

SUMMARY
Type 1 interferon (T1IFN) signaling promotes inflammation and lupus pathology, but its role in autoreactive B cell development in the antibody-forming cell (AFC) and germinal center (GC) pathways is unclear. Using a lupus model that allows for focused study of the AFC and GC responses, we show that T1IFN signaling is crucial for autoreactive B cell development in the AFC and GC pathways. Through bone marrow chimeras, DNA-reactive B cell transfer, and GC-specific Cre mice, we confirm that IFNαR signaling in B cells promotes autoreactive B cell development into both pathways. Transcriptomic analysis reveals gene expression alterations in multiple signaling pathways in non-GC and GC B cells in the absence of IFNαR. Finally, we find that T1IFN signaling promotes autoreactive B cell development in the AFC and GC pathways by regulating BCR signaling. These data suggest value for anti-IFNαR therapy in individuals with elevated T1IFN activity before clinical disease onset.
In Brief
The B-cell-intrinsic mechanisms of type 1 interferon (T1IFN) signaling in regulating B cell tolerance is unclear. Domeier et al. show that T1IFN signaling in B cells causes loss of B cell tolerance, promoting autoreactive B cell development into the antibody-forming cell and germinal center pathways by regulating BCR signaling.
Graphical Abstract
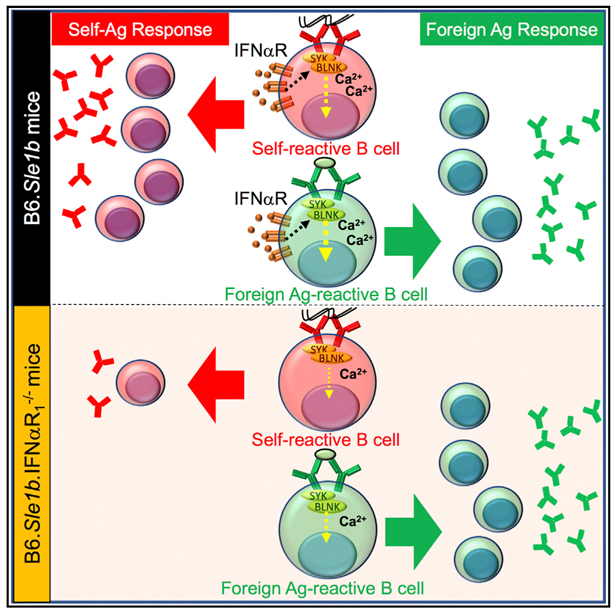
INTRODUCTION
Systemic lupus erythematosus (SLE) is a multifactorial autoimmune disease characterized by the production of DNA- and RNA-based autoantibodies (autoAbs). Loss of self-tolerance initiates SLE, but further amplification of autoimmune responses is required for the progression to end-stage organ pathology, which includes kidney glomerulonephritis (Nguyen et al., 2002). The autoimmune responses and pathology in SLE-prone mice and SLE patients are accelerated by pro-inflammatory cytokines, including interferons (Agrawal et al., 2009; Bennett et al., 2003; Crow, 2014; Liu and Davidson, 2013; Lourenço and La Cava, 2009; Mathian et al., 2005; Nickerson et al., 2013; Santiago-Raber et al., 2003). Several type 1 interferon (T1IFN)- targeted therapies (sifalimumab, rontalizumab, and anifrolumab) have been proposed for SLE but without an understanding of mechanisms by which T1IFN signaling may promote SLE development. The contribution of T1IFN signaling in the initial loss of tolerance is not clear. In SLE patients, an elevated T1IFN activity is detected before disease onset and continues to rise with disease progression (Bennett et al., 2003; Munroe et al., 2016); thus, T1IFN is likely involved both in initial loss of tolerance and subsequent disease progression.
Multiple tolerance checkpoints during B cell development in the bone marrow and in the periphery maintain B cell tolerance. The extra-follicular antibody-forming cell (AFC) and follicular germinal center (GC) pathways are two major peripheral B cell tolerance checkpoints (Cappione et al., 2005; Vinuesa et al., 2009; William et al., 2002). Unlike microbial antigen-induced AFC and GC responses that generate anti-microbial antibodies (Abs), in autoimmunity, AFCs and GCs spontaneously develop in the absence of detectable microbial infections or purposeful immunization (called spontaneous AFCs, Spt-AFCs, and spontaneous GCs [Spt-GCs]) and produce autoantibodies (autoAbs) (Cappione et al., 2005; Domeier et al., 2017; Luzina et al., 2001; Tiller et al., 2010; Vinuesa et al., 2009; Woods et al., 2015). Autoimmune-prone mice and humans exhibit elevated Spt-AFC and Spt-GC responses (Domeier et al., 2017; Luzina et al., 2001; Tiller et al., 2010; Wong et al., 2015; Woods et al., 2015; Rahman et al., 2007; Fukuyama et al., 2005) that correlate well with the increased autoAb production and numbers of autoAb-producing AFCs.
Spt-AFCs and Spt-GCs are known to play a role in autoimmunity (Domeier et al., 2017; Luzina et al., 2001; Rahman et al., 2007; Fukuyama et al., 2005), but the mechanisms that drive autoreactive B cell development in the AFC and GC pathways are poorly understood. Although previous studies revealed elevated short-lived AFC and GC B cell responses in pre-autoimmune lupus-prone mice upon IFNα treatment (Mathian et al., 2011, Liu et al., 2011), the mechanisms by which T1IFN signaling alters B cell tolerance and promotes autoreactive B cell development through the AFC and GC pathways are not defined. To study the role of T1IFN signaling in loss of B cell tolerance and autoreactive B cell development in the AFC and GC pathways, we used the B6.Sle1b model, which harbors the SLAM locus from the lupus-prone NZM2410 strain (Kumar et al., 2006; Wandstrat et al., 2004; Wong et al., 2012,2015). Mutations in the SLAM family genes within the Sle1b sublocus promote elevated Spt-AFC, Spt-GC, and autoAb responses (Wong et al., 2012, 2015). However, B6.Sle1b mice do not develop a phenotype of chronic SLE without other immune-activating loci (i.e., Sle2, Sle3, yaa, and lpr), making this model particularly well suited for studying loss of B cell tolerance and autoreactive B cell development in the AFC and GC pathways without the confounding effects of systemic inflammation (Morel et al., 2000; Nguyen et al., 2002).
Here, we report that T1IFN signaling via IFNαR plays an important role in elevated Spt-AFC, Spt-GC, and autoAb responses in B6.Sle1b mice. Using B-cell-specific bone marrow chimeras and a transgenic DNA-reactive HKIR B cell transfer system, we confirm that B-cell-intrinsic IFNαR signaling is required for altered B cell selection and autoreactive B cell development in the AFC and GC pathways. Transcriptional data indicate that T1IFN signaling in non-GC and GC B cells promotes autoreactive B cell development at the AFC and GC stages and does so by regulating genes in several crucial signaling pathways, such as cytokine signaling, B cell receptor (BCR) signaling, and transcriptional regulation (including nuclear factor κB [NF-κB] signaling). Our data further confirm that IFNαR signaling regulates BCR signaling at the naive and GC B cell stages. Additionally, using a Cre-lox-based approach, we showed that IFNαR signaling in B cells at the GC stage is required for elevated Spt-GC responses and the production of IgG2c autoAbs in B6.Sle1b mice. Collectively, these results indicate that IFNαR signaling in B cells maintains BCR signaling beyond the threshold required for effective tolerance, causing loss of B cell tolerance and driving autoreactive B cell development into the AFC and GC pathways in autoimmune-prone B6.Sle1b mice.
