

Abstract
Building cells from their component parts will hinge upon our ability to reconstitute biochemical compartmentalization and exchange between membrane-delimited organelles. By contrast with our understanding of other cellular events, the mechanisms that govern membrane trafficking has lagged because the presence of phospholipid bilayers complicates the use of standard methods. This chapter describes in vitro methods for purifying, reconstituting and visualizing membrane remodeling activities directly by electron cryomicroscopy (cryoEM).
Keywords: Membrane remodeling, electron cryomicroscopy, cryoEM, endocytosis, mitochondria, dynamin, endophilin, bilayer, lipid-binding
Introduction to membrane remodeling
Chemical compartmentalization was as critical in the evolution of life as was the realization of self-replicating molecules. Robert Hooke’s use of the metaphorical “cell” draws our attention to the inhabitants of the “small rooms” he observed with his microscope; but also to the walls that outline a room, distinguish it from its neighbors, and that segregate inside from outside activities (1). Boundaries—and the cells that the boundaries define—arose when amphipathic molecules formed self-sealing but semi-permeable membranes to enclose chemical activities and to isolate them from dilution or admixture.
The goal expressed in this volume of Methods in Cell Biology, to build cells from their component parts, depends upon our understanding of biochemical compartmentalization. Specialized reactions occur more efficiently within confined, concentrated and chemically tailored spaces; but the benefits of compartmentalization require mechanisms for sorting and transporting molecules through membranes in order to maintain raw material supplies and remove byproducts. Cells also need to detect and respond to milieu variation outside their walls. The success of eukaryotic cells and multi-cellular cooperatives, moreover, depends upon robust mechanisms for moving and sorting molecules between organelles and between cells. Evolutionary forces have therefore fashioned protein modules that can reversibly mold membranes into cylindrical, spherical, and saddle-shaped surfaces (2). It takes significant amounts of energy to shape, fuse or fission membranes (3, 4). Nevertheless, cells constantly transform the sizes, shapes and connectivity of their membranes and such remodeling underlies cell division, migration, differentiation, and communication (5, 6). In addition, essentially every pathogen hijacks or disrupts membrane-associated protein complexes in order to infect or escape from host cells (7–9).
Despite such fundamental importance, we lack detailed views of the cellular machines that remodel the size, shape or topology of cellular membranes. In comparison with other cellular processes, our understanding of the mechanisms driving membrane remodeling has lagged because the presence of phospholipid bilayers precludes the use of many standard methods in cell biology. Until very recently, for example, we have had almost no structural information about membrane-bound protein assemblies (10–13). This chapter describes in vitro methods for purifying, reconstituting and visualizing membrane remodeling complexes directly in their membrane-bound states.
Although many proteins are recruited from the cytosol to coat, bend, and ultimately fission membranes, one major focus of research in this field centers on large GTPases of the dynamin family (14). This chapter focuses on methods for studying dynamin-family proteins and their binding partners. Dynamin proteins assemble into helical collars around the necks of budding vesicles and channel GTP energy into mechano-chemical constriction of the collar to promote fission (15, 16). Major unanswered questions now concern the mechanisms by which dynamin proteins are recruited to their target membranes and regulated by their binding partners (17, 18). All dynamin proteins engage in avid interactions with other proteins, many of which are membrane-binding or trans-membrane proteins themselves. Efforts in our lab are focused in part on testing the hypothesis that dynamins are recruited by binding partners that can co-assemble with dynamin and thereby modulate the properties of the helical collars in order to regulate or repurpose membrane fission reactions for different contexts (Figure 1).
Figure 1: Dynamin-family GTPases drive diverse membrane remodeling events.

A) In concert with a number of BAR-domain containing proteins, Dynamin-family GTPases shape and fission endocytic tubules from the plasma membrane. B) Dynamin-family GTPases partner with transmembrane proteins to help shape and fission mitochondria and other intracellular organelles.
The majority of work on dynamin proteins is focused on endocytosis at the plasma membrane, but dynamin-family proteins also function at intracellular organelles and some evidence suggests that the latter are the more ancient activity (Figure 1–2) (19–21). Chloroplasts, peroxisomes and mitochondria are the best known sites of intra-cellular activity for the dynamin-like proteins, with Dnm1 (yeast) and Drp1 (humans) receiving considerable attention for their role in regulating mitochondrial morphology and metabolism (22). How did dynamin GTPases adapt to catalyze fission in diverse contexts and on different spatial scales—from the <50 nm necks of clathrin-coated pits to the >1 micron tubules of peroxisomes, chloroplasts and mitochondria? Recent work indicates that the minimal machine that fragments large mitochondrial tubules comprises a dynamin and at least one transmembrane adaptor of the mitochondrial outer membrane (23). In this chapter we describe our protocols for purifying and reconstituting the co-complexes formed by mitochondrial outer membrane proteins MiD49/MiD51 and the dynamin-related protein Drp1 (Figure 1–2).
Figure 2: Domain architectures and X-ray crystal structures of the human proteins discussed in this chapter.

A) Dynamin1 (PDB: 3SNH) (47, 48). B) Dynamin related protein 1 or Drp1 (PDB: 4BEJ) (35). C) EndophilinA1 (PDB: 1X03) (49, 50). D) Mitochondrial Dynamics protein 49/51 or MiD49/51 (PDB: 4NXT) (33, 34).
We will also describe our approach to purifying, assembling and imaging a hetero-polymer of Dynamin1 and the BAR domain-containing protein EndophilinA1(18). The hetero-complex of Dynamin1 and EndophilinA1 assembles around narrow endocytic tubules, but its precise function remains poorly understood. This complex was implicated recently in a fast mode of clathrin-independent endocytosis that appears to be critical in migrating cells that internalize cell-surface signaling complexes from the leading edge (24). Genetic studies in many organisms, however, have indicated that EndophilinA1 and Dynamin1 null mutants have distinctly different phenotypes and, by contrast with Dynamin1 alone, this co-complex cannot mediate fission in vitro as assayed by electron microscopy (25–27). In this chapter we outline our approach to purifying, reconstituting and visualizing the Dynamin1 and EndophilinA1 copolymer in its membrane-bound state (Figure 5).
Figure 5: Membrane-remodeling complexes visualized with negative stain versus cryo-electron microscopy.

EndophilinA1 (A and D), Dynamin1 (B and E), or the hetero-complex formed by both proteins (C and F). In all cases, the purified proteins remodel spherical vesicles into high-curvature cylinders wrapped in an oligomeric protein coat. In the hetero-complex the adjacent turns of the Dynamin1 helix are separated by 2, 3 or 4 copies of an interleaving molecule of EndophilinA1. Bars 50nm.
The large sizes, the inherent heterogeneity of lipid mixtures, and the complexity of multi-component protein complexes make membrane-associated machines intractable for the mature techniques of X-ray crystallography or NMR spectroscopy (Figure 4–5). By contrast, electron microscopy is well suited to visualizing lipid bilayers and bound protein complexes directly (10–13, 28). Recent advances in electron microscopes, direct electron detectors, and new statistical approaches to image analysis have pushed the resolution limit for electron cryomicroscopy (cryo-EM) into the near-atomic regime (29–32). The application of these technologies to membrane-associated protein complexes will, in the near future, illuminate the protein-protein and protein-lipid interactions that determine membrane remodeling reactions and that enable biochemical compartmentalization or life within the walls of our little rooms.
Figure 4: Visualizing model membranes with transmission electron microscopy.
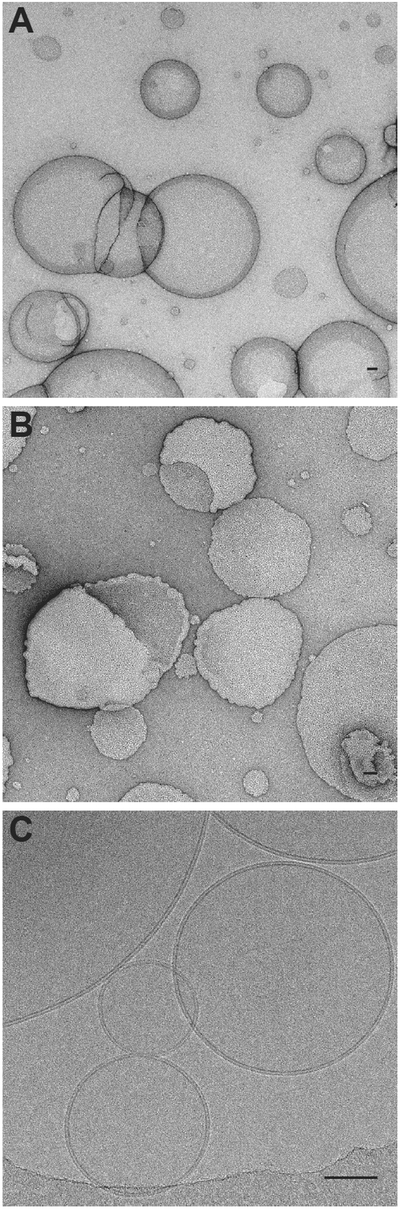
A) An electron micrograph of osmotically stable vesicles stained with uranyl acetate. B) An electron micrograph of “ruffled” vesicles due to an osmotic imbalance and volume shrinking during the negative staining procedure. C) Electron cryo-micrograph of vitrified liposomes with diverse diameters. Note the phospholipid headgroups that define the 5 nm bilayer. Bars 50 nm.
Methods
1.1. Purification of proteins from E. coli
1.1.1. BAR domain-containing protein over-expression in E. coli
This protocol is specifically adapted to express full-length and GST-tagged EndophilinA1 protein utilizing the pGEX6p1 expression vector (11). Alternative expression plasmids will also express other BAR domain-containing proteins. It is important to adapt this protocol to ensure that your expression vector and bacterial strain have been selected for the correct media and expression conditions.
1.1.1.1. Transforming E. coli cells
1.1.1.1.1. Reagents, equipment, and buffers
Human EndophilinA1 ORF cloned in pGEX6p1 plasmid. This vector introduces an N-terminal GST tag followed by a PreScission protease recognition site (GE, 88947, Little Chalfont, UK).
