

Abstract
Many genome-wide association studies (GWAS) have identified signals located in non-coding regions, and an increasing number of functional genomics annotations of regulatory elements and assays of regulatory activity have been used to investigate mechanisms. Genome-wide datasets that characterize chromatin structure help detect potential regulatory elements. Assays to experimentally assess candidate variants include transcriptional reporter assays, and recently, massively parallel reporter assays (MPRAs). Additionally, the effect of candidate regulatory elements and variants on gene expression and function can be evaluated using genomic editing with the CRISPR-Cas9 technology. We highlight some recent studies that employed these strategies to identify variant effects and elucidate molecular and/or biological mechanisms at GWAS loci for lipid traits and coronary artery disease.
Introduction
Human genome-wide association studies (GWAS) have identified hundreds of DNA variants associated with blood lipid levels and coronary artery disease. Blood lipid levels are a risk factor for cardiovascular disease, including increased low-density lipoprotein cholesterol (LDL-C), and increased triglycerides [1,2]. GWAS have been very successful at identifying genetic variants associated with these complex metabolic diseases [3,4] however, characterizing the molecular mechanisms responsible for these associations has been challenging.
Most variants identified by GWAS are located within non-coding regions of the genome [5], suggesting that these variants do not alter the structure or function of the encoded proteins. Variants located within regulatory elements, such as enhancer or silencer regions, may act to enhance or reduce gene expression. These regulatory regions may affect multiple genes and may regulate genes located hundreds of kilobases away [6,7]. Current challenges are to identify which GWAS variants have regulatory functions and to characterize the molecular mechanisms by which allelic differences affect gene activity and disease risk. Recently, identification of regulatory elements and variants has been facilitated by technological development of genome-wide functional assays.
Experimental assays are necessary to determine which of the variants located in regulatory regions have allelic effects on regulatory activity. For example, transcriptional reporter assays are used to identify variants that alter promoter or enhancer/silencer activity, and recently, high-throughput approaches have been used to test dozens to thousands of candidate variants in a massively parallel fashion. Notably, GWAS studies identify lead variants that are most strongly associated with a trait or disease; however, these lead variants are not necessarily the functional regulatory variants due to sampling variation, technical, or stochastic reasons. Variants that are strongly linked, or inherited together (in strong linkage disequilibrium) with the lead variants may have regulatory effects, which highlights the need to test a number of variants in experimental assays. Reporter assays are often performed by cloning variant-containing regions into vectors containing a reporter gene and transfecting biologically-relevant cell types including induced pluripotent stem cells (iPSCs) that can be differentiated into several target cell types [8]. Another recent strategy for experimental evaluation of regulatory elements and variants uses CRISPR-Cas9-mediated genome editing. Cells or organisms can be edited to delete or alter the effect of a regulatory element or to create specific allelic substitutions.
Functional genomics regulatory annotation data can be used to help guide the selection of candidate regulatory elements and variants for experimental testing. These data are generated through the use of high-throughput methods, including open chromatin, chromatin conformation, and chromatin immunoprecipitation assays followed by high-throughput sequencing. These assays can be helpful at detecting candidate regulatory elements, including enhancer or silencer regions, which may ultimately aid in elucidating the mechanisms underlying the relationships between the GWAS variants and trait [9–11]. In this review, we discuss some recent approaches used to identify functional variants and mechanisms at lipid or coronary artery disease GWAS loci.
High-throughput functional genomics assays
High-throughput functional genomics assays of chromatin structure identify genomic regions characteristic of regulatory elements. Several consortia, including the Encyclopedia of DNA elements (ENCODE) consortium, the National Institutes of Health (NIH) Roadmap Epigenomics Mapping Consortium, and others in the International Human Epigenome Consortium [9,10,12] have generated genome-wide maps in hundreds of cell types and tissues. Maps of open, or accessible, chromatin denote DNA regions devoid of histones and more accessible to transcription factors, as detected by DNase hypersensitivity (DNase HS) [13] formaldehyde-assisted isolation of regulatory elements (FAIRE) [14] or assays for transposase-accessible chromatin (ATAC)-sequencing [15,16] (Table 1). Maps of histone modifications, detected through chromatin immunoprecipitation (ChIP)-sequencing [17,18]), can be integrated to predict chromatin state, including promoter, enhancer, and silencer regions [19], and regions bound by transcription factors can be further annotated with sequence binding motifs and to detect transcription factor footprints [17,20]. Maps of chromatin interactions, higher-order chromatin structure, topologically associated domains (TADs), and frequently interacting regions (FIREs) are based on chromosome conformation capture methods, including Hi-C and chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) [21–23].
