

Abstract
Rat tail collagen solutions have been used as polymerizable in vitro three-dimensional (3D) extracellular matrix (ECM) gels for single and collective cell migration assays as well as spheroid formation. These 3D hydrogels are a relatively inexpensive, simple to use model system that can mimic the in vivo physical characteristics of numerous tissues within the body, namely the skin. While confocal imaging techniques such as fluorescence reflection and two-photon microscopy are able to visualize collagen fibrils during 3D imaging without fluorescence, other imaging modalities require direct conjugation of fluorescent dyes to collagen. Here we detail how to generate 3D collagen gels labeled with a fluorescent dye. Furthermore, we go through the steps required to reproducibly generate bright collagen hydrogels that are suitable for live cell 3D imaging techniques.
Keywords: Rat-tail collagen, Fibrils, Hydrogel, Fluorescent dye, 3D imaging
Background
The study of cell migration and cell interaction with its surrounding microenvironment has been started since the 1950’s when Paul Weiss and Beatrice Garber originally observed the effect of increasing plasma concentration (fibrin) on mesenchymal cell morphology (Weiss and Garber, 1952). In subsequent years and decades, biochemists started to delve into purifying extracts from rat tail collagen and started their use as a highly polymerizable 3D matrix ( Fitch et al., 1955 ; Gross et al., 1955 ; Chandrakasan et al., 1976 ). It wasn’t until the 1990’s that 3D matrices truly became useful to the cell biology community, especially for studying cell migration ( Friedl et al., 1995 ). Recently, a transition from simplified two-dimensional (2D) studies on ECMs to 3D has begun. This evolution has followed shortly behind the recent advances in fluorescence microscopy, especially super-resolution microscopy. While standard laser scanning confocal microscopes can utilize reflection microscopy or two-photon-based second harmonic generation to visualize collagen fibers in the absence of a fluorescent tag, both techniques do not always properly depict the ECM architecture due either to polarity issues or lack of a significant fibril thickness. A fluorescently-tagged ECM allows imaging of the smallest individual fibrils even with super-resolution techniques.
Unlike most proteins collagen cannot be simply tagged with a fluorophore when in solution because the numerous lysine residues are required for alpha helix formation with other monomers during polymerization ( Chandrakasan et al., 1976 ). For this reason, the labeling must be accomplished on preformed gels. This protocol describes how to label a polymerized gel, bring the collagen back into solution with acetic acid, and properly mix a minimal amount (2-4% of total protein) of labeled collagen with an unlabeled fraction to generate a bright, fluorescent collagen gel capable of sustaining cell viability while allowing observation of ECM architecture over multiple hours of fluorescence imaging.
Materials and Reagents
MatTek Dishes (35 mm, #1.5 coverslip, 20 mm opening: MATTEK, catalog number: P35G-1.5-20-C)
ColorpHast pH-indicator-strips with pH range 6.5-10 (Merck, catalog number: 109543)
10-100 and 1,000 μl Gilson MICROMAN® positive displacement pipette tips (Gilson, catalog numbers: FD10004, FD10006)
10 cm tissue culture dish (Thermo Fisher Scientific, catalog number: 150350)
Cell lifters (Corning, catalog number: 3008)
Aluminum foil
Plastic wrap
Scintillation vial (Sigma-Aldrich, catalog number: Z190527)
1.5 ml microfuge tubes (Thermo Fisher Scientific, catalog number: AM12400)
Dubecco’s Minimal Essential Medium (DMEM) powder, Phenol red (Sigma-Aldrich, catalog number: D2429)
NaOH pellets (Sigma-Aldrich, catalog number: S8045)
Phosphate Buffered Saline with Calcium and Magnesium (PBS++) chilled to 4 °C and at room temperature (GE Healthcare, HycloneTM, catalog number: SH30264.02)