BL21 (DE3) RIPL competent cells (Agilent Technologies, 230280, Santa Clara, CA). This strain of E. coli has been engineered to have an expanded set of tRNA genes that improve heterologous protein expression. Other competent bacterial strains with similar features may also work.
Autoclave
LB liquid media (Sigma-Aldrich, L2542, St. Louis, MO)
Bunsen burner
Inoculation loop
10ml culture tubes (Kimble Chase, 73500–13100, Vineland, NJ)
Temperature controlled water bath (42 °C) or equivalent
Temperature controlled shaking incubator (37 °C)
LB agar plates with ampicillin and chloramphenicol (Teknova, L5204, Hollister, CA)
1.1.1.1.2. Detailed procedure
Thaw stock of BL21 competent cells on ice (50 μl aliquot).
Add 50–100 ng of EndophilinA1 expression plasmid to thawed cells.
Incubate on ice for 30 minutes, with gentle mixing every 5–10 minutes.
Heat shock cells in 42 °C water bath for 45 seconds.
Place the heat shocked cells on ice for 5 minutes.
Transfer cells to 900 μl of LB liquid media and allow cells to incubate at 37 °C with shaking at 100 rpm for 45–60 minutes in a 10 ml culture tube.
Allow LB agar plates with ampicillin (100 μg/ml) and chloramphenicol (34 μg/ml) to equilibrate to room temperature.
Plate 50–100 μl of the starter culture onto the LB agar selection plates using sterile technique with a Bunsen burner and an inoculation loop.
Use sterile glass beads or a sterile bacterial spreader to absorb the culture onto the plate.
Incubate plates at 37 °C until individual bacterial colonies are observed (8–16 hours). Glycerol stocks stored at −80 °C can be made from cultures of individual colonies from these plates to preserve this specific expression strain of E. coli.
1.1.1.2. Protein over-expression
1.1.1.2.1. Reagents, equipment, and buffers
BL21 (DE3) RIPL cells expressing EndophilinA1
Autoclave
Inoculation loop
LB liquid media (Sigma-Aldrich, L2542, St. Louis, MO)
Ampicillin (Gold Biotechnology, A-301–5, St. Louis, MO)
Chloramphenicol (Gold Biotechnology, C-105–25, St. Louis, MO)
500 ml Erlenmeyer flask (Thermo Fisher Scientific, FB-500–500, Waltham, MA)
Temperature controlled shaking incubator (37 °C)
Spectrophotometer with 600 nm absorbance setting
Cuvettes
2.8 L Fernbach flasks (Corning, 4424–2XL, Corning, NY)
- ZY auto-induction media (Studier et al., 2005)
- ZY- 10g Bacto-tryptone, 5g yeast extract, 925ml water
- 50X 5052- 0.5% Glycerol, 0.05% Glucose, 0.2% α-Lactose
- 20X NPS- 0.5 M (NH4)2SO4, 1 M KH2PO4, 1 M Na2HPO4
- Trace metals mix- 50 mM FeCl3.6H2O, 20 mM CaCl2.2H2O, 10 mM MnCl2.4H2O, 10 mM ZnSO4.7H2O, 2 mM CoCl2.6H2O, 2 mM CuCl2.2H2O, 2 mM NiCl2.6H2O, 2 mM Na2MoO4.2H2O, 2 mM H3BO4.
- Before use, mix per liter- 928 ml ZY, 1 ml 1 M MgSO4, 1 ml Trace metals mix, 20 ml 50X 5052 solution, 50 ml 20X NPS solution.
Temperature controlled shaking incubator (19 °C)
Balance
Centrifuge with Avanti JLA 8.1 rotor (Beckman Coulter, Indianapolis, IN) or equivalent
1 L centrifuge bottles (Beckman Coulter, 363676, Indianapolis, IN)
1.1.1.2.2. Detailed procedure
Prepare 100 ml of LB media with ampicillin (100 μg/ml) and chloramphenicol (34 μg/ml) in a 500 ml Erlenmeyer flask. Ensure that culture media and glassware have been autoclaved.
Utilize a sterile inoculation loop to pick an E. coli colony from the agar plate and inoculate 100 ml LB media plus antibiotics. Following proper sterile techniques during all bacterial expression steps is crucial to prevent contamination.
Shake the culture at 150 rpm at 37 °C overnight (8–12 hours).
Measure culture density utilizing optical density (OD600) reading in a spectrophotometer - Culture density = dilution factor * OD600 reading.
Prepare 500 ml of sterile ZY auto-induction media with 100 μg/ml ampicillin and 34 μg/ml chloramphenicol in a 2.8 L Fernbach flask.
Add enough starter culture to 500 ml flasks to obtain an OD600 of 0.05. Volume to add = (Desired OD600 * expression culture Volume) / OD600 reading of starter culture.
Shake the culture at 150 rpm at 37 °C until bacteria is in mid log phase (OD600 = 0.7 to 0.8).
Shift the culture to 19 °C and shake-incubate overnight (8–20 hours). We have found that the 19°C temperature shift aids in protein stability during overexpression and leads to an increased yield of stable protein.
Harvest the cultures using 1 L centrifuge tubes.
Centrifuge the cells at 4000 × g for 10 minutes at 4 °C in an Avanti JLA 8.1 rotor.
Collect the cell pellet in a disposable 50 ml centrifuge tube. Typical yields are 5–20 g of cell culture pellet per 1 L of liquid culture.
You may proceed immediately to the next step or freeze and store cell pellets in 50 ml disposable centrifuge tubes at −80 °C until ready for the next step.
1.1.2. BAR domain-containing protein purification and storage
1.1.2.1. Reagents, equipment, and buffers
BL21 (DE3) RIPL EndophilinA1 cell pellet
250 mL Griffin stainless steel beaker (Sigma-Aldrich, Z155527, St. Louis, MO)
Aprotinin (Roche, 10236624001, Mannheim, Germany)
Phenylmethylsulfonyl fluoride (PMSF) (Roche, 10837091001, Mannheim, Germany)
Leupeptin (Roche, 11034626001, Mannheim, Germany)
Pepstatin (Roche, 11524488001, Mannheim, Germany)
DNase I (Roche, 10104159001, Mannheim, Germany)
Lysozyme (Sigma-Aldrich, L6876, St. Louis, MO)
Branson sonifier with 0.5 inch tip (Thermo Fisher Scientific, 22309783, Waltham, MA) or equivalent
50ml Oak Ridge centrifuge tubes (Thermo Fisher Scientific ,3139–0030, Waltham, MA)
Centrifuge with Avanti JA 25.50 rotor (Beckman Coulter, Indianapolis, IN) or equivalent
0.45 μm syringe filter (Thermo Fisher Scientific, 190–9945, Waltham, MA)
Glutathione sepharose 4 fast flow affinity beads (GE, 17513202, Little Chalfont, UK)
Glass gravity columns 25 mm ID; 200 mm length (Kimble Chase Kontes, K420400–2520, Vineland, NJ)
Vivaspin 3 kDa MWCO centrifugal concentrator (Sartorius Stedim Biotech VS2092, New York, NY)
PreScission protease (GE, 88947, Little Chalfont, UK)
Dialysis tubing 12–14 kDa MWCO (Thermo Fisher Scientific, 2115214, Waltham, MA)
SDS PAGE supplies
Bio-Rad NGC chromatography system (Bio-Rad, 788–0003, Hercules, CA) or equivalent
HiLoad 16/600 superdex 200 column (GE, 28989335, Little Chalfont, UK)
Liquid Nitrogen
BAR-buffer A: 300 mM KCl, 50 mM Tris pH 7.4, 5 mM Imidazole, 1 mM DTT, 1% Triton X-100.
BAR-buffer B: 150 mM KCl, 50 mM Tris pH 7.4, 1 mM DTT
1.1.2.2. Detailed procedure
Thaw and resuspend the cell pellet in BAR-buffer A containing protease inhibitors PMSF (1 mM), pepstatin (1 μM), leupeptin (10 μM) and aprotinin (0.5 μM). Use 50 ml BAR-buffer A per 20 g cell pellet and place the homogenized mixture into a 250 ml Griffin stainless steel beaker pre-chilled on ice. Remainder of purification steps should be performed on ice unless otherwise noted.
Add 500 units of DNase I per 50 ml lysate. DNase I digests the bacterial DNA present in the lysate. The removal of cellular DNA prevents partial insolubility of the over-expressed protein by preventing entanglement of the two.
Add 15 mg powdered lysozyme to the lysate and incubate on ice for 30 minutes with gentle stirring every 5 minutes.
Sonicate the resuspended cell pellet at 90% duty cycle for 30 minutes with 30 seconds sonication and 1 minute rest cycles.
Clear the lysate with a 20000 × g spin for 1 hour at 4 °C in 50 ml oak ridge centrifuge tubes.
Decant the supernatant into a pre-chilled 250 ml beaker and filter the solution with a 0.45 μm syringe filter and a 60 ml syringe into a clean pre-chilled beaker.
Prepare an affinity column by loading 5 ml (bed volume) of glutathione sepharose bead slurry into a glass gravity column pre-filled with deionized water.
Wash beads with ≥200 ml of deionized water.
Equilibrate beads with three 100 ml washes of BAR-buffer A.
Apply the clarified filtered lysate onto the glutathione sepharose beads and incubate at 4 °C for 4–12 hours with gentle rocking.
Let the unbound lysate flow through the gravity column.
Wash unbound protein impurities with 40 bed volumes BAR-buffer A.
Resuspend the GST tagged BAR protein bound to sepharose beads into 10 bed volumes of BAR-buffer B and add 20 units of PreScission protease.
Incubate at 4 °C for 8 hours
Elute the cleaved protein from the affinity column into a 50 ml centrifuge tube on ice.
Evaluate cleaved protein purity and molecular weight with SDS-PAGE followed by Coomassie blue staining.
Dialyze the protein against 2 L of BAR-buffer B using a 12–14 kDa MWCO dialysis tubing for 8–12 hours.
Concentrate the eluate with 3 kDa MWCO centrifugal concentrators to <2 ml final volume.
Inject the concentrated protein solution over a HiLoad 16/600 superdex 200 column pre equilibrated with BAR-buffer B. EndophilinA1 elutes at ~85 ml.