Table 1. Acronyms for terms and assays used to study noncoding, regulatory regions.
| Acronym | Full name | Description |
|---|---|---|
| DNase | DNase I hypersensitivity | assay used to detect open, accessible chromatin regions |
| FAIRE | Formaldehyde-assisted isolation of regulatory elements | assay used to detect open, accessible chromatin regions |
| ATAC-seq | Assay for transposase-accessible chromatin with high-throughput sequencing | assay used to detect open, accessible chromatin regions |
| ChIP | Chromatin immunoprecipitation | assay used to detect protein binding or histone modifications |
| TAD | Topologically associated domain | term used to describe three-dimensional chromatin organization and interacting regions |
| FIRE | Frequently interacting regions | term used to describe three-dimensional chromatin organization |
| 3C, 4C, 5C, HiC, ChIA-PET | Chromosome conformation capture, “ -on-chip,” -carbon copy, chromatin interaction analysis by paired-end tag sequencing | assays used to detect chromatin contacts, chromatin organization |
| EMSA | Electrophoretic mobility shift assay | assay used to detect proteins bound to a nucleotide sequence |
| MPRA | Massively parallel reporter assay | assay used to test candidates for effects on transcriptional activity |
| CRISPR | Clustered regularly interspaced short palindromic repeats | used typically with Cas9 protein and guide RNAs for genomic editing |
Integrating these data and maps from trait-relevant cell types can be used to guide the identification of trait-associated variants located in regulatory regions. Data sets of chromatin accessibility and histone marks have differing signal strength compared to background and may be influenced by cellular environment, so multiple lines of evidence (i.e. peaks) can provide greater evidence of a regulatory element. Specific histone marks tend to be observed at different types of elements, such as H3K27ac marks at active enhancers. However, neither individual marks nor the absence of regulatory evidence from an assay is definitive, and mere presence of a variant in a region of histone marks does not indicate that the variant alleles alter regulatory activity [24]. While evidence of chromatin interactions between variant positions and a transcription start site supports the potential for a regulatory effect of variants on a gene, the interaction alone is not definitive, and absence of interactions may be due to assay resolution or cellular environment. Therefore, combining one or more pieces of evidence from high-throughput regulatory assay data with other functional genomics experiments, such as reporter assays and genomic editing, enhances the likelihood of identifying candidate regulatory variants.
Allelic differences in transcriptional activity
A common approach to examine variants for effects on transcriptional activity is a reporter assay. DNA segments of tens to thousands of base pairs containing individual allele(s) of a variant or haplotype are cloned into a vector containing a reporter gene, such as luciferase, whose activity is easily measured, and transfected into cells expected to express relevant transcription factors. The relative luciferase activity is compared between alleles and to a control lacking the inserted DNA segment (Figure 1).
Figure 1. Recent strategies for identifying regulatory variants and elements using functional genomics assays.