Rat tail collagen solution (dissolved in 20 mM acetic acid, commercial brands are fine, but in-house preparations are usually cleaner and polymerize faster) at a concentration greater than 5 mg/ml (6 mg/ml used here)
Sodium bicarbonate (Sigma-Aldrich, catalog number: S5761)
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (Sigma-Aldrich, catalog number: H4034)
Boric acid (powder 99.5%, Sigma-Aldrich, catalog number: B6768)
Acetic acid (Sigma-Aldrich, catalog number: 27221-1L) (chilled to 4 °C, less than 50 ml of 16.65 M is required)
Atto-488 NHS-ester 1 mg (Sigma-Aldrich, catalog number: 41698)
DMSO (Sigma-Aldrich, catalog number: D2650)
Sircol Collagen Assay kit (available from Accurate Chemical & Scientific, catalog number: CLRS1000)
Slide-A-Lyzer Dialysis cassette (G2) with 20,000 MW cutoff (Thermo Fisher Scientific, catalog number: 87735)
1 M Tris (pH 7.4, KD Medical, catalog number: RGF-3340)
NaCl (Sigma-Aldrich, catalog number: S7653)
HCl (37%, Sigma-Aldrich, catalog number: H1758-500ML)
10x DMEM (see Recipes)
10x reconstitution buffer (10x RB; see Recipes)
1 N NaOH (500 μl in a microfuge tube; see Recipes)
1 N HCl (500 μl in a microfuge tube; see Recipes)
50 mM Borate buffer pH 9.0 (see Recipes)
5 mg/ml Atto-488 NHS-ester dye in DMSO (see Recipes)
Sodium Chloride (NaCl), 8% solution (see Recipes)
50 mM Tris buffer (see Recipes)
Equipment
10-100 and 1,000 μl Gilson MICROMAN® positive displacement pipette (Gilson, catalog numbers: F148314, F148180)
Lab timer
2-4 L beaker
Ultra-clear 8 x 20 mm centrifuge tubes (Optional: Beckman Coulter, catalog number: 345843)
Rectangular ice bucket packed with ice
Water bath
Large Magnetic stir bar to large beaker (Sigma-Aldrich, SP Scienceware - Bel-Art Products - H-B Instrument, catalog number: Z284491)
Small Magnetic stir bar for scintillation vial (Sigma-Aldrich, SP Scienceware - Bel-Art Products - H-B Instrument, catalog number: Z126942)
Stir plate
Mini centrifuge (i.e., VWR, model: Galaxy Mini)
Biological hood (any brand)
Brightfield microscope with 10 and 20x phase contrast objectives
Cooled centrifuge capable of 20,000 × g and 4 °C (i.e., TOMY, model: MX-307)
Lab Rocker (i.e., Denville Scientific, model: 110, catalog number: S2110)
Airfuge ultra centrifuge (optional: Beckman Coulter, catalog number: 340400)
Laser scanning or spinning disk confocal microscope (i.e., Nikon Instruments, model: A1R or Yokogawa, model: CSU-X1)
Procedure
Notes: If you are planning on repeatedly making collagen gels, it is highly recommended to invest in a 10-100 and a 100-1,000 μl positive displacement pipette. These greatly help accurate pipetting of viscous solutions such as collagen. The greater your accuracy in pipetting, the better the overall experimental reproducibility.
-
Collagen gel calculations
-
Prior to any collagen gel generation, it is important to accurately calculate the volumes of the different components needed from stock solutions. Because only a small fraction (between 2-4%) of labeled collagen is used for gel generation, a relatively small amount of collagen is used for the labeling process. Typically, a heavy week of experiments will entail generating 20-24 dishes containing 150 μl of collagen and use approximately 2,000 μl of stock concentration collagen. This amounts to a range of 40 to 80 μl or 120 to 240 μg of labeled collagen being used per week. Here, the 5 ml of labeled 3 mg/ml collagen should last between 37 and 75 weeks of similar usage. Smaller volumes of collagen can be labeled, but it is recommended to use no less than 2.5 ml due to normal losses in the protocol. The following steps walk you through the initial calculations.
-
Calculate the amount of collagen:
-
Divide by 10 to calculate the amount of 10x RB and 10x DMEM :
10x DMEM:
10x RB:
-
The volume of 1 N NaOH will vary based on the pH of your stock collagen concentration which can vary between 1.8 and up 3.0. A good starting point is to add 3 μl per ml of collagen, then adjust accordingly. We find 16 μl is suitable for our homemade collagen.