Pool the peak fractions and check the concentration using a spectrophotometer. Concentrate the protein using 3 kDa MWCO centrifugal concentrators to a final concentration >5 mg/ml.
Aliquot into 10–50 μl aliquots (single use). This prevents exposing the purified protein to freeze thaw cycles that cause degradation.
Flash freeze the protein with liquid nitrogen and store at −80 °C.
1.1.3. Soluble MiD49/51 adaptor protein truncation over-expression in E. coli
The protocol described below is for the expression and purification of MiD49 from E. coli. The protocol also can be used to purify MiD51. The soluble domain of MiD49 (amino acids 126–454) was used for this purification. Variations of this protocol have previously been described (33, 34).
1.1.3.1. Reagents, equipment, and buffers
Human MiD49 soluble domain (amino acids 126–454) ORF cloned in pGEX6p1 vector. This vector introduces an N-terminal GST tag followed by a PreScission protease recognition site.
Same reagents as protocol step 1.1.1.1.1 for transformation
Same reagents as protocol step 1.1.1.2.1 for protein over-expression
1.1.3.2. Detailed procedure
Same protocol as BAR domain protein over-expression in E. coli. Follow protocol steps 1.1.1.1.2 for transformation and protocol steps 1.1.1.2.2 for protein over-expression.
1.1.4. Soluble MiD adaptor protein truncation purification and storage
1.1.4.1. Reagents, equipment and buffers
All protein purification reagents and equipment as described for section 1.1.2.1
MiD-buffer A: 50 mM Tris pH 8.0, 500 mM NaCl, 5%Glycerol, 1 mM DTT, 0.1%, Triton X-100
MiD-buffer B: 50 mM Tris pH 8.0, 500 mM NaCl, 5% Glycerol, 1 mM DTT
MiD-buffer C: 20 mM Tris pH 8.0, 200 mM NaCl, 5% glycerol, 1 mM DTT
1.1.4.2. Detailed procedure
Thaw and resuspend the cell pellet in MiD-buffer A containing protease inhibitors PMSF (1 mM), pepstatin (1 μM), leupeptin (10 μM) and aprotinin (0.5 μM). Use a volume of 50 ml per 20 g of pellet. Transfer the resuspended pellet to a 250 ml Griffin stainless steel beaker pre-chilled on ice. Remainder of purification steps should be performed on ice unless otherwise noted.
Add 500 units DNase I per 50 ml lysate.
Add 15 mg powdered lysozyme to the lysate and incubate the resuspended pellet on ice for 30 minutes with gentle stirring every 5 minutes.
Sonicate the resuspended cell pellet at 90% duty cycle for 30 minutes with 30 seconds sonication and 1 minute rest cycles.
Spin at 20000 × g for 1 hour at 4 °C in 50 ml oak ridge centrifuge tubes to separate the soluble and insoluble portions.
Decant the supernatant into a pre-chilled 250 ml beaker and filter the solution with a 0.45 μM syringe filter and a 60 ml syringe into a clean pre-chilled beaker.
Prepare an affinity column by adding 5 ml of the glutathione sepharose bead slurry into a glass gravity column pre-filled with deionized water.
Incubate the clarified lysate from step 6 for 12 hours with glutathione sepharose beads equilibrated with MiD-buffer A at 4 °C.
Allow the unbound protein to flow through and wash the beads with 40 times the bed volume with MiD-buffer A followed by MiD-buffer B.
Incubate the beads overnight with 20 units of PreScission protease at 4 °C with gentle rocking for 12 hours.
Pass the supernatant over a gravity column and obtain the flow through. The flow through should contain the cleaved MiD49. Evaluate the molecular weight and percentage purity of the cleaved protein with SDS-PAGE followed by Coomassie blue staining.
Concentrate the cleaved protein using a 3 kDa MWCO centrifugal concentrator to a volume < 2 ml and load on a HiLoad 16/600 superdex 200 column pre-equilibrated with MiD-buffer C for size exclusion. The column volume for the superdex 200 is 120 ml and the MiD49 peak appears at ~72 ml.
Evaluate protein purity and molecular weight with SDS-PAGE followed by Coomassie blue staining.
Collect the fractions positive for MiD49 and concentrate using a 3 kDa MWCO centrifugal concentrator. Concentration of the protein preparation can be estimated at this stage using a spectrophotometer. Concentrate the protein to ~5 mg/ml.
Divide into 20 μl aliquots (single use) and flash freeze using liquid nitrogen and store at −80 °C until further use.
1.1.5. Drp1 over-expression in E. coli
Full length dynamin proteins have been challenging to purify historically, owing to their propensity to oligomerize and poor stability/solubility when over-expressed in bacteria. As a result, multiple protocols have been developed to purify dynamins from yeast and insect cell based expression systems. This protocol describes over-expression and purification of human Drp1 from E. coli. The combination of specialized buffers utilized here results in highly pure, stable and assembly-competent Drp1 protein. Variations of this protocol have been reported (23, 35).
1.1.5.1. Reagents, equipment and buffers
1.1.5.2. Detailed procedure
Same protocol as BAR domain protein over-expression in E. coli. Follow protocol steps 1.1.1.1.2 for transformation and protocol steps 1.1.1.2.2 for protein over-expression.
1.1.6. Drp1 purification and storage
1.1.6.1. Reagents, equipment and buffers
BL21 (DE3) RIPL cells expressing human Drp1
All protein purification reagents and equipment as described for section 1.1.1.1.2
Qiagen Ni-NTA Agarose beads. (Qiagen 30230, Venlo, Netherlands)
Drp1-buffer A: 50 mM HEPES/NaOH pH 7.5, 400 mM NaCl, 20 mM imidazole, 5 mM MgCl2 and 1 mM DTT
Drp1-buffer B: 50 mM HEPES/NaOH pH 7.5, 800 mM NaCl, 5 mM MgCl2, 20 mM imidazole, 1 mM DTT, 1 mM ATP, 10 mM KCl
Drp1-buffer C: 50 mM HEPES/NaOH pH 7.5, 400 mM NaCl, 20 mM imidazole, 5 mM MgCl2 and 1 mM DTT, 0.5% CHAPS
Drp1-buffer D: 50 mM HEPES/NaOH pH 7.5, 400 mM NaCl, 300 mM imidazole, 5 mM MgCl2 and 1 mM DTT
Drp1-buffer E: 20 mM HEPES/NaOH pH 7.5, 300 mM NaCl, 2.5 mM MgCl2 and 1 mM DTT
1.1.6.2. Detailed procedure
Thaw and resuspend the cell pellet in Drp1-buffer A containing protease inhibitors PMSF (1 mM), pepstatin (1 μM), leupeptin (10 μM) and aprotinin (0.5 μM). Use a volume of 50 ml per 20 g of pellet. Transfer the resuspended pellet to a stainless steel beaker.
Add 0.5 mg DNase1 and 50 mg powdered Lysozyme. Incubate the mixture on ice for 30 minutes with constant stirring every 5 minutes.
Sonicate the resuspended cell pellet at 90% duty cycle for 30 minutes with 30 seconds sonication and 1 minute rest cycles.
Spin the lysate at 20000 × g for 1 hour at 4 °C to separate the soluble and insoluble portions.
Decant the supernatant in a pre-chilled 250 ml beaker and filter the solution with a 0.45 μm syringe filter and a 60 ml syringe into a clean pre-chilled beaker.
Prepare an affinity column by equilibrating 5 ml of the Qiagen nickel-NTA beads in Drp1-buffer A.
Incubate the beads with the clarified lysate for 1 hour at 4 °C with gentle rocking.
Allow the unbound portion of the lysate to flow through using a gravity column.
Wash the beads with 40 bed volumes of Drp1-buffer A, followed by Drp1-buffer B and Drp1-buffer C (40 bed volumes each).
Wash with 40 bed volumes of Drp1-buffer A once more.
Elute the protein with 10 bed volumes of Drp1-buffer D.
Cleave the eluted protein overnight at 4 °C using 20 units of PreScission protease while dialyzing to Drp1-buffer A (2 L) for 12 hours.
Rebind cleaved protein to fresh beads equilibrated with Drp1-buffer A.
The protein can bind to the beads despite cleavage of the His tag. After washing the beads with 40 bed volumes of Drp1-buffer A, elute with 10 bed volumes of Drp1-buffer D.
Concentrate the eluate using a 3 kDa MWCO centrifugal concentrator and reduce the volume to < 2 ml.
Inject the concentrated protein over a HiLoad 16/600 superdex 200 column pre equilibrated with Drp1-buffer E for size exclusion.
Elute from the size exclusion column with 1 column volume of Drp1-buffer E. The column volume for the superdex 200 is 120 ml and the Drp1 peak appears at ~60 ml.
Collect the fractions from the protein peak, estimate percentage purity and molecular weight using SDS-PAGE followed by Coomassie blue staining.
Concentrate to 9 mg/ml using a 3 kDa MWCO centrifugal concentrator. Divide into 20 μl single use aliquots. Flash freeze using liquid nitrogen and store at −80 °C
1.2. Purification of SH3 domains from E. coli
This protocol is for the preparation of recombinant purified GST-Amphiphysin2 SH3 domain protein. The application for this protein is to prepare an SH3 affinity resin that can be utilized to purify untagged full length Dynamin1 protein from any source. GST tagged Amp2SH3 (residues 494–588) binds to glutathione sepharose beads and creates a Dynamin1 affinity resin. This allows you to affinity purify Dynamin1 using the Amphiphysin SH3 domain and Dynamin1 proline rich domain interaction (36).
1.2.1. SH3 domain protein over-expression in E. coli
The same protocol and reagents as the BAR domain protein over-expression in E. coli can be followed for the transformation and over-expression of SH3 domain (section 1.1.1).
1.2.1.1. Reagents, equipment, and buffers
Human Amphiphysin SH3 (Amp2SH3) gene cloned in pGEX6p1 expression vector. This vector introduces an N-terminal GST tag followed by a PreScission protease recognition site.