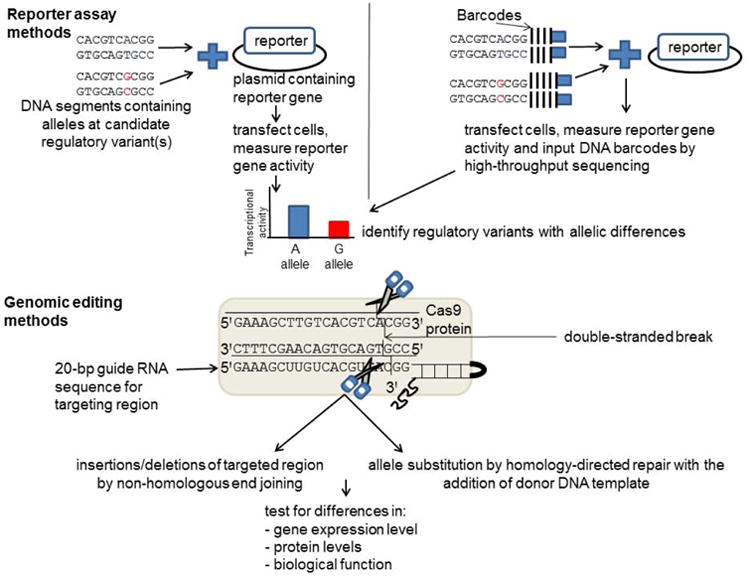
DNA segments can be cloned into a vector containing a reporter gene, one at a time or in a library. Reporter activity is then measured and compared to the reporter activity of a control vector or input DNA. These assays can be used to identify variants that show allelic differences in transcriptional activity. Candidate regulatory variants and their effect on gene expression and function can be further investigated by genomic editing methods. The DNA segment containing the candidate regulatory variant(s) can be targeted using CRISPR-Cas9 methods and a guide RNA to generate double-stranded breaks. Insertions or deletions can be generated by non-homologous end joining, or allelic substitutions can be generated by homology-directed repair with the addition of a donor DNA template. The effects of these edits can be tested by evaluating gene expression levels, protein levels, and biological function.
Recently, individual or multiple regulatory variants have been shown to influence allelic differences in transcriptional activity at GWAS loci for lipid traits or coronary artery disease. At an association signal for triglycerides near TMEM241, the rs17259126-A allele showed higher transcriptional activity than the rs17259126-G allele and also stronger HNF4A protein binding [25]. The authors proposed that the rs17259126-G allele is associated with lower TMEM241 expression, leading to higher triglyceride levels. At an association signal for high density lipoprotein cholesterol (HDL-C) near ANGPTL8, the rs12463177-C allele was associated with lower ANGPTL8 expression, showed lower transcriptional activity in reporter assays, and decreased protein binding in electrophoretic mobility shift assays (EMSAs) [26]. At an association signal for coronary artery disease near GUCY1A3, the rs7692387-A allele showed higher transcriptional activity than the rs7692387-G coronary artery disease risk allele. Individuals homozygous for the risk allele showed lower expression of GUCY1A3 (encoding [alpha]1 subunit of soluble guanylyl cyclase (sGC)) in whole blood. In addition, human platelet-rich plasma samples homozygous for the risk allele showed a reduced effect on inhibition of both sGC stimulation and induced platelet aggregation [27]. Finally, at an association signal for coronary artery disease near PPAP2B (also known as PLPP3, encoding LPP3 protein), stimulating primary human macrophages with oxidized LDL led to more strongly increased transcriptional enhancer activity for the rs72664324-A allele compared to the rs72664324-G allele [28]. In addition, the rs72664324-G risk allele was associated with lower PPA2B expression (induced by oxidized LDL) in primary human macrophages. The authors hypothesized that lower expression results in reduced enzymatic function, which affects pro-inflammatory signaling in atherosclerotic plaques. For all of these examples, one or more chromatin annotations were used to select variants to test for regulatory activity, and further studies of regulatory and biological mechanisms would increase the rigor of the conclusions.
Transcriptional activity assays have also shown that more than one variant on the same haplotype can have allelic effects. At an HDL-C association signal near GALNT2, candidate variants were tested for allelic or haplotype differences in transcriptional activity regardless of chromatin annotation [24]. Two variants, rs2281721 and rs4846913, that are in strong linkage disequilibrium with each other (r2=0.96) and located ∼2 kb apart, showed strong allelic differences in transcriptional enhancer activity that matched the direction of variant association with GALNT2 expression level in adipose and liver. The alleles associated with increased HDL-C levels were associated with increased GALNT2 expression level and increased transcriptional enhancer activity. These data are consistent with subsequent evidence in rodents, non-human primates and humans that loss of the glycoprotein modifications by GALNT2 on target proteins, including phospholipid transfer protein (PLTP), led to lower HDL-C levels [29]. These variants also showed differential binding to USF1 and CEBPB in EMSA and ChIP assays. These findings suggest that multiple variants may act together to contribute to transcriptional activity and the underlying molecular mechanism(s) at GWAS lipid loci.