RESULTS
Type 1 Interferon Signaling Is Essential for Elevated Spt-AFC and Spt-GC Responses in B6.Sle1b Mice
Given the increase in T1IFN expression prior to the onset of SLE (Munroe et al., 2016), we asked whether T1IFN signaling contributed to increased autoAb-producing Spt-AFC and Spt-GC responses in B6.Sle1b mice (Wong et al., 2012, 2015). We first measured surface expression of IFNαR on B and T cells. Whereas IFNαR expression was higher on B220+FashiGL-7hi GC B cells than B220+FaslowGL-7low non-GC B cells, we found reduced expression of IFNαR on CD4+PD-1hiCXCR5hi follicular helper T (Tfh) cells compared to CD4+PD-1lowCXCR5low naive T cells and GC B cells in 3-month-old female B6 and B6.Sle1b mice (Figure 1A). Using 3- and 6-month-old B6.Sle1b female mice deficient in IFNαR1 (named Sle1b.IFNαR1−/−), we found that Sle1b.IFNαR1−/− mice had reduced percentages of B220+PNAhiFashi GC B cells and CD4+PD1hiCXCR5hi Tfh cells than B6.Sle1b mice (Figures 1B and 1C); in contrast, no differences were found between B6.IFNαR1−/− and B6 mice. Histological analysis of IgDnegGL7+ GC structures confirmed that Sle1b.IFNαR1−/− mice had smaller Spt-GCs at both 3- and 6-month time points than B6.Sle1b mice (Figure 1D). Additionally, Sle1b.IFNαR1−/− mice exhibited a reduced number of splenic immunoglobulin G (IgG)+ AFCs than B6.Sle1b control mice, but the number of IgM+ AFCs was not significantly different among the strains (Figures 1E and 1F). We also observed decreased total serum IgG titers in Sle1b.IFNαR1−/− mice (Figure 1H), including a reduction in IgG2c (Figure 1J) compared to B6.Sle1b mice. No significant differences in IgM (Figure 1G) and IgG1 (Figure 1I) serum titers were observed between B6.Sle1b and Sle1b.IFNαR1−/− mice. These reduced responses in B6.Sle1b mice in the absence of IFNαR signaling were not due to a major defect in primary B cell development in the bone marrow and in the periphery (Figure S1). These data indicate that IFNαR signaling in autoimmune-prone B6.Sle1b mice is required for the development of elevated Spt-GC response, increased numbers of IgG+ Spt-AFCs, and increased production of IgG and IgG2c antibodies in vivo.
Figure 1.

IFNαR1 Is Required for Dysregulated Spt-AFC and Spt-GC Formation in B6.Sle1b Mice (A) Flow cytometric analysis of surface expression of IFNαR1 on B220+FaslowGL-7low non-GC B cells (red), B220+FashGL-7hi GC B cells (blue), CD4+PD-1low CXCR5low non-Tfh (orange), and CD4+PD-1hiCXCR5hi Tfh (green) in female B6 and B6.Sle1b mice. Error bars represent mean ± SEM.
(B and C) The percentages of GC B cells (B) and Tfh cells (C) in splenocytes from 3- and 6-month-old female mice.
(D) Spleen sections from 6-month-old B6, B6.IFNαR1−/−, B6.Sle1b, and B6.Sle1b.IFNαR1−/− female mice (5 mice per group) were stained with anti-CD4 Ab (red) GL-7 (green) and anti-IgD Ab (blue), and GC areas were measured for 10 GCs per spleen section (right panels). The scale bars represent 50 μm.
(E and F) Each symbol represents a measured GC. IgM+ (E) and IgG+ (F) AFCs in the spleens of 6-month-old B6, B6.IFNαR1−/−, B6.Sle1b, and B6.Sle1b.IFNαR1−/− female mice are shown.
(G-J) Analysis of serum titers of IgM (G), IgG (H), IgG1 (I), and IgG2c (J) antibodies in 6-month-old mice by ELISA.
Each symbol represents a mouse, and these data represent at least two-three independent experiments. Statistical significance was determined by one-way ANOVA with a follow-up Tukey multiple-comparison test (NS, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
We next asked whether decreased Spt-GC and Spt-AFC responses in Sle1b.IFNαR1−/− mice caused a reduction in anti-nuclear antibody (ANA) reactivity. We observed a significant decrease in the number of double-stranded DNA (dsDNA), histone, and nucleosome-specific AFCs (Figures 2A–2D) in the spleens and reduced serum autoAbs to these self-antigens in Sle1b.IFNαR1−/− mice compared to B6.Sle1b mice (Figures 2E–2G). Together, these data show that IFNαR signaling is required for the regulation of Spt-GC and Spt-AFC responses, leading to autoAb production in lupus-prone B6.Sle1b mice.
Figure 2.

IFNαR1 Is Required for ANA Production in B6.Sle1b Mice
(A) ANA detection from sera of 6-month-old B6, B6.IFNαR1−/−, B6.Sle1b, and B6.Sle1b.IFNαR1−/− female mice by fluorescent Hep-2 assay. Representative images are shown with pie charts that quantitate the distribution of the serum samples as negative staining (non-Hep-2 reactive), reactive to cytoplasmic antigens (cytoplasmic), reactive to nuclear and cytoplasmic antigens (nuclear and cytoplasmic), and reactive to nuclear and/or nucleolar antigens (nucleolar/nuclear). The scale bars represent 75 μm.
(B-D) Quantification of dsDNA-specific (B), histone-specific (C), and nucleosome-specific (D) AFCs in total splenocytes from 6-month-old mice of the indicated genotypes.
(E-G) Analysis of serum titers of dsDNA-reactive (E), histone-reactive (F), and nucleosome-reactive (G) ANAs in the sera of 6-month-old female mice of the indicated genotypes (key at bottom of figure) by ELISA.
These data are representative of two independent experiments, and each symbol represents a mouse. Statistical significance was determined by one-way ANOVA with a follow-up Tukey multiple-comparison test (**p < 0.01, ***p < 0.001, ****p < 0.0001).
Previous studies in the BXD2 model of SLE demonstrated that T1IFN increases follicular entry of marginal zone B cells (MZBs) carrying apoptotic cell-derived self-antigens and causes loss of marginal zone macrophages (MZMs) (Li et al., 2015; Wang et al., 2010). Subsequently, T1IFN was shown to promote elevated Spt-GC responses in BXD2 mice (Li et al., 2015; Wang et al., 2010). Thus, we asked whether MZM and MZB populations were altered in B6.Sle1b and Sle1b.IFNαR1−/− mice. We found no significant differences in MARCO+SIGNR1+MZM (Figure S2A) and B220+CD93−IgMhiCD23− (Figure S2B) and IgMhiIgD− (Figure S2C) MZB populations. These results indicate that elevated Spt-GC and Spt-AFC responses in B6.Sle1b mice were not due to altered regulation of the MZ compartment.
IFNαR Signaling Does Not Overtly Affect Induced-GC and Ab Responses or B Cell Affinity Maturation to T-Dependent Antigen Immunization
We next asked whether IFNαR signaling contributes to overall induced-GC responses and the quality of Ab responses generated against T-dependent antigens. We observed no significant differences in the percentage of total GC B cells at 14 days post-immunization with 4-hydroxy-3-nitrophenylacetyl-conjugated chicken γ-globulin (NP-CGG) (Figures S3A and S3B). Although not statistically significant, five out of six Sle1b.IFNαR1−/− mice had a higher percentage of NP+ GC B cells than B6.Sle1b mice (Figure S3C). The percentage of NPneg GC B cells in Sle1b.IFNαR1−/− mice was lower than in B6.Sle1b control mice (Figure S3D), consistent with reduced Spt-GC responses in Sle1b.IFNαR1−/− mice. Immunohistology and the quantitative analysis of the GC size of these two strains were comparable (Figure S3E).