Note: This will vary greatly depending on the original pH of your collagen and can even vary between commercial lots and preparations. Initial testing may be required for determining the actual amount needed per dish.
-
Calculate the amount of PBS++:
or 1.984 ml
-
-
-
Collagen Gel preparation
-
Pre-chill all gel components on ice (collagen stock solution, 10x DMEM, 10x RB, 1 N NaOH, 1 N HCl, PBS++) and the 10 cm tissue culture dish. Add the tissue culture dish last: be sure no water condenses on the inner dish surface prior to adding gel components.
Note: To reduce collagen solution loss when transferring from a conical tube to the 10 cm tissue culture dish we find it helpful to mix directly in the dish.
Pipette the proper calculated amount of stock collagen solution (2.5 ml) into the pre-cooled tissue culture dish. Be sure to keep the dish on ice.
Add the 10x DMEM (0.25 ml) and then the 10x RB (0.25 ml). After adding each slowly triturate with the 1 ml positive displacement pipette, making sure not to introduce any bubbles. Alternatively, you can use a cell scraper to homogenize.
Add the calculated 1 N NaOH to the solution and triturate/mix with a positive displacement pipette or cell scraper. Once the solution is mixed well and shows a consistent, single color take a 2 μl sample and test the pH with a pH strip, waiting 1-2 min to determine the pH. The pH should be between 7.0 and 7.4, and the solution color should be peach (pinkish orange: Figure 1). If the solution is over a pH of 7.4, adjust with 1 N HCl, 1 μl at a time. Be sure to note the changes and adjust the PBS++ accordingly including for the 2 μl sample (i.e., if 2 μl of HCl were added, reduce the PBS++ by the same amount).
Add PBS++ (adjusted if necessary) to the tissue culture dish until fully mixed and place again in the ice.
Allow gravity to help spread the collagen evenly over the dish by tilting the dish to 45-degree and rotating the dish.
Cover and place the dish on the bench top and allow the collagen to polymerize at room temperature (approximately 21 °C). Check the collagen gel by eye using a 10x or 20x phase contrast objective on an inverted brightfield microscope after 30 min and then every fifteen minutes thereafter. Small, intertwined fibrils should be visible (Figure 1). Again, the polymerization process can vary preparation to preparation based on the collagen used. Too short an incubation time can lead to loss of collagen gel integrity and an overall loss of collagen monomers and will result in a greatly reduced end-product concentration. Longer incubations have no major effect (always side on longer incubations). Testing in small 150 μl batches is suggested if you are unsure. Also, taking a brightfield image before and after adding the PBS++ will help determine if the structure has dramatically changed.
-
-
Collagen gel labeling with NHS-ester dye
Once the solution is completely polymerized and formed a gel, add 10 ml of 50 mM borate buffer (pH 9.0) and incubate for 15 min at room temperature.
-
Meanwhile, calculate the amount of dye needed to properly label the amount of protein within the gel using the following equation:
-
The above equation is for 1 mg of Atto-488 NHS-ester diluted in 200 μl (5 mg/ml) of DMSO using a 2-molar excess which is recommended by the company.
Note: Each dye has a different molar-excess that works the best for NHS-conjugation. Do not assume the above will work for all dyes. Over labeling can lead to issues with gel formation later on.
Add 45.28 μl of Atto-488 NHS-ester dye to a 15 ml conical tube and bring the volume up to 5 ml with 50 mM borate buffer and vortex quickly.
Carefully aspirate the borate buffer from the tissue culture dish (bring the dish to a 45-degree angle and siphon off at the bottom edge with an aspiration pipette).
-
Add the dye solution to the collagen gel and wrap the culture dish with aluminum foil to protect from light. Allow the dye to conjugate to the collagen gel for 1 h at room temperature or 4 h at 4 °C (can do overnight) while rocking.
Note: At room temperature the majority of the dye will conjugate within the first 20 min.
Aspirate dye and add 10 ml of 50 mM Tris buffer (pH 7.5) to quench the dye reaction. Incubate with rocking for 10 min. Keep the gel covered with foil.