Overall, transcriptional reporter assays are valuable to demonstrate that specific nucleotides can increase or decrease transcriptional activity, especially in enhancer regions. At some loci, including a stimulus can better capture allelic differences in regulatory activity and simulate how variants act in vivo. Transcriptional reporter assays for individual variants are feasible to implement in a research laboratory with moderate cost and minimal computational requirements. However, these assays examine DNA segments and variants subcloned into vectors, removing the region from its natural chromatin conformation and cellular context. In addition, these assays are low-throughput, allowing only a few DNA variants to be analyzed concurrently.
High-throughput screens for regulatory variants
The advent of high-throughput, massively parallel reporter assays (MPRAs) have enabled multiple variants and regulatory regions to be analyzed more rapidly and efficiently. These methods typically use a barcode or tag at the end of the reporter gene to facilitate high-throughput sequencing and quantification of expressed sequence reads [30–33]. In another design, expression level of the regulatory region itself is assayed [34,35].
Recent studies have applied MPRA methods to lipid and coronary artery disease loci. Tewhey and colleagues used MPRA in lymphoblastoid cell lines to test variants associated with gene expression levels. Tested variants included 9,664 variants at 163 GWAS loci for a wide range of traits; of these, 248 variants (2.6%) exhibited allelic differences. For example, at an association signal for coronary artery disease near UBE2Z, a screen of 105 variants in MPRAs detected eight variants that showed allelic differences, one of which, rs4378658, is also located in an ENCODE-annotated regulatory region [36]. This study also reported allelic differences in MPRA for rs342468 near AFF1 at a triglycerides locus. In another study, Pashos and colleagues used MPRAs in NIH 3T3 fibroblasts to screen candidate functional variants at lipid GWAS loci that were also associated with gene expression levels in hepatocyte-like cells. They screened 525 variants across three loci and reported the single variant at each locus that showed the strongest evidence of allelic differences [37]. Among the variants at the three loci, rs10872142 also overlapped regulatory chromatin annotations in human liver and adipose cells (Figure 2). rs10872142 is in strong linkage disequilibrium (r2=0.97, Europeans) with a lead variant associated with LDL-C, rs11153594, and the rs11153594-C allele associated with increased LDL-C pairs with the rs10872142-C allele. Induced pluripotent stem cells homozygous for the rs10872142-C allele showed higher FRK expression compared to cells heterozygous (A/C) at rs10872142. Variants detected by MPRA screens still require further experimental validation.
Figure 2. Chromatin regulatory annotations predict rs10872142 is located within an enhancer.