Same reagents as protocol step 1.1.1.1.1 for transformation
Same reagents as protocol step 1.1.1.2.1 for protein over-expression
1.2.1.2. Detailed protocol
Same protocol as BAR domain protein over-expression in E. coli. Follow protocol steps 1.1.1.1.2 for transformation and protocol steps 1.1.1.2.2 for protein over-expression.
1.2.2. SH3 domain protein purification and storage
The same protocol and reagents as the BAR domain protein purification and storage (section 1.1.2) can be followed for the purification of the Amp2SH3 protein with the following alterations to the protocol.
1.2.2.1. Reagents, equipment, and buffers
Same reagents as protocol step 1.1.2.1
BL21 (DE3) RIPL Amp2SH3 cell pellet
Reduced glutathione (Sigma-Aldrich, G4251, St. Louis, MO)
SH3-buffer A: 300 mM KCl, 50 mM Tris pH 8.0, 5 mM imidazole, 1 mM DTT
SH3-buffer B: 300 mM KCl, 50 mM Tris pH 8.0, 5 mM imidazole, 1 mM DTT, 20 mM glutathione
1.2.2.2. Detailed procedure
Follow protocol steps 1.1.2.2.1 to 1.1.2.2.10 except with BL21 (DE3) RIPL Amp2SH3 cell pellet as the starting material.
Bind the clarified filtered Amp2SH3 lysate to the glutathione sepharose beads for 4–12 hours at 4 °C.
Allow the unbound protein to flow through a gravity column.
Wash unbound protein impurities with 40 bed volumes of SH3-buffer A.
Elute the Amp2SH3 with 10 bed volumes of SH3-buffer B containing 20 mM reduced glutathione.
Concentrate the eluate with 3 kDa MWCO centrifugal concentrators to a final volume of 5 ml.
Dialyze the Amp2SH3 protein using a 3 kDa MWCO dialysis tubing against 2 L SH3-buffer B for 12 hours at 4 °C.
Evaluate protein purity and molecular weight with SDS-PAGE followed by Coomassie blue staining.
Calculate protein concentration using a spectrophotometer.
Divide the purified protein into 10 mg aliquots (single use) using microcentrifuge tubes. Flash freeze the tubes in liquid nitrogen and store at −80 °C. These aliquots contain sufficient protein for the generation of an SH3 affinity resin (section 1.2.3).
1.2.3. Preparation of an SH3 affinity resin
This protocol is to be followed for the purification of untagged Dynamin1 (protocol step 1.3.3).
1.2.3.1. Reagents, equipment, and buffers
Amp2SH3 purified protein
Glutathione sepharose 4 fast flow affinity beads (GE, 17513202, Uppsala, Sweden)
Glass gravity columns 25 mm ID; 200mm length (Kimble Chase, K420400–2520, Vineland, NJ)
SH3-buffer A: 300 mM NaCl, 50 mM Tris pH 8.0, 5 mM Imidazole, 1 mM DTT
1.2.3.2. Detailed Procedure
Thaw a 10 mg aliquot of Amp2SH3 protein on ice.
Prepare an affinity column by loading 5 ml (bed volume) of glutathione sepharose bead slurry into a glass gravity column pre-filled with deionized water.
Wash beads with ≥200 ml of deionized water. Elute 95% of the buffer from the column ensuring that the beads remain in solution and are kept at 4 °C for all affinity column steps.
Equilibrate beads with three 100 ml washes of SH3-buffer A.
Apply 10 mg purified Amp2SH3 onto the glutathione sepharose beads and incubate at 4 °C for 2–4 h with gentle rocking.
Allow the unbound protein to flow through the gravity column.
Wash the beads with 40 bed volumes of SH3-buffer A to remove any unbound Amp2SH3.
The column is ready for the Purification of Dynamin1 via the SH3 affinity resin (section 1.3.3).
1.3. Expression of full-length, untagged Dynamin-1 in Insect cells
1.3.1. Preparation of viruses
This protocol is to produce high-titer recombinant baculovirus stock of Dynamin1 (37). This protocol is prepared according to the Expression Systems protocol. All information detailed here is also found in the Expression Systems protocols.
1.3.1.1. Reagents, equipment, and buffers
Human Dynamin1 gene cloned in pVL1392 Baculovirus transfer vector.
125 ml Erlenmeyer culture flasks (Corning, 431143, Corning, NY)
BSL2 biological fume hood
Spodoptera frugiperda (Sf9) cells
1.5 ml sterile microcentrifuge tubes
Lipofectamine 3000 (Life Technologies, L3000–015, Carlsbad, CA)
24 deep well block (Life Technologies, CS15124, Carlsbad, CA)
ESF 921 Insect Cell Culture Medium, Protein-Free (Expression Systems, 96–001, Davis, CA)
Transfection-buffer A: 2 ug recombinant transfer vector, 0.5 ug linearized viral DNA, 100 μl transfection medium
Transfection-buffer B: 6 μl lipofectamine 3000, 100 μl transfection medium
1.3.1.2. Detailed protocol
All cell transfer and reagent mixing steps need to be performed in a BSL2 certified biological tissue culture hood.
1.3.1.2.1. Generation of P0 viral stock.
Inoculate 1×106 SF9 cells in 0.5 ml ESF 921 media per well of a 24 deep well block.
Mix solution A and incubate for 5 minutes.
Mix solution B and incubate for 5 minutes.
Combine transfection-buffer A and B and incubate at room temperature for 30 minutes.
Add 1 ml of transfection media to the well of SF9 cells and incubate at 27 °C for 4–5 hours.
Add 3 ml ESF 921 to the well.
Cover the wells and incubate at 27 °C with 120–150 rpm orbital shaking for 4–5 days.
Clear cell debris with a 2500 rpm room temperature spin for 5 minutes and collect and save the supernatant as the P0 viral stock. Store all viral stock solutions in light protected containers and keep at 4 °C.
1.3.1.2.2. Generation of P1 viral stock
In a 125 ml Erlenmeyer culture flask transfer SF9 cells grown at mid log phase to a final concentration of 1×106 cells/ml with a final volume of 30 ml.
Grow cells for 24 hours at 27 °C with gentle shaking.
Inoculate the culture with 0.5 ml of P0 viral stock.
Incubate infected cells for 4–5 days at 27 °C with gentle shaking.
Clear cell debris with a 2500 rpm room temperature spin for 5 minutes and collect and save supernatant at P1 stock.
Titer virus to determine multiplicity of infection (MOI) by performing Viral Plaque Assay (Refer to Expression System protocol here: www.expressionsystems.com/documents/Plaque%20Assay.pdf).
Typical P1 titers are 1×106 – 1×108 infectious units/ml. P1 virus can be stored at −80 °C in aliquots sufficient to generate P2 viral stocks.
1.3.1.2.3. Generation of P2 viral stock
Follow the same protocol as for the P1 viral stock but infect the cells at an MOI of 0.1.
Use the following equation to determine the volume of P1 stock to obtain a specific MOI.
Inoculum required (ml) = Desired MOI (pfu/cell) × number of cells / titer of viral stock (pfu/ml).
Typical P2 titers are 5×108 infectious units per ml.
1.3.1.2.4. Generation of P3 viral stock.
Follow the same protocol as for the P1 viral stock using P2 stock to infect. Excessive passage of baculovirus can cause unwanted mutations or undesirable variants of baculovirus amplifying in your stock culture. Therefore, it is recommended to use P2 or P3 stocks for infection of expression cultures. It is better to regenerate the P2 and P3 viral stocks from saved P1 viral stocks when additional virus is required.
1.3.2. Infection and culture of SF9 cells
1.3.2.1. Reagents, equipment, and buffers
P2 or P3 Dynamin1 virus
125 ml Erlenmeyer culture flasks (Corning, 431143, Corning, NY)
2.8 L unbaffled Fernbach flasks (Corning, 4420–2XL, Corning, NY)
Gentamicin (Life Technologies, 15750–060, Carlsbad, CA)
25 ml transfer pipette
Centrifuge with Avanti JLA 8.1 rotor (Beckman Coulter, Indianapolis, IN) or equivalent
1 L centrifuge bottles (Beckman Coulter, 363676, Indianapolis, IN)
ESF 921 Insect Cell Culture Medium, Protein-Free (Expression Systems, 96–001, Davis, CA)
1.3.2.2. Detailed procedure
In a 2.8 L unbaffled Fernbach flask, transfer SF9 cells grown at mid log phase to a final concentration of 1.8×106 to 2.0×106 cells/ml with a final volume of 500 ml with ESF 921 media. Add gentamicin to a final concentration of 10 μg/ml to prevent bacterial contamination.
Grow cells at 27 °C with gentle shaking until cells have reached a culture density of 1.0×106 cells/ml.
Inoculate the culture with 10 ml of P2 viral stock.
Incubate the infected cells for 48 hours at 27 °C with 150 rpm shaking. 48 hours infection time optimal for Dynamin1 purification as longer incubations tend to lead to increased degradation of Dynamin1. The C-terminal proline rich domain is susceptible to cleavage from the protein during longer viral incubations.
Collect the cells with a 2500 × g spin for 10 minutes at 4 °C in 1 L centrifuge tubes and Avanti JLA 8.1 rotor.
Remove the supernatant and resuspend the cells in 50 ml fresh ESF 921 media per 500 ml cell culture pellet with gentle shaking for 5 minutes.
Collect the resuspended cell pellet in a disposable 50 ml centrifuge tube.
Spin the tubes in a benchtop swinging bucket centrifuge at 2500 rpm at 4 °C.
Remove the supernatant. The cells can be stored at −80 °C until the purification steps. Typical yields are 5–10 g of cell pellet per 500 ml culture.
1.3.3. Purification of Dynamin1 via SH3 affinity resin
This procedure utilizes an affinity resin comprised of glutathione sepharose beads bound to GST amphiphysin-2 SH3 domain (36). The Amp2SH3 domain captures full length Dynamin1 through a high affinity interaction with its PRD.