Whereas we found reduced serum titers of high-affinity NP4-specific IgG (Figure S3F) in Sle1b.IFNαR1−/− mice as compared to B6.Sle1b control mice, serum titers of low-affinity NP29-specific IgG did not differ between Sle1b.IFNαR1−/− and B6.Sle1b mice at 14-day time point (Figure S3G). Next, we evaluated NP-specific serum IgG titers in B6.Sle1b and Sle1b.IFNαR1−/− mice 30 days post-immunization to determine whether difference in NP4 IgG titers between B6.Sle1b and Sle1b.IFNαR1−/− mice continued over time. Both high-affinity NP4- and low-affinity NP29-specific serum IgG titers did not differ between Sle1b.IFNαR1−/− and B6.Sle1b mice at this time point (Figures S3H and S3I). We also found no difference in high- and low-affinity NP-specific IgG Ab responses at 14-day (not shown) and 30-day time points (Figures S3J and S3K) between the B6 and B6.IFNαR1−/− strains. Collectively, these data suggest that T1IFN signaling does not play a major role in the GC and Ab responses or B cell affinity maturation to T-dependent foreign antigen in both autoimmune and non-autoimmune mice.
IFNαR Signaling in B Cells Is Required for AutoAb-Producing AFC Development and AutoAb Production in B6.Sle1b Mice
To determine the B-cell-intrinsic requirement of IFNαR signaling in Spt-GC, Spt-AFC, and autoAb responses in B6.Sle1b mice, we generated mixed bone marrow (BM) chimeras by reconstituting lethally irradiated mu mutant (uMT) mice, which lack mature B cells, with a mixture of BM (80% uMT and 20% derived from either B6.Sle1b or Sle1b.IFNαR1 BM). The expression of IFNαR1 on B cells following reconstitution was confirmed by flow cytometry (Figure 3A). We found significantly reduced numbers of ANA-reactive AFCs in the spleen (Figures 3D and 3E), reduced titers of total IgG2c subtype antibodies (Figure 3H), and reduced serum titers of IgG2c ANAs (Figures 3I and 3J) in mice that were reconstituted with Sle1b.IFNαR1−/− BM compared to B6.Sle1b control BM. However, mice that were reconstituted with Sle1b.IFNαR1−/− BM did not exhibit significant changes in the percentages of GC B cells (Figure 3B) or Tfh (Figure 3C) or in total IgM+ or IgG+ AFCs (data not shown). Interestingly, a significantly reduced number of dsDNA- and nucleosome-specific AFCs was seen in the BM of recipient mice that were reconstituted with Sle1b.IFNαR1−/− BM than in B6.Sle1b control BM (Figures 3F and 3G). These data indicate that IFNαR signaling in B cells is required for autoAb-producing splenic and BM AFC responses and autoAb production, suggesting the cell-intrinsic role of IFNαR signaling in autoreactive B cell development into autoAb-producing AFCs, potentially via the extra-follicular AFC and the GC-AFC pathways.
Figure 3.

B-Cell-Intrinsic IFNαR1 Is Required for ANA-Producing AFC Responses in B6.Sle1b Mice
(A) Flow cytometric analysis of surface expression of IFNαR1 on B220+ B cells in B6.Sle1b and B6.Sle1b.IFNαR1−/− chimeric mice 3 months after BM cell transfer. (B and C) The percentages of B220+GL-7hiFashi GC B cells (B) and CD4+CXCR5hiPD-1hi Tfh cells (C) in total splenocytes of the chimeras.
(D and E) Numbers of dsDNA-specific (D) and nucleosome-specific (E) splenic AFCs in chimeric mice described in (A)–(C).
(F and G) Numbers of dsDNA-specific (F) and nucleosome-specific (G) long-lived bone marrow AFCs in chimeric mice described in (A)–(C).
(H) Analysis of serum titers of total IgG2c antibodies in these mice.
(I and J) Analysis of dsDNA-reactive (I) and nucleosome-reactive IgG2c (J) in the sera of these mice.
These data represent one experiment of four or five mice of each genotype. Statistical significance was determined using an unpaired, nonparametric Mann-Whitney Student’s t test (NS, not significant, *p < 0.05, **p < 0.01, ***p < 0.001).
IFNαR Signaling Promotes the Accumulation of DNA-Reactive B Cells in the GC Compartment of B6.Sle1b Mice
Using a p-azophenyl arsonate (Ars) and DNA dual-reactive heavy chain knockin (named HKIR) B cell transfer system (Alabyev et al., 2007; Rahman et al., 2007), we previously showed that HKIR B cells with DNA reactivity were negatively regulated by the GC tolerance mechanism, but expression of the Sle1b sublocus in HKIR B cells (named HKIR.Sle1b) rendered these cells capable of accumulating in the GCs (Wong et al., 2012). To determine whether B-cell-intrinsic IFNαR signaling is important for autoreactive B cell accumulation in the GC compartment of B6.Sle1b mice, we transferred B6.Sle1b. HKIR and Sle1b.IFNαR1−/−.HKIR B cells into syngeneic B6.SJL (CD45.1+) recipients 7 days post-immunization with Ars-KLH. Recipient mice were boosted with a second dose of Ars-KLH at the time of B cell transfer and their spleens harvested 5 days after transfer to quantitate HKIR B cells (detected by E4 antibody) within the GC (Figure 4A). We observed a reduced percentage of B220+GL7hiCD95hiE4+ HKIR GC B cells in mice that received Sle1b.IFNαR1−/−.HKIR B cells compared to mice that received B6.Sle1b.HKIR B cells (Figure 4C). However, the overall percentage of B220+GL7hiCD95hi total GC B cells between the two groups did not differ significantly, confirming a consistent total GC response to immunization (Figure 4B). Histological analysis for HKIR E4+ cells within IgDnegGL7+ GCs (Figure 4D), quantitative analysis for E4+ cells/GC area (Figure 4E), and E4 fluorescence intensity in GCs (Figure 4F) confirmed that the recipients of Sle1b.IFNαR1−/−.HKIR cells had quantitatively reduced HKIR E4+ GC responses compared to mice that received B6.Sle1b.HKIR B cells, although overall GC areas did not differ between the two groups (Figure 4G).
Figure 4.

B-Cell-Intrinsic IFNαR1 Maintains Self-Reactive B Cells in the GC
(A) Schematic of the immunization and transfer of HKIR CD45.2+ B cells to 8- to 10-week congenic B6.SJL (CD45.1+) female recipients. Donor CD45.2+ HKIR B cells were from 8- to 10-week-old B6.Sle1b.HKIR (Sle1b.HKIR) and B6.Sle1b.IFNαR1−/−.HKIR (Sle1b.IFNαR1−/−.HKIR) female mice.
(B and C) Flow cytometric analysis of the percentages of B220+GL-7hiFashi total (B) and B220+CD45.2+E4+GL7hiFashi donor-derived (C) GC B cells in the recipient mice. Each symbol represents a mouse.
(D)Representative spleen sections from recipient mice (n = 5 per group) were stained with anti-E4 monoclonal antibody (mAb) specific for HKIR B cells (red), GL-7 (green), and anti-IgD (blue). Representative images are shown. The scale bars represent 100 μm.
(E-G) The number of E4+ cells per μm2 GC area (E), the fluorescence intensity of E4 staining (F), and the total GC areas (G) were measured for 10 GCs per spleen section. Each symbol represents a germinal center.
(H) HKIR E4+ B-cell-produced serum Ab titers were measured 14 days post-B cell transfer.
(I) HKIR E4+ AFCs in the bone marrow were quantified 30 days post-B cell transfer. Each symbol represents a mouse.
