point Sciences, Curevac, Neon and Inception, and a consultant for Genentech. All potential conflicts of interest are being managed by the Conflict of Interest Office, Icahn School of Medicine at Mount Sinai. M.S., S.B. and V.R. declare no competing interests.
References
- 1.Rezvani K, Rouce R, Liu E & Shpall E Mol. Therapy 25, 1769–1781 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D’Aloia MM, Zizzari IG, Sacchetti B, Pierelli L & Alimandi M Cell Death Dis. 9, 282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheever MA & Higano CS Clin. Cancer Res 17, 3520–3526 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Leaf RK et al. J. Immunotherapy 40, 315–322 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Doebel T, Voisin B & Nagao K Trends Immunol. 38, 817–828 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Villani AC et al. Science 356, eaah4573 (2017).28428369 [Google Scholar]
- 7.Alcantara-Hernandez M et al. Immunity 47, 1037–1050.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palucka K & Banchereau J Immunity 39, 38–48 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenblatt J et al. Sci. Transl. Med 8, 368ra171 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Tendeloo VF et al. Proc. Natl Acad. Sci. USA 107, 13824–13829 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schreibelt G et al. Clin. Cancer Res 22, 2155–2166 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Saxena M & Bhardwaj N Trends Cancer 4, 119–137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garg AD et al. Oncoimmunology 6, e1328341 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anguille S, Smits EL, Lion E, van Tendeloo VF & Berneman ZN The Lancet Oncology 15, e257–e267 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Saxena M & Bhardwaj N Curr. Opin. Immunol 47, 35–43 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Lint S et al. Cancer Immunol. Res 4, 146–156 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Van der Jeught K et al. Oncotarget 6, 1359–1381 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hutten TJ et al. J. Immunol 197, 2715–2725 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Goyvaerts C et al. Oncotarget 5, 704–715 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manches O et al. Haematologica 90, 625–634 (2005). [PubMed] [Google Scholar]
- 21.Kranz LM et al. Nature 534, 396–401 (2016). [DOI] [PubMed] [Google Scholar]
- 22.De Vries J & Figdor CI Nature 534, 329–331 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Hartung E et al. J. Immunol 194, 1069–1079 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Cauwels A et al. Cancer Res. 78, 463–474 (2018). [DOI] [PubMed] [Google Scholar]
- 25.De Munck J, Binks A, McNeish IA & Aerts JL J. Leukocyte Biol 102, 631–643 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Harrington KJ et al. Expert Rev. Anticancer Therapy 15, 1389–1403 (2015). [DOI] [PubMed] [Google Scholar]
- 27.van den Bijgaart RJ et al. Cancer Immunol. Immunotherapy 66, 247–258 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pardi N, Hogan MJ, Porter FW & Weissman D Nat. Rev. Drug Discov. 17, 261–279 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sabado RL, Balan S & Bhardwaj N Cell Res. 27, 74–95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prue RL et al. J. Immunotherapy 38, 71–76 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Tel J et al. Cancer Res. 73, 1063–1075 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Balan S, Finnigan J & Bhardwaj N Cancer J. 23, 131–137 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carreno BM et al. Science 348, 803–808 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiang CL et al. Clin. Cancer Res 19, 4801–4815 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Lint S et al. Cancer Immunol. Immunotherapy 63, 959–967 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sundarasetty BS et al. Gene Therapy 22, 707–720 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Squadrito ML, Cianciaruso C, Hansen SK & De Palma M Nat. Methods 15, 183–186 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balan S & Dalod M Methods Mol. Biol. 1423, 19–37 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Yewdall AW, Drutman SB, Jinwala F, Bahjat KS & Bhardwaj N PLoS ONE 5, e11144 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Avigan DE et al. J. Immunotherapy 30, 749–761 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Balan S et al. J. Immunol. 193, 1622–1635 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang J, Lau KY, Jung J, Ravindran P & Barrat FJ Eur. J. Immunol. 44, 1130–1136 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Eisenberg E & Levanon EY Trends Genet. 29, 569–574 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Sawasdikosol S, Zha R, Yang B & Burakoff S Immunologic Res. 54, 262–265 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Hobo W et al. Blood 116, 4501–4511 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Pen JJ et al. Gene Therapy 21, 262–271 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Wilgenhof S et al. J. Clin. Oncol. 34, 1330–1338 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Wilgenhof S et al. Cancer Immunol. Immunotherapy 64, 381–388 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilgenhof S et al. Ann. Oncol. 24, 2686–2693 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Daenthanasanmak A et al. Mol. Therapy Methods Clin. Dev 1, 14060 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kreiter S et al. Cancer Res. 70, 9031–9040 (2010). [DOI] [PubMed] [Google Scholar]
- 52.Kheshtchin N et al. Cancer Immunol. Immunotherapy 65, 1159–1167 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]