1.3.3.1. Reagents, equipment, and buffers
SH3 affinity resin as prepared in 1.2.3.2
Protease inhibitor cocktail tablets (Roche, 05892791001, Mannheim, Germany)
Glass gravity columns 25 mm ID; 200 mm length (Kimble Chase, K420400–2520, Vineland, NJ)
100 ml glass dounce tissue homogenizer (Kimble Chase 885303, Vineland, NJ)
32 ml Tube, thick wall, polycarbonate (Beckman Coulter, 355631, Indianapolis, IN)
Centrifuge and 50.2 Ti fixed angle rotor (Beckman Coulter, 337901, Indianapolis, IN)
Vivaspin 50 kDa MWCO centrifugal concentrator (Sartorius Stedim Biotech VS2031)
0.45 μm syringe filter (Thermo Fisher Scientific, 190–9945, Waltham, MA)
Rocker shaker
SDS PAGE supplies
Dynamin-buffer A: 150 mM NaCl, 20 mM HEPES pH 7.4, 1 mM EGTA, 1 mM DTT, 5% (v/v) glycerol, Protease inhibitor cocktail tablets (Roche)
Dynamin-buffer B: 150 mM NaCl, 20 mM HEPES pH 7.4, 1 mM EGTA, 1 mM DTT, 5% (v/v) glycerol
Dynamin-buffer C: 1.2 M NaCl, 20 mM PIPES pH 6.5, 1 mM DTT, 5% glycerol
Dynamin-buffer D: 100 mM NaCl, 20 mM HEPES pH 7.4, 50% glycerol
1.3.3.2. Detailed Procedure
Thaw Dynamin1 SF9 cell pellets on ice.
Add 50 ml of Dynamin-buffer A per 10 ml of cell pellet.
Obtain a homogeneous mixture with gentle vortex mixing.
Lyse the cells by tissue grinding the solution in a 100 ml glass dounce.
Initial 10 plunges with course (smaller) pestle and final 10–40 plunges with fine pestle.
A small sample may be directly visualized under a compound light microscope to ensure all cells have been lysed.
Clear the lysate with ultra-centrifugation at 45000 × g for 1 h at 4 °C.
Filter the cleared lysate supernatant through a 0.45 μm syringe filter (PN) and collect into a clean beaker on ice.
During the centrifugation step prepare the SH3 affinity resin according to procedure steps 1.2.3.
Add the cleared filtered Dynamin1 lysate to the column and allow protein binding for 4 hours with gentle rocking at 4°C.
Allow the supernatant to flow through the column and collect this entire fraction.
Wash the beads with 40 bed volumes of Dynamin-buffer B. Elute Dynamin1 off of the column with 10 bed volumes of Dynamin-buffer C.
Evaluate the purity and molecular weight of the eluate with SDS-PAGE followed by Coomassie blue staining.
Concentrate the eluate to 1–5 ml using centrifugal concentrators.
Dialyze the concentrated eluate into Dynamin-buffer D for 8–16 hours at 4 °C.
Flash freeze the dialyzed protein in 50 μl (single use) aliquots with liquid nitrogen and store the protein at −80 °C.
1.4. Making model membranes
1.4.1. Approximating target membrane lipid species distribution
Membranes that outline different cellular compartments have evolved to be comprised of different lipid compositions (38). For example, the lipid composition of the plasma membrane differs markedly from that of the mitochondria. When one designs in vitro reactions that involve the use of model membranes, care should be taken to reflect the composition of the relevant target membrane in the cell and its lipid specificities. For example, in the steady state, the mitochondrial membrane has the relatively rare lipid cardiolipin, amongst other more abundant “structural” lipids such as phophatidylcholine (PC) and phosphatidylethanolamine (PE). Phosphatidylinositol (PI)-3-phosphate (PI3P) is enriched on the early endosomes, PI(3,5)P2 on late endosomes, PI(4,5)P2 on the plasma membrane and PI(4)P on the golgi apparatus, among other examples. In most cases, the molar percentage of cholesterol also varies between various membranes within the cell.
1.4.2. Use of defined biological sources
1.4.2.1. Reagents, equipment, and buffers
Brain extract from bovine type 1 Folch fraction 1 (Sigma-Aldrich, B1502, St. Louis, MO)
Phosphatidylethanolamine (PE) (Avanti Polar Lipids, 840022, Alabaster, AB)
Phosphatidylserine (PS) (Avanti Polar Lipids, 840032, Alabaster, AB)
1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS) (Avanti Polar Lipids, 840034, Alabaster, AB)
1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS) (Avanti Polar Lipids, 840035, Alabaster, AB)
L-α-phosphatidylinositol (PI) (Avanti Polar Lipids, 840042C, Alabaster, AB)
L-α-phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) (Avanti Polar Lipids, 840046, Alabaster, AB)
1,2-dioctanoyl-sn-glycero-3-(phosphoinositol-3-phosphate) (PI3P) (Avanti Polar Lipids, 850187, Alabaster, AB)
1,2-dioleoyl-sn-glycero-3-phospho-(1’-myo-inositol-5’-phosphate) (PI5P) (Avanti Polar Lipids 850152P, Alabaster, AB)
L-α-phosphatidylcholine (PC) (Avanti Polar Lipids, 840051C, Alabaster, AB)
Cholesterol (Avanti Polar Lipids, 700000P, Alabaster, AB)
There are multiple sources of lipids used as model membranes in the laboratory. Popular examples include lipid mixtures like the Folch fractions which are extracts from tissues using organic solvent-based procedures (39). These fractions are subsets of the total lipid makeup of membranes from a specific tissue, such as the brain in the case of the bovine brain lipid extract. Other sources include soybean, heart muscle and semi-purified mitochondrial lipid extracts. The major components of these mixtures are phospholipids including PC, PE and PS.
In other cases, the lipid composition desired may be a simpler mix of defined components. For example, if you are testing whether a protein binds to a specific lipid species you will need to make a defined model membrane that contains or omits this lipid at a given molar ratio. Many individual lipid species are available as pure powders or dissolved in chloroform from commercial vendors. It is convenient to know the molarity of your stocks beforehand so that you can add components in the molar ratios that you desire. Some commonly available lipids that are major structural components of cellular membranes are POPS, DOPS, PI, Cardiolipin, PI(4,5)P2, PI3P, PI4P, PI5P and PC amongst others. Cholesterol is also a major structural component in most membranes that should be included between 20 and 30 molar percent.
1.4.3. Preparation of vesicles
1.4.3.1. Reagents, equipment, and buffers
Lipid stocks dissolved in chloroform
Glass vials (Agilent 5182–0714, Santa Clara CA)
PFA-Teflon tips (Elemental Scientific, ES-7000–1001, Omaha NE)
Vortexer
Gaseous Nitrogen/Argon
Vacuum source
N-hexane (Sigma-Aldrich, 139386, St. Louis, MO)
10 mm filter supports (Avanti Polar Lipid, 610014, Alabaster, AB)
Whatman nucleopore track-etch membrane (Avanti Polar Lipids, 800319, Alabaster, AB)
Resuspension buffer- 20mM HEPES pH 7.5, 150mM KCl.
Bath sonicator
Avanti liposome mini-extruder setup (Avanti Polar Lipids, 610000, Alabaster, AB)
Hamilton gastight syringe (Avanti Polar Lipids 610017, Alabaster, AB)
1.4.3.2. Detailed procedure
Add the required lipid components in the desired molar ratios to a glass vial. The final volume of each lipid species added to the mix depends upon the molar stock concentration. Long exposure of plastics to organic solvents can result in leaching of plastic and contamination of lipid stocks. Therefore it is essential to use glass micropipettes or PFA-Teflon tips with lipid solutions in chloroform.
Take the glass vial to a fume hood. Gently vortex to mix the components. While vortexing, introduce a steady flow of an inert gas such as Nitrogen or Argon at the mouth of the tube. The purpose of doing this is to create an inert atmosphere to prevent oxidation of lipids while they are dried.
Keep vortexing under the steady flow of nitrogen/argon till the liquid dries out and the lipid forms a film all around the sides of the glass vial.
Attach the glass tube to a vacuum source such as a lyophilizer to pump out residual chloroform for one hour.
Resuspend the lipid film in 500μl absolute n-hexane to remove residual chloroform trapped within the lipid film. Repeat the vortexing step under inert gas until you obtain the thin lipid film again.
Attach the glass tube to a vacuum source such as a lyophilizer to pump out residual chloroform for 4 hours or overnight.
Resuspend the lipid film in your resuspension buffer to obtain a final concentration of no more than 2 mg/ml total lipid.
Shake lightly to resuspend most of the lipid film in the buffer. If there is some lipid film left on the walls of the tube, use gentle vortexing to remove it from the wall.
Incubate overnight at 4 °C with gentle rocking.
Membranes can be aliquoted (50–100 ul aliquots) and stored at −80 °C after flash freezing in liquid nitrogen.
In the method described above, liposomes are formed by the hydration of lipid films that detach from the walls of the glass vial on agitation, simultaneously forming closed spherical vesicles. This generally leads to a majority of the liposomes being multilamellar, meaning that there are vesicles within vesicles (“onions”). For most downstream experiments such as flotation or reverse sedimenation assays (protocol step 1.5.2) or in vitro membrane remodeling reactions with BAR domain-containing proteins or dynamins (protocol step 1.6.2), unilamellar vesicles are essential. There are 2 major methods available to increase the percentage of unilamellar vesicles in the mixture:
-
11.1.Sonication: Sonication disrupts the large multilamellar vesicles (LMVs) formed after rehydration of lipid films and breaks them up into very small and predominantly unilamellar vesicles (SUVs) that are curvature limited (15–50 nm in diameter). Following sonication, repeated freeze/thaw cycles (up to 10) after the formation of SUVs results in spontaneous fusion of these small vesicles and results in larger unilamellar vesicles (LUVs).
-
11.1.1)Sonicate membranes for 5 minutes in a water bath sonicator at maximum power.
-
11.1.2)Flash freeze the membranes, thaw by keeping in a water bath at room temperature.
-
11.1.3)Repeat the freeze and thaw cycle 10 times.
-
11.1.4)Flash freeze membranes and keep at −80 °C until further use.
-
11.1.1)
-
11.2.Extrusion: using the Avanti liposome extruder, a liposome solution can be repeatedly made to pass through polycarbonate membranes of defined pore sizes. This process, like sonication, breaks and reseals vesicles and increases the number of unilamellar vesicles in the solution. This also introduces an upper size limit on the vesicles. In our hands, forcing a liposome suspension through a polycarbonate membrane of pore size 1 μm ~20 times yields a unilamellar vesicle population with a mean diameter of ~200 nm.