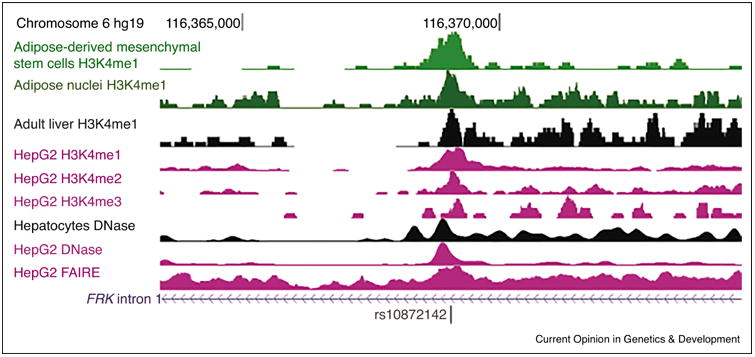
, rs10872142 is strongly linked with LDL-C-associated lead GWAS variant rs11153594 and was reported as a regulatory variant associated with FRK gene expression in liver [37]. H3K4me1 ChIP-seq peaks are shown for adipose-derived mesenchymal stem cells and primary adipose nuclei (top green tracks) from the NIH Epigenomics Roadmap Consortium's Human Epigenome Atlas [10]. H3K4me1, H3K4me2, or H3K4me3 represent mono-, di- or tri-methylation of lysine 4 of histone H3, and are modifications commonly observed in regulatory enhancer or promoter regions [18,19]. ENCODE H3K4me1,2,3 ChIP-seq, DNase, and FAIRE peaks are shown for HepG2 hepatocellular carcinoma cells (purple tracks), and H3K4me1 ChIP-seq and DNase peaks are shown for human adult liver and hepatocytes (black tracks) [9]. The DNase and FAIRE peaks represent open, accessible chromatin regions devoid of histones, and the peaks corresponding to H3K4me1, H3K4me2 and H3K4me3 marks on the adjacent histones are commonly observed at regulatory enhancer and/or promoter regions. The multiple tracks provide greater evidence that the variant is located within a regulatory region.
Taken together, MPRAs can be useful at narrowing down and identifying GWAS regulatory variants. These assays can be especially beneficial if there are numerous candidate functional variants at an association signal, because they enable a large number of variants to be screened in an experiment. MPRAs require a greater cost per experiment but cost substantially less per variant than assays of individual variants. However, limitations to MPRAs remain, as study designs may not successfully capture all potential regulatory variants, leading to false negatives, and the assays still remove putative regulatory regions from their natural chromatin conformation and cellular context, which may be necessary to observe functional consequences. MPRAs have a larger computational requirement to design reagents and to analyze high-throughput sequencing reads compared to individual transcriptional reporter assays. Additionally, while MPRAs can help detect regulatory variants that affect transcriptional activity, they would not identify variants that affect integrated processes such as mRNA splicing.
Genomic editing to characterize regulatory variants and elements
Another exciting method to examine the effect of regulatory elements and variants at GWAS loci is genomic editing followed by assays of gene expression and/or gene function (Figure 1). Genomic editing can be achieved through using clustered regularly interspaced short palindromic repeats (CRISPR) technology, guide RNAs, and Cas9 nuclease protein to create double-stranded breaks at target DNA sequences [38–40]. The double-stranded breaks can be repaired by non-homologous end-joining (NHEJ), which is more error-prone and often results in insertions or deletions of the targeted sequence, or by homology-directed repair, which repairs the break using template DNA, yet is less efficient [41]. Using the CRISPR-Cas9 system, one can disrupt or delete a regulatory element or substitute variant allele(s). Isolated clones of cells containing heterozygous or homozygous edits, or in some cases, pools of edited cells or organisms, can be analyzed to assess the effects on gene expression and/or gene function [42,43].
Genome editing has been used recently to delete regulatory elements at GWAS signals for lipid traits and coronary artery disease. At ANGPTL3/DOCK7, a well-established locus that alters blood cholesterol and triglyceride levels [44,45], Pashos and colleagues deleted ∼40 bp spanning candidate regulatory variant rs10889356 in pluripotent stem cell lines. In undifferentiated cells, deletion of the putative regulatory element reduced expression of DOCK7, and after differentiation into hepatocyte-like cells, deletion-containing cells showed both decreased DOCK7 expression and increased ANGPTL3 expression [37]. Nonsense mutations in ANGPTL3 have been found in individuals with combined hypolipidemia [44]. At an association signal for multiple traits including coronary artery disease, Gupta and colleagues deleted ∼90 bp spanning rs9349379 in pluripotent stem cells. After differentiation into endothelial and vascular smooth muscle cells, deletion-containing cells showed increased expression of EDN1 compared to wild-type cells [46]. Both experiments served to validate the target gene and direction of effect of the predicted regulatory element.