Add 10 ml of PBS++. Rinse gel with PBS++ 6 x over the next 4 h to wash out the excess dye.
-
Liquefying the collagen gel into a solution
-
Aspirate PBS++ and invert dish with one side raised on its lid in a tissue culture hood for 10-15 min to reduce the amount of fluid within the gel.
Note: The remaining steps in this section should be performed at 4 °C.
-
Add 500 to 1,000 μl of 500 mM acetic acid to the gel. Bring the gel into a cold room and rock slowly for 1 h.
Note: The larger the volume of acetic acid you add the easier it is to get the gel to go into solution. However, this will decrease your final concentration. We suggest starting with 500 μl and adding extra incrementally over time if needed.
After 1 h, use a cell scraper to mix the gel. Scrape gel to one side of the dish (on an angle) and pipette up gel solution with a 1,000 μl positive displacement pipette set to 750 μl. If using a regular pipette, you may need to cut off the pipette tip at about the 100 μl mark for a larger tip opening because of the gel viscosity.
Transfer the collagen solution to a scintillation vial wrapped in aluminum foil and add a small magnetic stir bar. Stir gel at 4 °C overnight. Check periodically to see if the gel has gone into solution. You should not see any ‘chunks’ of polymerized collagen. If you do, add more acetic acid followed by further stirring at 4 °C for several hours. Alternatively, take 5 to 10 μl and smear this in a 35 mm tissue culture dish and check for solution consistency under a microscope at 10x magnification.
Check the volume of the gel. Based on the starting volume (5 ml) and concentration (3 mg/ml) you can make an educated guess at the concentration. If you need a more concentrated solution proceed to the salt precipitation protocol (Procedure F). If the guesstimate is fine, then continue the next step below.
-
-
Dialyzing the dye-labeled collagen solution
Pre-cool 8 L of 20 mM acetic acid at 4 °C. This requires 9.6 ml of a 16.65 M solution (standard purchased concentration).
Add 4 L of 20 mM acetic acid to a large beaker. Wet the Slide-A-Lyzer G2 cassette in the acid. Pipette the collagen into the cassette being sure to remove any air bubbles before closing.
Add a large stir bar and the cassette containing the labeled collagen to the beaker and cover with plastic wrap and use a rubber band to firmly secure the plastic wrap. Cover the upper of the beaker with aluminum foil to protect the labeled collagen from excess light. Stir for 4 h.
Change acetic acid once and let dialyze further overnight.
Remove collagen solution from the cassette to several 1.5 ml centrifuge tubes. Place tubes in a cooling centrifuge. Spin at 20,000 × g for 1 h at 4 °C. Remove and save the supernatant, being careful not to pull up any of the pellet.
Follow the Sircol collagen assay to determine the collagen’s concentration.
Fluorescently labeled collagen can be stored at 4 °C protected from light for more than one year or indefinitely at -80 °C.
-
Concentration of collagen solutions using salt precipitation (modified from Chandrakasan et al., 1976 )
Perform all steps at 4 °C.
-
Estimate the current volume of the collagen to be concentrated (should still be in the scintillation vial) and add an equal volume of 8% NaCl.
Note: A precipitate should begin forming in several minutes.
Stir the solution at 4 °C for 4 h or overnight.
Once the liquid portion is clear with no apparent collagen remaining, transfer the collagen to a 15 ml centrifuge tube.
Spin down the solution at 13,000 × g for 20 min at 4 °C.
Aspirate the supernatant and keep the pellet. Add 2 ml of 500 mM acetic acid (approximately concentrating by two times).
Add a small magnetic stir bar to the 15 ml tube and stir the solution at 4 °C until collagen goes into solution, 3-4 h.
Go to Procedure E for dialysis.