These data are obtained from three independent experiments. Statistical significance was determined using an unpaired, nonparametric Mann-Whitney Student’s t test (NS, not significant, *p < 0.05, **p<0.01, ***p<0.001).
E4 serum Ab titers 14 days post-B cell transfer (21 days postprimary immunization) were lower in mice receiving Sle1b. IFNαR1−/−.HKIR B cells than Sle1b.HKIR B cells (Figure 4H). We also found two-fold fewer long-lived E4+ AFCs in mice receiving Sle1b.IFNαR1−/−.HKIR B cells than Sle1b.HKIR B cells 30 days after B cell transfer (Figure 4I), although the difference did not reach statistical significance due to an outlier in each group. These results show that B-cell-intrinsic IFNαR signaling is required for autoreactive B cell development in the GC pathway.
Transcriptomic Analysis of Non-GC and GC B Cells Shows Differentially Expressed Genes in Multiple Signaling Pathways in the Absence of IFNαR
To determine how IFNαR signaling in B cells may promote autoreactive B cell development into the AFC and GC pathways at the molecular signaling level, we sorted non-GC and GC B cells from B6.Sle1b and B6.Sle1b.IFNαR1−/− mice using fluorescence activated cell sorting (FACS) and analyzed the transcripts of these cell subsets by RNA sequencing. We first generated a heatmap for differentially expressed genes showing ≥2-fold differences in GC B cells and non-GC B cells between the strains after hierarchical clustering (Figure 5A). We then used ingenuity pathway analysis to identify major signaling pathways that represented the differentially expressed genes (see Table S1). We subsequently used interferome to identify interferon-regulated genes associated with T1IFN signaling (Rusinova et al., 2013). Genes differentially regulated between B6.Sle1b and Sle1b.IFNαR1−/− non-GC and GC B cells were associated with cytokine signaling (Figure 5B), BCR signaling (Figure 5C), transcriptional regulation (including NF-κB signaling; Figure 5D), DNA repair (Figure 5E), actin cytoskeleton (Figure 5F), and inducible T-cell costimulator (ICOS) ligand signaling (Figure 5G).
Figure 5.

Transcriptomic Comparison of B6.Sle1b and B6.Sle1b.IFNαR1−/− Non-GC and GC B Cells
(A) Sorted B220+PNA−Fas− non-GC B cells and B220+PNAhiFashi GC B cells from 3-month-old B6.Sle1b and B6.Sle1b.IFNαR1−/− female mice were processed for RNA sequencing in 3 independent experiments. Values as shown by the scale bar are quantified by relative values that reflectZ score (difference in expression divided by SD) relative to the mean values. Gene groups from four patterns of gene expression were collected, and interferon-associated gene expression was confirmed by interferome and follow-up pathway analysis using ingenuity pathway analysis as listed in Table S1.
(B–G) Heatmaps were constructed from hits that were identified by interferome.The represented pathways include cytokine signaling (B), B cell receptor signaling (C), transcription factors (D), DNA repair (E), actin cytoskeleton (F), and ICOS ligand signaling (G).
Data shown are average values from three independent experiments.
To determine whether changes in expression of genes associated with BCR signaling in RNA sequencing (RNA-seq) analysis may impact overall B cell signaling, we first measured calcium flux at the naive B cell stage. Stimulation of purified B cells with anti-IgM caused a lower Ca2+ flux in B6.IFNαR1−/− and Sle1b.IFNαR1−/− B cells than in their respective control B6 or B6.Sle1b B cells (Figure 6A). Consistent with our previous report (Wong et al., 2015), we observed intermediate levels of Ca2+ flux in B6.Sle1b B cells compared to B6 control B cells. To measure BCR signaling events in GC B cells, we transferred B6.Sle1b. HKIR and Sle1b.IFNαR1−/−.HKIR B cells into Ars-KLH immunized syngeneic B6.SJL (CD45.1+) recipients (Figure 4A). We compared the relative expression of p-Syk and BLNK in B220+GL7hiCD95hiE4+ HKIR GC B cells in mice that received Sle1b.IFNαR1−/−.HKIR B cells as compared to mice that received B6.Sle1b.HKIR B cells at day 5 post-transfer (Figure 6B). We saw reduced phosphorylation of Syk (Figure 6C) and reduced expression of BLNK (Figure 6D) in B220+GL7hiCD95hiE4+ GC B cells of the HKIR. Sle1b.IFNαR1−/− genotype than in B220+GL7hiCD95hiE4+ GC cells of the HKIR.Sle1b genotype. These results indicate that B-cell-intrinsic IFNαR signaling helps maintain BCR signaling in non-GC and GC B cells in B6.Sle1b mice at a level that is above the threshold for tolerance induction, resulting in the development of autoreactive B cells into the AFC and GC pathways.
Figure 6.

IFNαR1 Signaling in Non-GC and GC B Cells Sustains the Level of BCR Signaling Required for Autoimmune Responses in B6.Sle1b Mice
(A) Calcium flux in purified B cells from B6 (black), B6.IFNαR1−/− (blue), B6.Sle1b (gray), and B6.Sle1b.IFNαR1−/− (orange) mice after anti-IgM stimulation. (Left) Representative flow plot shows relative Fluo3/FuraRed ratio flux plot after 5 μg/mL anti-IgM stimulation. (Right) Flux ratio (peak flux/baseline flux; left), time to peak (middle), and recovery time from peak to 50% flux value (right) for the indicated groups in combined data from 3 independent experiments are shown.
(B) Flow cytometric gating of the transferred CD45.2+ Sle1b.HKIR and Sle1b.IFNαR1−/−.HKIR B cells within the GCs of B6.SJL (CD45.1+) mice.
(C and D) Flow cytometric analysis to evaluate the phosphorylation of SYK (C) and total BLNK protein level (D) in Sle1b.HKIR and Sle1b.IFNαR1−/−.HKIR donor-derived B220+CD45.2+E4+GL-7hiCD95hi GC B cells. Representative histograms are shown on the left, and geometric mean fluorescence intensity (gMFI) values are plotted on the right.
These data are obtained from two independent experiments, each involving three mice per genotype. Statistical significance was determined using an unpaired, nonparametric Mann-Whitney Student’s t test (*p < 0.05).
GC B-Cell-Intrinsic IFNαR Signaling Maintains Spt-GCs and Enhances Class-Switched ANA Production
Changes in the epigenetic landscape of B cells due to transcriptional differences in multiple signaling pathways between the B6.Sle1b and Sle1b.IFNαR1−/− non-GC B cells prior to GC entry may complicate the interpretation of the B-cell-intrinsic role of T1IFN signaling in altering GC checkpoint. To circumvent this potential caveat and to specifically determine whether IFNαR signaling in GC B cells is required for the maintenance of Spt-GCs and altered B cell selection, we crossed B6.Sle1b.IFNαR1fl/fl mice to a GC-specific Cre mouse line (Cg-Ighg1-Cre mice, called GCCre; Casola et al., 2006) to generate B6.Sle1b.IFNαR1fl/flGCCre mice (designated Sle1-b.IFNαR1fl/flGCCre mice). We confirmed that IFNαR1 expression in GC B cells was lower in GC B cells of Sle1b. IFNαR1fl/flGCCre female mice than in B6.Sle1b.IFNαR1fl/fl control female mice (Figure 7A). Sle1b.IFNαR1fl/flGCCre mice had a smaller fraction of GC B cells than B6.Sle1b.IFNαR1fl/fl control mice at 6 months age (Figure 7B).Tfh percentage did not significantly differ between the strains (Figure 7C). Spleens of 6-month-old Sle1b.IFNαR1fl/flGCCre mice also had smaller IgDnegGL7+ Spt-GC structures than B6.Sle1b.IFNαR1fl/fl mice (Figure 7D). Sle1b.IFNαR1fl/flGCCre mice had similar total IgG titers (Figure 7E) but reduced IgG2c titers than B6.Sle1b.IFNαR1fl/fl control mice (Figure 7F). The decreased Spt-GC responses in Sle1b.IFNγR1fl/flGCCre mice were strongly associated with reduced serum anti-dsDNA and anti-nucleosome (Figures 7G and 7H) IgG2c autoAbs and reduced IgG2c ANA reactivity (Figure 7I). Altogether, these data indicate a GC B-cell-intrinsic role of T1IFN signaling in autoreactive B cell development and production of class-switched autoAbs in B6.Sle1b mice.