Editing cells or organisms to create allele substitutions can provide an unambiguous test of a variant's effect. Pashos and colleagues replaced the rs10872142-C allele with the rs10872142-A allele in an induced pluripotent stem cell line and observed decreased FRK expression [37]. Gupta and colleagues performed two steps of genome editing to create stem cell lines homozygous for either the rs9349379-A or G allele to further validate the variant effect on EDN1 gene and protein levels. The authors found that endothelial cells homozygous for the coronary artery disease risk allele rs9349379-G showed higher EDN1 expression and higher levels of the encoded vasoconstrictor protein, which they hypothesize results in the increased disease risk [46].
Overall, genome editing provides a valuable and precise tool to elucidate effects of genetic variation on genes. Generating deletions and/or allelic substitutions allows variant or element effects to be evaluated in their genomic and cellular context, in contrast to approaches that employ transient transfection or transduction of exogenous DNA and reporter gene vectors. The ability to create deletions and substitutions allows multiple types of variants, including splicing variants, to be examined. Despite the utility of studying variant substitutions, the relatively inefficient homology-directed repair pathway still presents challenges in evaluating allelic effects of individual or multiple variants.
Conclusions and perspective
The discovery of hundreds of GWAS loci has provided an unparalleled opportunity to better understand the molecular basis of complex disease, yet for many loci, the underlying genes and mechanisms remain unknown. The many noncoding disease risk variants may not necessarily affect expression of the nearest gene, may act on more than one gene, and may increase or decrease gene expression. These points highlight the value of functional assays to investigate the molecular mechanisms responsible for GWAS associations. Functional genomics regulatory annotation data combined with experimental assays, such as transcriptional reporter assays in a cellular context and genome editing, can pinpoint regulatory regions and/or variants at GWAS signals.
Recent functional studies have demonstrated the complexity of regulatory variant contributions to GWAS loci. Transcriptional reporter assays, especially MPRAs, have demonstrated that many variants located in annotated regulatory elements do not exhibit effects on transcriptional activity and that variants outside annotated elements can show effects on activity [36]. These results suggest that many potential regulatory variants do not have functional consequences and/or transcriptional assays may be imperfect. While chromatin annotations can be useful in guiding the selection of candidate regulatory variants to test in experimental assays, these annotations shouldn't be used exclusively to define which variants are regulatory. In addition, while single functional variants have been implicated at some loci, MPRAs often detect multiple variants that exhibit significant allelic or haplotype differences in transcriptional activity [35,36], and rigorous functional studies have demonstrated that more than one variant on the same haplotype can affect gene regulation [26,24]. Furthermore, variants on different haplotypes, often detected as different association signals, can affect regulation or function of the same or potentially different genes.
An important study design consideration for functional assays to characterize GWAS loci is cell type. Cell lines are readily available, relatively straight-forward to maintain, and proliferate, enabling large-scale and repeatable studies. However, some regulatory elements and variants may only act in a specific cell or tissue, or under specific environmental conditions or stimuli [28,37,47], neither of which might be apparent from the phenotype or expression pattern of candidate genes. Testing candidate regulatory variants in pluripotent stem cells that can be differentiated into appropriate cell types is becoming more routine. Cell type may also be important for measuring the effects of altered gene expression on biological function such as cholesterol synthesis or atherosclerotic plaque formation.
Although we are starting to understand the roles of individual variant and genes at GWAS loci, many loci remain poorly understood. Additional and improved high-throughput methods are needed to more efficiently screen thorough sets of candidate variants, yet screens alone will likely remain insufficient, and rigorous validation of variant and gene effects will be required. Due to of the limitations of each individual assay, a combination of assays will be especially valuable to provide more lines of evidence and a clearer picture into the molecular and biological mechanisms responsible for lipid traits and coronary artery disease.