-
Calculating and mixing labeled rat tail collagen with unlabeled collagen and getting the proper amounts
Note: Many researchers have mixed fluorescently-labeled and unlabeled collagen solutions together at specific ratios (1:5, 1:4, etc.) in order to generate a fluorescently visible collagen gel capable of sustaining cell life. Collagen that is completely labeled will not polymerize and often is too bright for actual imaging. Too often the final collagen concentration is incorrectly determined because ratios do not take into consideration differences in collagen concentration between labeled and unlabeled fractions. Furthermore, batch-to-batch differences in the labeled collagen concentration make gel consistency an issue that can alter your experimental outcome. Here is described how to calculate and mix a 2-4% (4% shown) fluorescently-labeled gel based on protein weight to make up a 6 ml volume. Even if the change is minor, we suggest always using the calculated final concentration shown in step 6 below. Note, always keep collagen solutions at 4 °C or on ice to decrease the possibility of protein degradation.
-
Calculate what 4% of the unlabeled collagen (ULC) is for the amount you want to mix:
-
Multiple X by 0.04 to get 4% of this amount (Y).
-
Use this number to then calculate the volume (V) you need to take out from the unlabeled.
-
Perform the same for calculation for the fluorescently labeled collagen (FLC) as you did in #3 above.
Mix these together in a 15 ml conical tube slowly over 1 h at 4 °C. Transfer to 1.5 ml centrifuge tubes and spin at 20,000 × g for 30 min to remove any debris.
-
Calculate the ‘new’ amount (C; in mg) since the volumes removed and added in 3 and 4 will likely not be the same.
Note: Protein (in mg) should be identical to Step G1.
Then divide the new amount C by the total volume (TV = [total UL RTC – removed] + [added F RTC]).
Label the tube with the concentration and store at 4 °C.
-
-
Collagen clearing (Optional)
Note: To rid the collagen solution of excess debris, either from the original collagen processing or the labeling process, spinning at high RPMs using a Beckman Coulter Airfuge can rapidly clear the solution.
Using positive displacement pipettes, add 450 μl of 4% labeled collagen to four 8 x 20 mm centrifuge tubes.
Add tubes to the centrifuge rotor, lock the device and initiate the spin. Adjust the pressure to register between 25-30 PSI (approximately 90,000-10,000 rpm). Set the time for 30 min.
After 30 min, a colored streak will often be noticeable at the bottom edge of the centrifuge tubes. Being sure not to disturb the pelleted collagen, remove the supernatant to a 15 ml conical tube and store at 4 °C.
Repeat the process with the remaining collagen (this usually requires a total of 12 tubes).
This 4% fluorescently-labeled collagen can now be used for generating individual collagen gels for live cell microscopy using the calculations in Procedure I (below) and the protocol used in Procedure B.
-
Collagen gels for 20 mm diameter MatTek glass-bottomed dish
Note: For a 20 mm diameter glass-bottomed dish 150 μl of collagen solution will generate a 300 μm-thick gel. Due to the high viscosity of collagen solutions and some loss with a small volume/tube add 20 μl extra for each dish for calculations. Always polymerize a minimum of 1 extra dish over what you need. Below is an example calculation of 2 dishes per condition and at a final concentration of 3 mg/ml.
2 (dishes) x 170 (μl collagen per dish) = 340 μl of collagen solution
-
Calculate the amount of collagen:
-
Divide by 10 to calculate the amount of 10x RB and 10x DMEM:
10x DMEM:
10x RB:
The volume of 1 N NaOH: ~1 μl
-
Calculate the amount of PBS++:
or 0.135 ml
Follow Procedure B. Changing the polymerization temperature will alter the polymerization time and the architecture ( Doyle et al., 2015 ). Decreasing the polymerization temperature leads to a slower gel formation. We hypothesize that decreasing the temperature leads to fewer nucleation sites from which fibrils polymerize, hence the fibrils extend more and create larger pore sizes observed in Figure 2.
It is expected that following this protocol individual fibrils will be easily observable using laser scanning confocal, spinning disk confocal (Figure 2), or light sheet microscopy.
Figure 1. Fluorescent-labeling of polymerized collagen gels workflow.