Figure 7.

IFNαR1 Signaling in GC B Cells Is Required for GC Stability and IgG2c ANA Production in B6.Sle1b Mice
(A) Flow cytometric analysis of surface expression of IFNαR1 on B220+GL-7hiFashi GC B cells in 6-month-old B6.Sle1b.IFNαR1fl/fl (white) and B6.Sle1b.IFNαR1fl/flGCCre/+ (orange) female mice. (B and C) The percentages of B220+GL-7hiFashi GC B cells (B) and CD4+CXCR5hiPD-1hi Tfh cells (C) in total splenocytes of 6-month-old B6.Sle1b.IFNαR1fl/fl (white) and B6.Sle1b.IFNαR1fl/flGCCre/+ (orange) mice. Each symbol represents a mouse, and horizontal lines indicate mean values.
(D) Spleen sections from 5 mice per group were stained with anti-CD4 (red), GL-7 (green), and anti-IgD (blue). Representative images are shown in the left panels. GC areas were measured for 10 GCs per spleen section (right panel). The scale bars represent 100 μm.
(E and F) Total IgG (E) and IgG2c (F) serum titers in these mice.
(G and H) Serum titers of dsDNA-specific (G) and nucleosome-specific (H) IgG2c Abs in these mice.
(I) Serum IgG2c ANA reactivity was measured by Hep-2 slides. The scale bars represent 250 μm.
These data are obtained from two independent experiments, and each symbol represents a mouse. Statistical significance was determined using an unpaired, nonparametric Mann-Whitney Student’s t test (NS, not significant, *p < 0.05, **p < 0.01, ***p < 0.001).
DISCUSSION
T1IFN promotes SLE disease in both mouse and human, making this cytokine and its signaling pathway an attractive target for SLE therapeutics (Kiefer et al., 2012; Theofilopoulos et al., 2005). Whereas most studies in mouse and human have suggested T1IFN to be a therapeutic target for SLE patients with the disease activity, we focused our current studies on determining whether T1IFN signaling can be a target for an early intervention for SLE patients with elevated T1IFN activity prior to disease onset. For this purpose, we have used B6.Sle1b mice that develop high titers of autoAbs due to altered B cell tolerance, but they do not develop full-blown disease (Wong et al., 2012). Thus, the B6.Sle1b mouse model allows for the investigation of T1IFN signaling in the initial loss of tolerance without the confounding effects of systemic inflammation and severe disease in other SLE mouse models. Here, we show that T1IFN signaling elicits an important role in initiating SLE in addition to its established role in promoting end-stage pathology (Bagavant and Fu, 2009). In SLE-prone B6.Sle1b mice, we found that IFNαR expression is required for elevated Spt-GC and Spt-AFC responses and increased titers of serum autoAbs. Furthermore, B-cell-intrinsic IFNαR signaling within the GC is required for altered B cell selection that promotes the development of autoreactive B cells and class-switched autoAb-producing AFCs and autoAbs.
IFNαR signaling in BXD2 mice and B6.Sle1.Sle2.Sle3 mice results in aberrant localization of autoantigen-carrying MZBs to the B cell follicle (Li et al., 2015; Wang et al., 2010; Zhou et al., 2011). This mislocalization of MZBs correlates with a dissipation of the MZMs that regulate the entry of apoptotic cell-derived autoantigens into the B cell follicle (Li et al., 2015). Therefore, the direct role of B-cell-intrinsic T1IFN signaling in autoreactive B cell development in the AFC and GC pathways may not be determined by studies using the BXD2 model. In the B6.Sle1b model of SLE, we however did not observe any significant changes in the frequency or splenic localization of IgDnegIgM+ MZBs or a depletion of MARCO+SIGN-R1+ MZMs. Although MZMs and MZBs were not affected in B6.Sle1b mice in the steady state, TLR7 agonist (imiquimod [IMQ]) stimulation of B6.Sle1b mice or overexpression of TLR7 in B6.Sle1b mice is associated with the depletion of MZMs, disruption of the MZB compartment, and increased kidney pathology (S.B.C. and Z.S.M.R., unpublished data; Amano et al., 2003; Giltiay et al., 2013; Pisitkun et al., 2006). Whereas dissipation of MZBs and MZMs contribute to SLE development in BXD2 mice or in TLR7 overexpression SLE models, our findings show that autoreactive B cell development can occur without the disruption of MZBs and MZMs in B6.Sle1b mice.
Our RNA-seq data show differentially expressed genes between B6.Sle1b and Sle1b.IFNαR1−/− non-GC and GC B cells. Subsequent confirmatory experiments on BCR signaling at non-GC and GC stages indicate that IFNαR signaling in B cells may lower the BCR signaling threshold both outside and inside the GCs to promote elevated Spt-AFC, Spt-GC, and IgG2c autoAb responses in B6.Sle1b mice. Changes in BCR signaling in the absence of IFNαR, however, do not affect overall GC and Ab responses and B cell affinity maturation to T-dependent foreign antigen in both non-autoimmune B6 and autoimmune B6.Sle1b mice. Using the Wiskott-Aldrich syndrome protein (WASp) chimeric model of autoimmunity, it was reported that B-cell-intrinsic IFNαR signaling was not involved in Spt-GC regulation and subsequent SLE-associated pathology (Jackson et al., 2016). WASp-deficient chimeric mice develop autoimmunity as a result of an increased BCR signaling (Becker-Herman et al., 2011). We have found that IFNαR deficiency is associated with a reduction in calcium flux and BCR signaling in B6.Sle1b.IFNαR1−/− mice, suggesting that WASp deficiency could negate the requirement of IFNαR signaling in elevated Spt-GC responses because of the increased BCR signaling in WASp−/− B cells. In addition, our RNA sequencing results identified significant changes in several WAS-associated genes (jmy, mras, rdx, and nexn) and known regulators of the actin cytoskeleton, suggesting crosstalk between IFNαR and WASp signaling (Matsumoto et al., 1997; Ohtsuka et al., 1998; Vaiskunaite et al., 2000; Zuchero et al., 2009). Therefore, it is likely that IFNαR signaling is not required for GC dysregulation in models of SLE that feature enhanced BCR signaling.
Collectively, studies of different SLE mouse models that exhibit BCR hyperactivation, TLR7 overexpression and/or hyperactivation, and/or enhanced IFNαR signaling indicate the likelihood that there is a level of synergy between these signaling events. Together, they function to achieve a certain B cell activation threshold that is required for loss of tolerance and autoreactive B cell differentiation through the AFC and GC pathways. When other signals, such as BCR and TLR signaling, are robust, IFNαR signaling is not required to meet the activation threshold. However, when the signaling is lesser by these receptors, IFNαR signaling synergy is required to push B cells over the activation threshold. These differences in signal integration are likely to be present in human patients as well and further support personalized medicine approaches to the implementation of IFN-focused therapeutics.