-
11.2.1)Set up the extruder as per the instructions of the manufacturer. Use a 1 μm pore size membrane. (Refer to http://www.avantilipids.com/index.php?option=com_content&view=article&id=185&Itemid=193 for details of usage for the Avanti mini extruder). To minimize sample loss, pre wet the membrane and membrane supports.
-
11.2.2)Fill up the gastight syringe (syringe 1) with the liposome solution, attach it to one end of the mini-extruder. Attach an unfilled syringe (syringe 2) on the other available side.
-
11.2.3)Gently push the liquid through the extruder set up, into the syringe on the other side. Push it back to the syringe 1.
-
11.2.4)Repeat the process 20 times.
-
11.2.5)Collect the liposomes from syringe 2.
-
11.2.6)Aliquot into 100 μl aliquots, flash freeze and store at −80 °C.
-
11.2.1)
1.5. Measuring membrane binding
Once liposomes having the desired lipid composition have been prepared, protein binding to these membranes can be tested qualitatively by two major methods. Each method has strengths and weaknesses and we will discuss these in section 1.5.3 after describing the protocols (Figure 3).
Figure 3: Centrifugation assays for qualitative membrane binding.

A) Flotation or reverse sedimentation exploits the buoyancy of vesicles within a density gradient to separate bound from unbound protein and to avoid contamination with unstable protein aggregates. This assay rarely yields false-positives due to its stringency, but weak membrane interactions are typically missed. B) In a traditional sedimentation assay vesicles are pelleted rapidly at high G-forces along with bound proteins. If the target lipid or the bulk properties of the vesicles are chosen appropriately for the protein being investigated, this assay rarely results in false-negatives. However, unstable proteins or protein oligomers may pellet non-specifically.
1.5.1. Flotation or reverse sedimentation (Figure 3A)
1.5.1.1. Reagents, equipment, and buffers
Purified protein
Liposomes
1 ml ultracentrifuge tubes (Beckman Coulter, 343778, Indianapolis, IN)
Ultracentrifuge with TLS-55 rotor (Beckman Coulter, 346936, Indianapolis, IN)
Methanol (Sigma Aldrich, 179337, St. Louis, MO)
2x Laemmli buffer (Bio-Rad, 161–0737, Hercules, CA)
Chloroform (Sigma Aldrich, 319988, St. Louis, MO)
Balance
SDS PAGE supplies
Sucrose-buffer A: 2 M sucrose, 20 mM HEPES pH 7.5, 100 mM KCl
Sucrose-buffer B: 1 M sucrose, 20 mM HEPES pH 7.5, 100 mM KCl
Sucrose-buffer C: 0.5 M sucrose, 20 mM HEPES pH 7.5, 100 mM KCl
Buffer-D: 20 mM HEPES pH 7.5, 100 mM KCl
1.5.1.2. Detailed procedure
Mix protein and liposomes with a molar ratio of 1000:1 lipid:protein.
Incubate at 4 °C for 1 hour. In the case of Drp1 and MiD49, we noticed turbidity upon incubating Drp1 with liposomes at room temperature. Thus, to rule out any aggregation that may have caused the turbidity, the mixture was incubated at 4 °C for an hour to ensure optimal binding. The solution remained clear at 4 °C.
Transfer the mixture into an ultracentrifuge tube.
Homogenize the mixture with 300 μl of Sucrose-buffer A.
Mark the upper boundary of the layer on the outside of the tube with a marker. Carefully pipetting, add 150–300 μl Sucrose-buffer B followed by 300 μl Sucrose-buffer C. Mark the upper boundary of each layer.
Balance the tubes using buffer D.
Spin the tubes in a pre-chilled TLS-55 rotor at 175000 × g for 45 minutes at 4 °C.
Without disturbing other layers, carefully collect each sucrose layer separately and pipette into a microcentrifuge tube.
Methanol chloroform precipitation to estimate protein from sucrose layers (steps 9–16): To the sucrose layer that was collected (volume 150–300 μl), add 400 μl methanol, 100 μl chloroform followed by 300 μl water. Vortex and keep on ice for 5 minutes.
Spin at 12000 × g for 5 minutes in a benchtop centrifuge at room temperature.
Carefully remove the upper layer without disturbing the interface.
Add 300 μl methanol, vortex.
Spin at 12000 × g for 10 minutes at room temperature. Carefully remove the liquid phase without disturbing the protein pellet.
Add 30 μl 2x Laemmli sample buffer and mix well.
Analyze the amount of protein in each layer by SDS PAGE followed by Coomassie blue staining.
Analysis of the resulting SDS PAGE gel will identify if the protein bound the liposome membrane. Membrane bound protein will be detected in the 0.5 M sucrose layer and unbound proteins will be detected in the 2 M and 1 M sucrose layer. Comparison with positive controls (robust membrane binding proteins, e.g. EndophilinA1) and negative controls such as purified GST is essential.
1.5.2. Sedimentation (Figure 3B)
1.5.2.1. Reagents, equipment, and buffers
Liposomes
Purified protein
Benchtop centrifuge
2x Laemmli buffer (Bio-Rad, 161–0737, Hercules, CA)
Sedimentation-buffer A: 20 mM HEPES pH 7.5, 75 mM KCl
1.5.2.2. Detailed procedure
Mix the liposomes and protein with a molar ratio of 1000:1 lipid:protein.
Incubate the mixture for 1 hour at 4 °C.
Spin the mixture at 12000 × g for 5 minutes at room temperature to pellet liposomes.
Remove the supernatant and mix 1:1 with 2x Laemmli sample buffer. This is the unbound fraction.
Wash the pellet with the sedimentation-buffer A.
Spin again at 12000 × g for 5 minutes at room temperature. Collect the supernatant and mix 1:1 with 2x Laemmli sample buffer. This is the wash fraction.
Resuspend the pellet in Laemmli sample buffer. This is the bound fraction.
Analyze the amount of protein in each fraction by SDS-PAGE followed by Coomassie blue staining.
For a positive control like EndophilinA1, at a molar ratio of 1000:1 lipid to protein, >90% protein should pellet in the liposome fraction.
1.5.3. Special considerations and common problems
The solution conditions and lipid to protein ratios for the experiments described above may have to be determined empirically for each protein. For example, at a low lipid to protein ratio (40:1 molar ratio, which is approximately 1:1 by mass) and at low ionic strength (100 mM KCl), most of the vesicles in the reaction will be remodeled by EndophilinA1 and will be densely coated with protein (see Figure 5 for example). These dense structures will not “float” in a reverse sedimentation assay. However, using a vast excess of lipid reduces the number of densely decorated tubules and the more sparsely-bound liposomes will remain buoyant.
In general, the sedimentation assay should be avoided for proteins that have a tendency to oligomerize or aggregate under the buffer conditions of the assay. These proteins will pellet on spinning and can give false positive results for liposome binding. For this reason, aggregated or assembled fractions of these proteins should be removed prior to the experiment. This can be done by either pre-clearing the protein with a 20000 × g spin for 20 minutes prior to incubating it with liposomes, or using fresh protein off the size exclusion chromatography column.
The reverse sedimentation assay is a more stringent test for liposome binding proteins. For a protein to show positive binding in this assay, the strength of interaction between the lipid membrane and protein should be high enough to survive a high speed ultracentrifugation spin for 45 minutes and migration through protein-free layers of the gradient. For well characterized lipid binding proteins such as EndophilinA1, we have observed >90% binding at concentrations of ~2 μM. By contrast, proteins with a high rate of dissociation from the lipid are likely to dissociate during the centrifugation step. Such proteins will be observed in the intermediate layers or the bottom layer and this can lead to a false negative result. For this reason, binding conditions such as buffers, pH and salt conditions for optimal binding may have to be determined empirically for these proteins.
1.6. Reconstituting membrane remodeling machines
Once purified protein and lipid substrates have been generated the next step is to reconstitute in-vitro the assembly of these membrane remodeling factors. With these protocols, we describe the 1) membrane free co-polymer formation for Drp1 and MiD49 and 2) membrane bound assembly of Dynamin1 and EndophilinA1.
1.6.1. Co-polymer formation for Drp1 and MiD49
Drp1 localizes to the mitochondrial surface via adaptor proteins. We previously found that Drp1 not only interacts, but also co-assembles with the adaptor protein MiD49 in conditions of low ionic strength and in the presence of a non-hydrolyzable analog of GTP (23). Coassembly was characterized using electron microscopy. The following protocol is generalized for the coassembly of Drp1 with a soluble truncation of MiD49.
1.6.1.1. Reagents, equipment, and buffers
Purified Drp1 and MiD49 protein
Assembly buffer: 20 mM HEPES pH 7.5, 25 mM KCl, 200 mM GMP-PCP, 2 mM MgCl2, and 1 mM DTT
Slide-A-Lyzer mini dialysis units (Thermo Fisher 69570, Waltham MA)
Dilution buffer: 20 mM HEPES pH 7.5, 150 mM KCl
1.6.1.2. Detailed procedures
Thaw protein stocks on ice.
Pre-clear the protein by spinning at 12000 × g to remove any aggregate that may have formed upon thawing.
Measure the concentration of the protein.
In a microcentrifuge tube, add the proteins in 1:1 molar ratio. Dilute proteins as required using the dilution buffer.
Prepare the assembly buffer. Gently place the protein mixture into the dialysis unit and place the dialysis unit into the assembly buffer.
Dialyze overnight at room temperature.
Collect the solution from the dialysis unit with gentle pipetting for further analysis.
1.6.2. Coassembly of Dynamin1 with EndophilinA1 around membrane tubules
We have made recent progress in reconstituting the assembly of a hetero-polymer composed of Dynamin1 and EndophilinA1 that assembles around narrow endocytic tubules but whose function is largely unknown (Figure 5 (17, 24, 40)). To our surprise and by contrast with dynamin alone, this co-complex cannot mediate fission in vitro. Our initial studies hint at the mechanism underlying tubule elongation and fission inhibition: adjacent turns of the Dynamin-1 helix are spaced widely apart in comparison with the Dynamin-1 homo-polymer (Figure 5). This expanded spacing appears to be due to the interleaving EndophilinA1 molecules which prevent adjacent turns of the Dynamin spiral from interacting and thereby block mechano-chemical constriction caused by GTP hydrolysis (Figure 5).