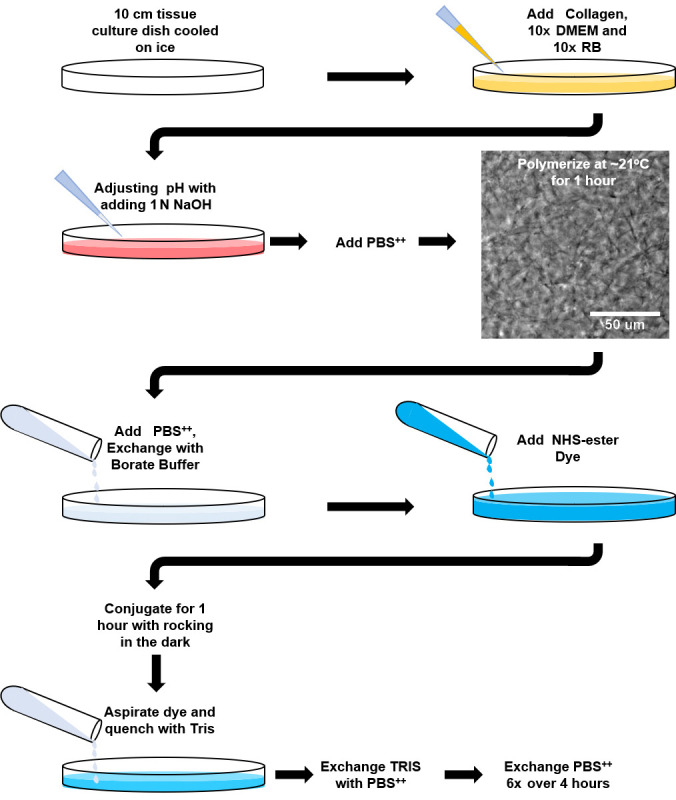
Schematic workflow for Procedure B and C of the protocol. Image shows 3 mg/ml rat tail collagen polymerized at 21 °C for 1 h (taken with a 20x objective). Note the appearance of discernable collagen fibrils.
Figure 2. Fluorescence images of a 4% labeled collagen.

A. Single confocal slice (XY) and a 10 μm YZ projection (right) of a 4% atto 488-labeled 3 mg/ml rat-tail collagen gel polymerized at 21 °C. B. 3D rendering of the collagen gel shown in panel A (X, Y, Z dimensions: 100 x 100 x 80 μm, respectively). C. High-resolution max intensity projections of an HT-1080 fibrosarcoma cancer cell transfected with EGFP-α-actinin (magenta) migrating through Atto 565-labeled 3 mg/ml rat-tail collagen (green) polymerized at 16 °C. Images show first frame (left) and 63rd frame (right) in a time-series (imaged every 0.5 μm in Z over 15 microns, every 30 sec). Time is in minutes.
Notes
While using rat tail collagen is a simple and easy way to use 3D hydrogel, it does require repetition to get gels to become consistent. Collagen type I 3D gels is highly dependent on 1) collagen concentration, 2) the ionic concentration, 3) pH, and of course, 4) temperature. Because each of these can affect the time to polymerization, which inevitably leads to the differences in ECM architecture, it is vital that each of the above parameters is consistent between experiments. pH is by far the most variable since it needs to be tested and adjusted to neutralize each batch of collagen made. It is suggested that pH should be checked for each batch and do not assume that the amount of 1 N NaOH will always be the same. Small aliquots of stocks solutions are used throughout the protocol to cut down on changes in concentration due to evaporation that can occur over time.
Other critical issues to be aware of are with the 10x DMEM solution: Due to the high salinity, a precipitate will always be present at 4 °C or when on ice. Trituration of the 10x DMEM solution is important to keep a consistent ionic concentration.