Both B6 and B6.Sle1b GC B cells exhibited a significant increase in IFNαR expression, and GC B-cell-specific deletion of IFNαR in B6.Sle1b mice resulted in reduced GC and IgG2c ANA responses. Together, these results suggest that IFNαR signaling controls the selection of B cells within the GC in a B-cell-intrinsic manner. Several potential sources of T1IFN have been proposed for promoting SLE development. Diphtheria-toxin-mediated depletion of plasmacytoid dendritic cells (pDCs), which are believed to be the major source for T1IFN, in BXSB mice suggested that pDCs are required for SLE development (Ganguly et al., 2013; Rowland et al., 2014). Further studies have proposed pDCs as the predominant source of T1IFN in SLE patients (Gujer et al., 2011; Huang et al., 2015). Although systemic overexpression of IFNα has been shown to promote changes in somatic hypermutation (Moisini et al., 2012), a functional block of pDC-derived IFNα with Siglec-H-targeted antibodies did not have a significant effect on B cell selection, suggesting the contribution of IFNα from another source to regulate B cell tolerance at the GC checkpoint. Follicular dendritic cells (FDCs) have recently been shown to produce IFNα within the GC microenvironment (Das et al., 2017). Collectively, these results suggest that, although pDC-derived IFNs promote SLE disease, local FDC-produced IFNα may directly contribute to loss of B cell tolerance in GCs.
Recent studies have shown that transitional type 1 (T1) B cells expressed elevated levels of ifnb, ifna1, ifna4, and ifna7 mRNA in autoimmune BXD2 mice to promote survival of these transitional B cells (Hamilton et al., 2017). Consistent with this recent report, we found a modest reduction in the percentage of T1 B cells in Sle1b.IFNαR1−/− mice compared to B6.Sle1b mice. Therefore, we do not rule out the role of IFNb in T1 B cell survival and potential differentiation of these cells into AFCs outside of GCs. Whereas the previous paper (Hamilton et al., 2017) showed dependence of T1 B cell survival on IFNb in a mixed bone marrow chimeric environment where IFNb+ and IFNbneg T1 B cells competed for the space and survival signal, T1 B cell survival in B6.Sle1b mice in a non-competitive environment only modestly depended on IFNb signaling through IFNαR. These two systems are fundamentally different; in their system, B cells lack the ability to produce IFNb, whereas in our system they lack the ability to sense it.
In summary, our current studies have revealed a greater understanding of how B-cell-intrinsic T1IFN signaling contributes to the initial loss of B cell tolerance and the development of autoreactive B cells into the AFC and GC pathways, driving autoimmunity in SLE models that do not exhibit hyperactive BCR and TLR signaling. Our studies on T1IFN signaling have implications in predicting SLE patients who might respond to anti-IFNαR therapy based on T1IFN activity. Our studies also identify applications of the anti-IFNαR therapy for an early intervention in SLE-susceptible individuals with an elevated T1IFN activity prior to disease onset, which may prevent autoreactive B cell development into the AFC and GC pathways and block disease progression.
EXPERIMENTAL PROCEDURES
Additional materials and methods can be found in the Supplemental Experimental Procedures.
Mice
C57BL/6 (B6), B6.129S2-Ighmtm1Cgn/J (B6.μMT), B6.IFNαR1−/−, B6(Cg)-IFNαR1tm1.1Ees/J (B6.IFNαR1flox/flox), B6.129P2(Cg)-Ighg1tm1(IRES-cre)Cgn/J (GCCre) and B6.SJLPtprc < b > /BoyJ (B6.SJL- CD45.1+) mice were purchased from the Jackson Laboratory and bred in house. B6.Sle1b and B6.HKIR mice were described previously (Morel et al., 2000; Notidis et al., 2002). Female mice of various ages (8–10 weeks, 3 months, and 6 months old) were used for all experiments. All animals were housed at Penn State Hershey Medical Center in specific pathogen-free animal facility. All procedures were performed in accordance with the guidelines approved by our Institutional Animal Care and Use Committee.
ELISpot Assays
ELISpot assays were performed as described (Wong et al., 2012). Briefly, splenocytes in RPMI containing 10% fetal bovine serum were plated at a concentration of 1 × 105 cells/well onto anti-IgM-, anti-IgG-, salmon sperm dsDNA- (Invitrogen, Grand Island, NY), calf thymus histone- (Sigma Aldrich, St. Louis, MO), or nucleosome-coated plates (Millipore, Bedford, MA). Serially diluted (1:2) cells were incubated for 6 hr at 37°C. IgM-producing AFCs were detected using biotinylated anti-mouse IgM (Jackson ImmunoResearch, West Grove, PA) and streptavidin (SA)-alkaline phosphatase (Vector Laboratories, Burlingame, CA). IgG-producing AFCs were detected using alkaline phosphatase-conjugated anti-mouse IgG (Molecular Probes, Grand Island, NY). dsDNA-, histone-, and nucleosome-specific AFCs were detected by biotinylated anti-kappa Ab (Invitrogen, Grand Island, NY) and SA-alkaline phosphatase or alkaline phosphatase-conjugated anti-mouse IgG (Molecular Probes, Grand Island, NY). Plates were developed using the Vector Blue Alkaline phosphatase Substrate Kit III (Vector Laboratories, Burlingame, CA). ELISpots were enumerated using a computerized ELISpot plate imaging and analysis system (Cellular Technology, Shaker Heights, OH).
Serology: Ig and ANA Titers
Ig and ANA titers were measured by ELISA as previously described (Wong et al., 2012). Briefly, for IgG or IgM detection, ELISA plates (Thermo Fisher Scientific, Waltham, MA) were coated with anti-IgM or anti-IgG (Invitrogen, Grand Island, NY) capture antibodies and detected using biotinylated antimouse IgM (Jackson ImmunoResearch, West Grove, PA) or alkaline-phosphatase conjugated anti-mouse IgG (Molecular Probes, Grand Island, NY). Total IgG autoAb titers were measured in plates coated with salmon sperm dsDNA (Invitrogen, Grand Island, NY), histone (Sigma Aldrich, St. Louis, MO), or nucleosome and detected with biotinylated anti-kappa Ab (Invitrogen, Grand Island, NY). IgG subtype-specific autoAb titers were detected by biotinylated-IgG1, -IgG2b, and AP-IgG2c Abs (Southern Biotech, Birmingham, AL). Biotinylated antibodies were detected by SA-alkaline phosphatase (Vector Laboratories, Burlingame, CA). The plates were developed by the PNPP (p-nitrophenyl phosphate, disodium salt; Thermo Fisher Scientific, Rockford, IL) substrates for alkaline phosphatase and read at γ405 nm.
Calcium Flux Assay
Purified B cells were incubated with Fura Red, AM (Molecular Probes) and Fluo-3, AM (Molecular Probes) Calcium dyes at 37°C in dye-free RPMI (Gibco). Prior to analysis, samples were rested in the dark at room temperature for 30 min and heated to 37° immediately prior to analysis. During flow cytometric analysis, a baseline reading was acquired from each sample for 1 min prior to stimulation of the cells with 1,5, or 10 μg/mL anti-IgM (Jackson ImmunoResearch) followed by 3 min of further acquisition on the BD LSR II flow cytometer.
Generation of Mixed BM Chimeric Mice
10- to 12-week-old female B6.μMT (B6.Ightm1Tim) recipient mice were lethally irradiated with two doses of 450 rads of X-rays (X-RAD 320iX Research Irradiator; Precision X-Ray) within a 4-hr interval. Within 24 hr of the second irradiation, each recipient received intravenously (i.v.) 107 T-cell-depleted BM cells isolated from B6.Sle1b and B6.Sle1b.IFNαR1−/− mice. Mice were analyzed at 3 months after transfer.