1.6.2.1. Reagents, equipment, and buffers
Purified Dynamin1 and EndophilinA1 protein
DOPS: Cholesterol (9:1 molar ratio) liposomes with 10 freeze/thaw cycles.
GTP (Jena Bioscience, NU-1012, Jena, Germany)
GDP (Jena Bioscience, NU-1172, Jena, Germany)
GMP-PCP (Sigma-Aldrich, M3509, St. Louis, MO)
GTPγS (Jena Bioscience, NU-412, Jena, Germany)
1.6.2.2. Detailed procedures
Thaw the protein and liposome stocks on ice.
Pre-clear protein stocks by spinning at 12000 × g to remove any aggregates.
Add liposomes, Dynamin1 and EndophilinA1 in the molar ratio 2:1:1. In general most reactions are conducted between 1 and 5 uM final protein concentrations in a final volume of 10–50 uL for TEM grid preparation. Remarkably, the Dynamin1 and EndophilinA1 copolymer forms on lipid templates regardless of the order of addition to the reaction.
Add desired Dynamin1 guanine nucleotide derivative (GDP, GTP, GMP-PCP, or GTPγS) to tubulation reaction. Final guanine nucleotide concentration of 0.5 to 5 mM.
Incubate for 20 minutes at room temperature. Longer incubations (>1 hour) at 4 °C will also generate membrane remodeled protein tubules. The tubulation reaction reaches equilibrium after 20 minutes at room temperature.
Collect the membrane-bound Dynamin1 and EndophilinA1 complex for further functional or structural characterizations by gentle pipetting.
1.7. Visualizing membrane remodeling
1.7.1. Preparation of samples for negative stain electron microscopy
The described protocol below is our preferred method for preparing negative stain grids. There are many variations in the preparation of negative stain TEM grids. These variations include the type of stain, timing of each step, addition or deletion of wash steps, and the type of filter paper utilized. Successfully stained TEM grids may be achieved with alternative methodologies (41). Figures 4A–B and 5A℃C are examples of negative stain grids prepared according to this procedure.
1.7.1.1. Reagents, equipment, and buffers
Membrane bound protein sample
Forceps
Formvar carbon film on 200 mesh copper grids (Electron Microscopy Sciences, FCF-200-Cu, Hatfield, PA)
2% w/v uranyl acetate (Structure Probe Inc., 02624-AB, West Chester, PA)
Pelco easiGlow glow discharge cleaning system (Ted Pella, 91000, Redding, CA)
#1 Whatman filter paper (GE, 1001–055, Little Chalfont, UK)
Storage grid box
Glass slide
Parafilm
Sample wash buffer
1.7.1.2. Detailed procedure
Prepare glow discharged carbon coated grids.
Using forceps, place the carbon coated grids onto a glass slide with their carbon side up.
Set glow discharge settings to the following conditions: Negative polarity, 30 seconds hold time, 30 seconds glow time, 15 mA current, and 0.39 mBar vacuum pressure.
Capture a grid with reverse force forceps by gripping the outermost edge of the grid.
Prepare sample washes and stain by pipetting 5–40 μl droplets onto the wax surface of a piece of parafilm. Two sample wash droplets and two 2% uranyl acetate droplets should be prepared. Sample wash buffer can be deionized water but we prefer to use the sample buffer.
Apply 2.5–5.0 μl of the membrane bound protein sample onto the carbon side of the grid and let it absorb for 30–60 seconds.
Wick away all moisture by blotting the edge of the grid against the surface of a piece of filter paper at a 90 degree angle.
Immediately immerse the carbon side of the grid onto the surface of the first wash droplet for 5 seconds while maintaining a hold of the grid with the forceps.
Move to the second wash droplet and hold for 5 seconds.
Move to the first stain droplet and hold for 5 seconds.
Wick away all excess stain by blotting the edge of the grid against the surface of a piece of filter paper at a 90 degree angle.
Immerse the carbon side of the grid onto the surface of the second stain droplet and hold for 30 seconds.
Wick away all excess stain by blotting the edge of the grid against the surface of a piece of filter paper at a 90 degree angle.
Allow the grid to air dry for 2 minutes and then place in a grid storage holder. The stained grid will usually remain stable for months in low humidity environments. To ensure stability of the negative stain grids use a vacuum desiccation chamber to store the grids.
1.7.2. Vitrification of samples for electron cryomicroscopy
1.7.2.1. Reagents, equipment, and buffers
Co-polymer protein assembly
Liquid Nitrogen
Compressed ethane gas
FEI Vitrobot Mark III (FEI, Hillsboro, OR)
Pelco easiGlow glow discharge cleaning system (Ted Pella, 91000, Redding, CA) or equivalent
Vitrobot pre-punched filter paper (Ted Pella, 47000–100, Redding, CA)
Tweezer assembly for Vitrobot Mark III (Ted Pella, 47000–500, Redding, CA)
Cryo storage grid box (Ted Pella, 160–40, Redding, CA)
Quantifoil R2/2 holey carbon on 200 mesh copper grids (Structure Probe Inc., 4420C-XA, West Chester, PA) or equivalent
Liquid nitrogen storage dewar
Forceps
Screwdriver
1.7.2.2. Detailed Procedure
The verification protocol described below is the generic protocol that should be utilized when vitrifying your sample for the first time. There is a large amount of variation in generating thin (100–50 um) vitreous ice. Check with your electron microscopy facility to determine successful blotting settings used for the particular vitrobot that will be used to generate your samples. Figure 4C and Figure 5D–F were generated using these detailed procedures.
Using forceps, place the grids to be vitrified onto a glass slide with their carbon side up.
Set glow discharge settings to the following conditions: Negative polarity, 30 seconds hold time, 30 seconds glow time, 15 mA current, and 0.39 mBar vacuum pressure. There is a wide host of grid treatments that can be utilized to optimize protein adsorption onto the cryo-EM grid.
Prepare the vitrobot apparatus by turning on the power to the machine, opening the FEI software on the computer, and ensuring that air is being supplied to the vitrobot at >90 psi.
Chill the blotting chamber to the desired temperature (4–25 °C) using the temperature settings on the FEI software.
Fill the water cylinder with deionized water and set the humidity to 100% on the FEI software.
Load pre-punched filter paper onto the blotting pads in the blotting chamber.
Chill the vitrobot cup and cryo storage box with liquid nitrogen.
Condense liquid ethane in the presence of liquid nitrogen.
Load a Quantifoil R2/2 holey carbon on 200 mesh grid onto the forceps by gripping the outermost edge of the grid ensuring that it is well centered. For protein samples that assemble into helical or filamentous polymers we prefer to utilize 2 μm holes with 2 μm spacing. This allows for longer filaments to sit across large areas of the vitreous ice contained within the holes. This is done so that straight segments of the polymers can be imaged for data collection.
Lock the forceps together with the forceps clamp.
Enter the desired vitrification settings into the software. Blot time: 4 seconds, blot offset: 0 mm, wait time: 30 seconds. These settings will need to be optimized upon visualization of the grid to obtain thin vitreous ice. Thick impenetrable ice over the majority of the grid indicates that the sample was under blotted. For this case, increase the blot time or decreasing the blot offset to achieve thinner ice. If the cryo-EM grid upon visualization lacks ice in the holes it indicates that the sample was over blotted. Decrease the blot time or increase the blot offset to amend this issue.
Load 2.5 – 5 μl of your sample onto the grid ensuring that the droplet is completely covering the grid surface. Go through the vitrification process that was set on the software.
Transfer the vitrified cryo-EM grid into the cryo storage box and secure the lid with a liquid nitrogen chilled screwdriver.
Store grids in a liquid nitrogen dewar. Vitrified grids must be stored at liquid nitrogen temperatures (−195 °C) at all times.
1.7.3. Low dose imaging
Imaging cryo-EM samples requires great care to prevent radiation damage to your biological sample. This protocol will go through the required microscope settings to image a vitrified protein sample and minimize exposure to a dose of ~ 20 electrons/Å2. For sample grid transfer and loading the holder in the microscope refer to the following cryo-EM resources (42, 43).
1.7.3.1. Reagents, equipment, and buffers
Liquid Nitrogen
Vitrified protein sample on holey carbon cryo-EM grid
Transmission electron microscope (FEI, Tecnai Polara, Hillsboro, OR) or equivalent
Cryo-transfer holder (Gatan, 626, Pleasanton, CA)
1.7.3.2. Detailed procedure
This procedure begins after you have transferred your cryo-EM grid from storage into a cryo-EM grid holder and have loaded the holder into the microscope.
Open the cryo holder shutter and the microscope column valves.
Activate low dose and select search mode.
Switch to a high spot size (6–10) at ~5000 x magnification and lower the beam intensity.
Set the eucentric height by activating the alpha-wobbler and adjust the z-height buttons to minimize image movement while looking at the phosphor screen.
Move the stage with the right track ball to an unwanted area of the grid with ripped or empty carbon.
Select the focus mode and switch to a spot size of 2 at 100 kx magnification or a magnification greater than your setting in the exposure mode. Switch the focus 2 settings to 0 μm offset at 0 degrees and set the focus 1 setting to 1.5 μm offset at 180 degrees. These two settings allow you to have one focus spot directly centered over the exposure spot and the second focus spot a specified distance away from the image collection area to limit beam exposure to your sample. The focus 2 setting then will primarily be utilized to verify your image shift settings as in detailed procedure step 1.7.3.2.9.
Ensure that the beam is properly aligned by performing all the direct alignments.
Select the exposure mode and switch to a beam spot size of 2 at the magnification at which you would like to collect your data.
Switch through all the low dose modes and ensure that the beam is properly aligned and centered.
Check the image alignment between search and exposure modes. To accomplish this, find a discernible feature on the carbon surface in exposure mode and center your field of view over this feature. Switch to search mode and center your field of view over the same identifiable feature using image shift. It is best to use a feature on the surface with high contrast that is unique in the area of the grid in which you are imaging.