Recipes
-
10x DMEM (50 ml)
Add 1 powdered DMEM with phenol red (Sigma-Aldrich) packet into 50 ml distilled water
Stir mixture with heat (approximately 50 °C) until DMEM powder goes into solution
Sterile filter while warm. Make four 10 ml and twenty 0.5 ml aliquots. The 0.5 ml size aliquots are for daily experiments
Store both sizes at -20 °C until use. Upon defrosting heat aliquots in a 37 °C water bath, then vortex and cool on ice
Can be kept indefinitely at -20 °C and 1 month at 4 °C
-
10x reconstitution buffer (100 ml)
2.2 g Sodium bicarbonate (Sigma-Aldrich)
4.8 g HEPES (Sigma-Aldrich) -or- 20 ml 1 M HEPES stock solution for 0.2 M final
Distilled water up to 100 ml
Filter sterilize and store at -20 °C in aliquots similar to 10x DMEM
Can be kept indefinitely at -20 °C and 1 month at 4 °C
-
Sodium Hydroxide solution (NaOH), 1 N
0.5 g NaOH pellets (Sigma-Aldrich)
Distilled water to 12.5 ml
Mix well and filter sterilize, divide into 500 μl aliquots and store indefinitely at -20 °C
-
1 N HCl
1 ml HCl (37% or 12 M, Sigma-Aldrich)
Distilled water to 12 ml
Filter sterilize, divide into 500 μl aliquots and store indefinitely at -20 °C
-
50 mM Borate buffer (pH 9.0)
1.55 g boric acid (powder 99.5%, Sigma-Aldrich)
Add distilled water to 400 ml
Add several solid NaOH pellets (Sigma-Aldrich) at a time while mixing until the pH is ~9.0
Add distilled water to 500 ml
Filter sterilize using a 0.2 μm filter
Store up to 1 year at room temperature
-
5 mg/ml fluorescent NHS-ester dye in DMSO (200 μl)
-
Atto-488 NHS-ester 1 mg (Sigma-Aldrich)
Note: Any NHS-ester dye is suitable and can be substituted.
Add 200 μl DMSO (Sigma-Aldrich) to dye tube and mix (wrapped in foil) on a rotational mixer for 1 h
Use immediately and store excess in 10-20 μl aliquots
Store at -80 °C
-
-
Sodium Chloride (NaCl), 8% solution
Dissolve 4 g NaCl (Sigma-Aldrich) in 500 ml distilled water, mix well
Filter sterilize using a 0.2 μm filter
-
50 mM Tris buffer (pH 7.4)
5 ml 1 M Tris (KD Medical)
Add distilled water to 100 ml
Filter sterilize using a 0.2 μm filter
Store up to 1 year at room temperature
Acknowledgments
I would like to thank Joshua Collin, Gavrel Pacheco, and Tomoko Ikeuchi for their helpful comments. This work was supported by the NIDCR Division of Intramural Research. This work was adapted from previous work (Doyle, 2016). The author declares no conflicts of interest.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1. Chandrakasan G., Torchia D. A. and Piez K. A.(1976). Preparation of intact monomeric collagen from rat tail tendon and skin and the structure of the nonhelical ends in solution. J Biol Chem 251(19): 6062-6067. [PubMed] [Google Scholar]
- 2. Doyle A. D., Carvajal N., Jin A., Matsumoto K. and Yamada K. M.(2015). Local 3D matrix microenvironment regulates cell migration through spatiotemporal dynamics of contractility-dependent adhesions. Nat Commun 6: 8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Doyle A. D.(2016). Generation of 3D collagen gels with controlled diverse architectures. Curr Protoc Cell Biol 72: 10. 20 11-10 20 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fitch S. M., Harkness M. L. and Harkness R. D.(1955). Extraction of collagen from tissues. Nature 176(4473): 163. [DOI] [PubMed] [Google Scholar]
- 5. Friedl P., Noble P. B., Walton P. A., Laird D. W., Chauvin P. J., Tabah R. J., Black M. and Zanker K. S.(1995). Migration of coordinated cell clusters in mesenchymal and epithelial cancer explants in vitro . Cancer Res 55(20): 4557-4560. [PubMed] [Google Scholar]
- 6. Gross J., Highberger J. H. and Schmitt F. O.(1955). Extraction of collagen from connective tissue by neutral salt solutions. Proc Natl Acad Sci U S A 41(1): 1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weiss P. and Garber B.(1952). Shape and movement of mesenchyme cells as functions of the physical structure of the medium: contributions to a quantitative morphology. Proc Natl Acad Sci U S A 38(3): 264-280. [DOI] [PMC free article] [PubMed] [Google Scholar]