Adaptive Transfer of HKIR Cells
B6.SJL (CD45.1+) recipient mice were immunized (intraperitoneally [i.p.]) with 500 μg/mouse p-azophenyl arsonate keyhole limpet hemocyanin (Ars-KLH) in Complete Freund’s Adjuvant (CFA) 1 week prior to transfer of 2 × 106 magnetic activated cell sorting (MACS) purified splenic B cells (i.v.) from either B6.Sle1b.HKIR or B6.Sle1b.IFNαR1−/−.HKIR donor mice. Immediately after transfer, mice were injected (i.p.) with 250 μg/mouse Ars-KLH (in Incomplete Freunds Adjuvant). Spleens and sera from the recipient mice were collected at different time points after cell transfer for immunohistochemical and flow cytometric analysis and for measuring serum E4 Ab titers specific for HKIR B cells. Bone marrow samples were harvested for quantifying the number of long-lived AFCs specific for HKIR B cells.
Phospho Flow
Phospho flow was performed as previously described (Khalil et al., 2012). Briefly, total splenocytes were isolated and immediately fixed in 1% paraformaldehyde in RPMI 1640 and incubated for 15 min at room temperature. Fixed splenocytes were washed in PBS solution prior to red blood cell (RBC) lysis. After RBC lysis, 1 × 106 total splenocytes were permeabilized in BD phosphor-flow perm buffer II and stained according to the manufacturer’s protocol.
Immunization
Mice were immunized with 200 μg/mouse with NP-CGG (Biosearch Technologies) in CFA (Sigma) followed by a booster immunization with 100 μg/mouse of NP-CGG in Incomplete Freunds Adjuvant 7 days later.
Statistical Analysis
Comparisons of two groups were analyzed using an unpaired, nonparametric, Mann-Whitney, Student’s t test, whereas one-way ANOVA, with a follow-up Tukey multiple-comparison test, was used to compare morethan two groups. GraphPad Prism 6 software (La Jolla, CA) was used for all analyses. Wherever indicated, NS indicates non-significant, *p < 0.05, **p < 0.01, and ***p < 0.001. Horizontal bars reflect mean values, and error bars reflect SD of the mean unless otherwise indicated.
Supplementary Material
Highlights.
Type 1 IFN (T1IFN) signaling in B cells promotes autoreactive B cell development
T1IFN signaling drives autoreactive B cell development by regulating BCR signaling
T1IFN signaling in GC B cells causes loss of tolerance and ANA production
T1IFN signaling is not required for immunization-induced B cell responses
ACKNOWLEDGMENTS
We thank Dr. Aron Lukacher and Adam J. Fike for the critical reading of the manuscript. We also thank the PSUHMC flow cytometry core facility for their assistance. This work was supported by the NIH grants RO1A1091670 to Z.S.M.R. and 5F31AI122608 to P.D.
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-seq data reported in this paper is GEO: GSE115349.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, three figures, and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.06.046.
AUTHOR CONTRIBUTIONS
P.P.D., S.B.C., and Z.S.M.R. designed and performed experiments; P.P.D. and Z.S.M.R. wrote the manuscript; and Y.I.K., C.S., S.L.S., and M.J.F. performed specific experiments and edited the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Agrawal H, Jacob N, Carreras E, Bajana S, Putterman C, Turner S, Neas B, Mathian A, Koss MN, Stohl W, et al. (2009). Deficiency of type I IFN receptor in lupus-prone New Zealand mixed 2328 mice decreases dendritic cell numbers and activation and protects from disease. J. Immunol. 183, 6021–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabyev B, Rahman ZS, and Manser T (2007). Quantitatively reduced participation of anti-nuclear antigen B cells that down-regulate B cell receptor during primary development in the germinal center/memory B cell response to foreign antigen. J. Immunol 178, 5623–5634. [DOI] [PubMed] [Google Scholar]
- Amano H, Amano E, Moll T, Marinkovic D, Ibnou-Zekri N, Martinez-Soría E, Semac I, Wirth T, Nitschke L, and Izui S, (2003). The Yaa mutation promoting murine lupus causes defective development of marginal zone B cells. J. Immunol. 170, 2293–2301. [DOI] [PubMed] [Google Scholar]
- Bagavant H, and Fu SM (2009). Pathogenesis of kidney disease in systemic lupus erythematosus. Curr. Opin. Rheumatol. 21, 489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker-Herman S, Meyer-Bahlburg A, Schwartz MA, Jackson SW, Hudkins KL, Liu C, Sather BD, Khim S, Liggitt D, Song W, et al. (2011). WASp-deficient B cells play a critical, cell-intrinsic role in triggering autoimmunity. J. Exp. Med. 208, 2033–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, and Pascual V (2003). Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 197, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappione A 3rd, Anolik JH, Pugh-Bernard A, Barnard J, Dutcher P, Silverman G, and Sanz I (2005). Germinal center exclusion of autoreactive B cells is defective in human systemic lupus erythematosus. J. Clin. Invest. 115,3205–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casola S, Cattoretti G, Uyttersprot N, Koralov SB, Seagal J, Hao Z, Waisman A, Egert A, Ghitza D, and Rajewsky K (2006).Tracking germinal center B cells expressing germ-line immunoglobulin gamma1 transcripts by conditional gene targeting. Proc. Natl. Acad. Sci. USA 103, 7396–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow MK (2014). Type I interferon in the pathogenesis of lupus. J. Immunol. 192, 5459–5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Heesters BA, Bialas A, O’Flynn J, Rifkin IR, Ochando J, Mittereder N, Carlesso G, Herbst R, and Carroll MC (2017). Follicular dendritic cell activation by TLR ligands promotes autoreactive B cell responses. Immunity 46, 106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domeier PP, Schell SL, and Rahman ZS (2017). Spontaneous germinal centers and autoimmunity. Autoimmunity 50, 4–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuyama H, Nimmerjahn F, and Ravetch JV (2005). The inhibitory Fcgamma receptor modulates autoimmunity by limiting the accumulation of immunoglobulin G+ anti-DNA plasma cells. Nat. Immunol. 6, 99–106. [DOI] [PubMed] [Google Scholar]
- Ganguly D, Haak S, Sisirak V, and Reizis B (2013). The role of dendritic cells in autoimmunity. Nat. Rev. Immunol. 13, 566–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giltiay NV, Chappell CP, Sun X, Kolhatkar N, Teal TH, Wiedeman AE, Kim J, Tanaka L, Buechler MB, Hamerman JA, et al. (2013). Overexpression of TLR7 promotes cell-intrinsic expansion and autoantibody production by transitional T1 B cells. J. Exp. Med. 210, 2773–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujer C, Sandgren KJ, Douagi I, Adams WC, Sundling C, Smed-Sören-sen A, Seder RA, Karlsson Hedestam GB, and Loré K (2011). IFN-α produced by human plasmacytoid dendritic cells enhances T cell-dependent naive B cell differentiation. J. Leukoc. Biol. 89, 811–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JA, Wu Q, Yang P, Luo B, Liu S, Hong H, Li J, Walter MR, Fish EN, Hsu HC, and Mountz JD (2017). Cutting edge: endogenous IFN-β regulates survival and development of transitional B cells. J. Immunol. 199,2618–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Dorta-Estremera S, Yao Y, Shen N, and Cao W (2015). Predominant role of plasmacytoid dendritic cells in stimulating systemic autoimmunity. Front. Immunol. 6, 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SW, Jacobs HM, Arkatkar T, Dam EM, Scharping NE, Kolhatkar NS, Hou B, Buckner JH, and Rawlings DJ (2016). B cell IFN-γ receptor signaling promotes autoimmune germinal centers via cell-intrinsic induction of BCL-6. J. Exp. Med. 213, 733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, Cambier JC, and Shlomchik MJ (2012). B cell receptor signal transduction in theGC is short-circuited by high phosphatase activity. Science 336,1178–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer K, Oropallo MA, Cancro MP, and Marshak-Rothstein A (2012). Role of type I interferons in the activation of autoreactive B cells. Immunol. Cell Biol. 90, 498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar KR, Li L, Yan M, Bhaskarabhatla M, Mobley AB, Nguyen C, Mooney JM, Schatzle JD, Wakeland EK, and Mohan C (2006). Regulation of B cell tolerance by the lupus susceptibility gene Ly108. Science 312 1665–1669. [DOI] [PubMed] [Google Scholar]