In search mode, find an area of the grid in which there is thin vitreous ice. Move the center of view to the middle of a hole.
Set your beam intensity so that you are detecting 20 electrons/physical pixel/second over your entire exposure time. For example if you collect your images with a 5 seconds exposure you will want 4.0 electron/physical pixel to hit one pixel of the detector every second.
Switch to search mode and center over a hole with thin vitreous ice that contains your protein sample.
Switch to focus mode and find eucentric focus using the FFT in digital micrograph by adjusting the z-height. Once focus is found reset the defocus on the microscope.
Set defocus to 0.5 – 4 μm under focus using the focus knob.
Switch to exposure mode and acquire the image.
Repeat steps 1.7.3.2.12 to 1.7.3.2.15 until no unexposed holes are left. Ensure that the imaging field of view has not been exposed in focus or exposure mode previously when moving to a new area to acquire an image. This is a crucial point that needs to be employed to prevent imaging radiation damaged protein sample.
Move to a new grid square when all the holes have been imaged and exposed to the beam. Continue in this manner until you have acquired the desired number of micrographs of your sample.
Perspective
This chapter has focused on biochemical methods for purifying and reconstituting membrane-remodeling factors for visualization by electron microscopy, rather than on subsequent image analysis and structure determination methods. In many ways, the rate-determining step in studying membrane remodeling phenomena is the biochemical reconstitution of the activity being investigated. Once purification and reconstitution are accomplished, modern electron microscopy and image analysis will almost certainly lead to new molecular and even atomic-resolution insights. The future of this approach is particularly bright. Beam induced motion was discovered to be the most significant factor limiting the resolution of cryo-EM images and 3D reconstructions, and the invention of direct electron detectors with individual electron detection and sub-second image acquisition has led to innovative software solutions for quantifying and correcting beam-induced motion and for restoring high resolution information (44–46). The use of direct electron detectors and modern image analysis algorithms is already revealing the mechanisms of protein-protein and protein-membrane interactions that underlie membrane remodeling phenomena.
Acknowledgements
Electron microscopy was performed at the University of Utah and the University of California. We thank David Belnap (Utah) and Michael Braunfeld (UCSF) for supervision of the electron microscopes. We thank Anita Orendt and the Utah Center for High Performance Computing and the NSF XSEDE consortium for computational support. We thank Aurelien Roux (University of Geneva), Pietro De Camilli (Yale University), and Janet Shaw (University of Utah) for sharing reagents and for critical discussions. The protocols described here were developed in part with support from the Searle Scholars Program, NIH/NIGMS grant 1DP2GM110772-01, the United States – Israel Binational Science Foundation BSF2013310, and the Herbert Boyer Junior Faculty Endowed Chair at UCSF. The authors declare no competing financial interests.
References
- 1.Hooke R, Micrographia: or some physiological descriptions of minute bodies made by magnifying glasses with observations and inquiries thereupon (1665; http://archive.nlm.nih.gov/proj/ttp/flash/hooke/hooke.html).
- 2.Frost A, Unger VM, De Camilli P, The BAR domain superfamily: membrane-molding macromolecules. Cell. 137, 191–196 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McMahon HT, Kozlov MM, Martens S, Membrane Curvature in Synaptic Vesicle Fusion and Beyond. Cell. 140 (2010), pp. 601–605. [DOI] [PubMed] [Google Scholar]
- 4.Kozlov MM, McMahon HT, Chernomordik LV, Protein-driven membrane stresses in fusion and fission. Trends Biochem. Sci 35 (2010), pp. 699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McMahon HT, Gallop JL, Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature. 438, 590–596 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Frost A, Unger VM, De Camilli P, Boomerangs, Bananas and Blimps: Structure and Function of F-{BAR} Domains in the Context of the {BAR} Domain Superfamily. Landes (2009). [Google Scholar]
- 7.Cossart P, Roy CR, Manipulation of host membrane machinery by bacterial pathogens. Curr. Opin. Cell Biol 22 (2010), pp. 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laliberté J-F, Sanfaçon H, Cellular remodeling during plant virus infection. Annu. Rev. Phytopathol 48, 69–91 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Moyer CL, Nemerow GR, Viral weapons of membrane destruction: Variable modes of membrane penetration by non-enveloped viruses. Curr. Opin. Virol 1, 44–49 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chappie JS et al. , A pseudoatomic model of the dynamin polymer identifies a hydrolysis-dependent powerstroke. Cell. 147, 209–222 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mim C et al. , Structural Basis of Membrane Bending by the N-BAR Protein Endophilin. Cell. 149, 137–145 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frost A et al. , Structural basis of membrane invagination by F-BAR domains. Cell. 132, 807–817 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Low HH, Sachse C, Amos LA, Löwe J, Structure of a Bacterial Dynamin-like Protein Lipid Tube Provides a Mechanism For Assembly and Membrane Curving. Cell. 139, 1342–1352 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doherty GJ, McMahon HT, Mechanisms of endocytosis. Annu. Rev. Biochem 78, 857–902 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Ferguson SM, De Camilli P, Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol (2012), , doi: 10.1038/nrm3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morlot S et al. , Membrane shape at the edge of the dynamin helix sets location and duration of the fission reaction. Cell. 151, 619–29 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meinecke M et al. , Cooperative recruitment of Dynamin and BAR domain-containing proteins leads to GTP-dependent membrane scission. J. Biol. Chem, M112.444869- (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daumke O, Roux A, Haucke V, BAR domain scaffolds in dynamin-mediated membrane fission. Cell. 156 (2014), pp. 882–892. [DOI] [PubMed] [Google Scholar]
- 19.Osteryoung KW, Nunnari J, The division of endosymbiotic organelles. Science. 302, 1698–1704 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Rahaman A, Elde NC, Turkewitz AP, A Dynamin-Related Protein Required for Nuclear Remodeling in Tetrahymena. Curr. Biol 18, 1227–1233 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elde NC, Morgan G, Winey M, Sperling L, Turkewitz AP, Elucidation of clathrin-mediated endocytosis in tetrahymena reveals an evolutionarily convergent recruitment of dynamin. PLoS Genet. 1 (2005), doi: 10.1371/journal.pgen.0010052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lackner LL, Nunnari JM, The molecular mechanism and cellular functions of mitochondrial division. Biochim. Biophys. Acta - Mol. Basis Dis 1792 (2009), pp. 1138–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koirala S et al. , Interchangeable adaptors regulate mitochondrial dynamin assembly for membrane scission. Proc. Natl. Acad. Sci. U. S. A 110, 1300855110- (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boucrot E et al. , Endophilin marks and controls a clathrin-independent endocytic pathway. Nature. 517, 460–465 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Schuske KR et al. , Endophilin is required for synaptic vesicle endocytosis by localizing synaptojanin. Neuron. 40, 749–762 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Milosevic I et al. , Recruitment of endophilin to clathrin-coated pit necks is required for efficient vesicle uncoating after fission. Neuron. 72, 587–601 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verstreken P et al. , Endophilin mutations block clathrin-mediated endocytosis but not neurotransmitter release. Cell. 109, 101–112 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Szwedziak P, Wang Q, Bharat TAM, Tsim M, Löwe J, Architecture of the ring formed by the tubulin homologue FtsZ in bacterial cell division. Elife. 3, e04601 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henderson R, Structural biology: Ion channel seen by electron microscopy. Nature. 504, 93–4 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Kim J et al. , Subnanometre-resolution electron cryomicroscopy structure of a heterodimeric ABC exporter. Nature. 517, 396–400 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liao M, Cao E, Julius D, Cheng Y, Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 504, 107–12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu P et al. , Three-dimensional structure of human γ-secretase. Nature (2014), doi: 10.1038/nature13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richter V et al. , Structural and functional analysis of MiD51, a dynamin receptor required for mitochondrial fission. J. Cell Biol 204, jcb.201311014- (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Losón OC et al. , The Mitochondrial Fission Receptor MiD51 Requires ADP as a Cofactor. Structure. 22, 367–77 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fröhlich C et al. , Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J. 32, 1280–1292 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Owen DJ et al. , Crystal structure of the amphiphysin-2 SH3 domain and its role in the prevention of dynamin ring formation. EMBO J. 17, 5273–5285 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Warnock DE, Terlecky LJ, Schmid SL, Dynamin GTPase is stimulated by crosslinking through the C-terminal proline-rich domain. EMBO J. 14, 1322–1328 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Meer G, Voelker DR, Feigenson GW, Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol 9, 112–124 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Folch J, Foch lipid extraction. 55, 999–1033 (1987). [Google Scholar]
- 40.Farsad K et al. , Generation of high curvature membranes mediated by direct endophilin bilayer interactions. J. Cell Biol 155, 193–200 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Booth DS, Avila-Sakar A, Cheng Y, Visualizing Proteins and Macromolecular Complexes by Negative Stain EM: from Grid Preparation to Image Acquisition. J. Vis. Exp (2011), , doi: 10.3791/3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cabra V, Samsó M, Do’s and Don’ts of Cryo-electron Microscopy: A Primer on Sample Preparation and High Quality Data Collection for Macromolecular 3D Reconstruction. J. Vis. Exp, 1–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grassucci RA, Taylor DJ, Frank J, Preparation of macromolecular complexes for cryo-electron microscopy. Nat. Protoc 2, 3239–46 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li X et al. , Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–90 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bai XC, Fernandez IS, McMullan G, Scheres SHW, Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles. Elife. 2013, e00461 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campbell MG et al. , Movies of ice-embedded particles enhance resolution in electron cryo-microscopy. Structure. 20, 1823–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faelber K et al. , Crystal structure of nucleotide-free dynamin. Nature. 477 (2011), pp. 556–560. [DOI] [PubMed] [Google Scholar]
- 48.Ford MGJ, Jenni S, Nunnari J, The crystal structure of dynamin. Nature. 477, 561–6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Masuda M et al. , Endophilin BAR domain drives membrane curvature by two newly identified structure-based mechanisms. EMBO J. 25, 2889–97 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gallop JL et al. , Mechanism of endophilin N-BAR domain-mediated membrane curvature. EMBO J. 25, 2898–910 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]