- Li H, Fu YX, Wu Q, Zhou Y, Crossman DK, Yang P, Li J, Luo B, Morel LM, Kabarowski JH, et al. (2015). Interferon-induced mechanosensing defects impede apoptotic cell clearance in lupus. J. Clin. Invest. 125, 2877–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, and Davidson A (2013). IFNa inducible models of murine SLE. Front. Immunol. 4, 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Bethunaickan R, Huang W, Lodhi U, Solano I, Madaio MP, and Davidson A (2011). Interferon-α accelerates murine systemic lupus erythematosus in a T cell-dependent manner. Arthritis Rheum. 63, 219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenço EV, and La Cava A (2009). Cytokines in systemic lupus erythematosus. Curr. Mol. Med. 9, 242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzina IG, Atamas SP, Storrer CE, daSilva LC, Kelsoe G, Papadimitriou JC, and Handwerger BS (2001). Spontaneous formation of germinal centers in autoimmune mice. J. Leukoc. Biol. 70, 578–584. [PubMed] [Google Scholar]
- Mathian A, Weinberg A, Gallegos M, Banchereau J, and Koutouzov S (2005). IFN-alpha induces early lethal lupus in preautoimmune (New Zealand Black x New Zealand White) F1 but not in BALB/c mice. J. Immunol. 174, 2499–2506. [DOI] [PubMed] [Google Scholar]
- Mathian A, Gallegos M, Pascual V, Banchereau J, and Koutouzov S (2011). Interferon-α induces unabated production of short-lived plasma cells in pre-autoimmune lupus-prone (NZB×NZW)F1 mice but not in BALB/c mice. Eur. J. Immunol. 41, 863–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K, Asano T, and Endo T (1997). Novel small GTPase M-Ras participates in reorganization of actin cytoskeleton. Oncogene 15, 2409–2417. [DOI] [PubMed] [Google Scholar]
- Moisini I, Huang W, Bethunaickan R, Sahu R, Ricketts PG, Akerman M, Marion T, Lesser M, and Davidson A (2012). The Yaa locus and IFN-α fine-tune germinal center B cell selection in murine systemic lupus erythematosus. J. Immunol. 189, 4305–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel L, Croker BP, Blenman KR, Mohan C, Huang G, Gilkeson G, and Wakeland EK (2000). Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc. Natl. Acad. Sci. USA 97, 6670–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munroe ME, Lu R, Zhao YD, Fife DA, Robertson JM, Guthridge JM, Niewold TB, Tsokos GC, Keith MP, Harley JB, and James JA (2016). Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann. Rheum. Dis. 75, 2014–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen C, Limaye N, and Wakeland EK (2002). Susceptibility genes in the pathogenesis of murine lupus. Arthritis Res. 4 (Suppl 3), S255–S263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson KM, Cullen JL, Kashgarian M, and Shlomchik MJ (2013). Exacerbated autoimmunity in the absence of TLR9 in MRL.Fas(lpr) mice depends on Ifnar1. J. Immunol. 190, 3889–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notidis E, Heltemes L, and Manser T, (2002). Dominant, hierarchical induction of peripheral tolerance during foreign antigen-driven B cell development. Immunity 17, 317–327. [DOI] [PubMed] [Google Scholar]
- Ohtsuka T, Nakanishi H, Ikeda W, Satoh A, Momose Y, Nishioka H, and Takai Y (1998). Nexilin: a novel actin filament-binding protein localized at cellmatrix adherens junction. J. Cell Biol. 143, 1227–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, and Bolland S (2006). Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science 312, 1669–1672. [DOI] [PubMed] [Google Scholar]
- Rahman ZS, Alabyev B, and Manser T (2007). FcgammaRIIB regulates autoreactive primary antibody-forming cell, but not germinal center B cell, activity. J. Immunol. 178, 897–907. [DOI] [PubMed] [Google Scholar]
- Rowland SL, Riggs JM, Gilfillan S, Bugatti M, Vermi W, Kolbeck R, Unanue ER, Sanjuan MA, and Colonna M (2014). Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J. Exp. Med 211, 1977–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, and Hertzog PJ (2013). Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 41, D1040–D1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago-Raber ML, Baccala R, Haraldsson KM, Choubey D, Stewart TA, Kono DH, and Theofilopoulos AN (2003). Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J. Exp. Med. 197, 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theofilopoulos AN, Baccala R, Beutler B, and Kono DH (2005). Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 23, 307–336. [DOI] [PubMed] [Google Scholar]
- Tiller T, Kofer J, Kreschel C, Busse CE, Riebel S, Wickert S, Oden F, Mertes MM, Ehlers M, and Wardemann H (2010). Development of selfreactive germinal center B cells and plasma cells in autoimmune Fc gammaRIIB-deficient mice. J. Exp. Med. 207, 2767–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaiskunaite R, Adarichev V, Furthmayr H, Kozasa T, Gudkov A, and Voyno-Yasenetskaya TA (2000). Conformational activation of radixin by G13 protein alpha subunit. J. Biol. Chem. 275, 26206–26212. [DOI] [PubMed] [Google Scholar]
- Vinuesa CG, Sanz I, and Cook MC (2009). Dysregulation of germinal centres in autoimmune disease. Nat. Rev. Immunol. 9, 845–857. [DOI] [PubMed] [Google Scholar]
- Wandstrat AE, Nguyen C, Limaye N, Chan AY, Subramanian S, Tian XH, Yim YS, Pertsemlidis A, Garner HR Jr., Morel L, and Wakeland EK (2004). Association of extensive polymorphisms in the SLAM/CD2 gene cluster with murine lupus. Immunity 21, 769–780. [DOI] [PubMed] [Google Scholar]
- Wang JH, Li J, Wu Q, Yang P, Pawar RD, Xie S, Timares L, Raman C, Chaplin DD, Lu L, et al. (2010). Marginal zone precursor B cells as cellular agents for type I IFN-promoted antigen transport in autoimmunity. J. Immunol. 184, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- William J, Euler C, Christensen S, and Shlomchik MJ (2002). Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science 297, 2066–2070. [DOI] [PubMed] [Google Scholar]
- Wong EB, Khan TN, Mohan C, and Rahman ZS (2012). The lupus-prone NZM2410/NZW strain-derived Sle1b sublocus alters the germinal center checkpoint in female mice in a B cell-intrinsic manner. J. Immunol. 189, 5667–5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong EB, Soni C, Chan AY, Domeier PP, Shwetank Abraham, T. Limaye, N. Khan T.N., Elias MJ, Chodisetti SB, et al. (2015). B cell-intrinsic CD84 and Ly108 maintain germinal center B cell tolerance. J. Immunol. 194, 4130–4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods M, Zou YR, and Davidson A (2015). Defects in germinal center selection in SLE. Front. Immunol. 6, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Niu H, Zheng YY, and Morel L (2011). Autoreactive marginal zone B cells enter the follicles and interact with CD4+ T cells in lupus-prone mice. BMC Immunol. 12, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchero JB, Coutts AS, Quinlan ME, Thangue NB, and Mullins RD (2009). p53-cofactor JMY is a multifunctional actin nucleation factor. Nat. Cell Biol. 11, 451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.