Acknowledgments
Funding: The authors were supported by National Institutes of Health Grant Numbers R01DK072193, U01DK105561, and R01DK093757 (to K.L.M.). T.S.R. was also supported by T32HL069768.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
*of special interest
**of outstanding interest
- 1.Sharrett AR, Ballantyne CM, Coady SA, Heiss G, Sorlie PD, Catellier D, Patsch W Atherosclerosis Risk in Communities Study Group. Coronary heart disease prediction from lipoprotein cholesterol levels, triglycerides, lipoprotein(a), apolipoproteins A-I and B, and HDL density subfractions: The Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2001;104:1108–13. doi: 10.1161/hc3501.095214. [DOI] [PubMed] [Google Scholar]
- 2.Talayero BG, Sacks FM. The role of triglycerides in atherosclerosis. Curr Cardiol Rep. 2011 doi: 10.1007/s11886-011-0220-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, Yang J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet. 2017 doi: 10.1016/j.ajhg.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manolio TA. In Retrospect: A decade of shared genomic associations. Nature. 2017 doi: 10.1038/546360a. [DOI] [PubMed] [Google Scholar]
- 5.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci. 2009 doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L, Wingett SW, Andrews S, Grey W, Ewels PA, et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet. 2015 doi: 10.1038/ng.3286. [DOI] [PubMed] [Google Scholar]
- 7.Acemel RD, Maeso I, Gómez-Skarmeta JL. Topologically associated domains: a successful scaffold for the evolution of gene regulation in animals. Wiley Interdiscip Rev Dev Biol. 2017 doi: 10.1002/wdev.265. [DOI] [PubMed] [Google Scholar]
- 8.Hankowski KE, Hamazaki T, Umezawa A, Terada N. Induced pluripotent stem cells as a next-generation biomedical interface. Lab Investig. 2011 doi: 10.1038/labinvest.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.ENCODE Project Consortium. Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012 doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015 doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Javierre BM, Sewitz S, Cairns J, Wingett SW, Várnai C, Thiecke MJ, Freire-Pritchett P, Spivakov M, Fraser P, Burren OS, et al. Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell. 2016 doi: 10.1016/j.cell.2016.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stunnenberg HG, Abrignani S, Adams D, de Almeida M, Altucci L, Amin V, Amit I, Antonarakis SE, Aparicio S, Arima T, et al. The International Human Epigenome Consortium: A Blueprint for Scientific Collaboration and Discovery. Cell. 2016 doi: 10.1016/j.cell.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 13.Song L, Crawford GE. DNase-seq: A high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb Protoc. 2010 doi: 10.1101/pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007 doi: 10.1101/gr.5533506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013 doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017 doi: 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007 doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 18.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011 doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ernst J, Kellis M. ChromHMM: Automating chromatin-state discovery and characterization. Nat Methods. 2012 doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boyle AP, Song L, Lee BK, London D, Keefe D, Birney E, Iyer VR, Crawford GE, Furey TS. High-resolution genome-wide in vivo footprinting of diverse transcription factors in human cells. Genome Res. 2011;21:456–464. doi: 10.1101/gr.112656.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H. An oestrogen-receptor-α-bound human chromatin interactome. Nature. 2009 doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Berkum NL, Lieberman-Aiden E, Williams L, Imakaev M, Gnirke A, Mirny LA, Dekker J, Lander ES, Hi CA. J Vis Exp. 2010. A Method to Study the Three-dimensional Architecture of Genomes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmitt AD, Hu M, Jung I, Xu Z, Qiu Y, Tan CL, Li Y, Lin S, Lin Y, Barr CL, et al. A Compendium of Chromatin Contact Maps Reveals Spatially Active Regions in the Human Genome. Cell Rep. 2016 doi: 10.1016/j.celrep.2016.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24*.Roman TS, Marvelle AF, Fogarty MP, Vadlamudi S, Gonzalez AJ, Buchkovich ML, Huyghe JR, Fuchsberger C, Jackson AU, Wu Y, et al. Multiple Hepatic Regulatory Variants at the GALNT2 GWAS Locus Associated with High-Density Lipoprotein Cholesterol. Am J Hum Genet. 2015 doi: 10.1016/j.ajhg.2015.10.016. At an HDL-cholesterol GWAS signal, this study demonstrated that at least two variants on the same haplotype exhibited strong allelic differences in enhancer activity and transcription factor binding consistent with the direction of a colocalized association with gene expression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodríguez A, Gonzalez L, Ko A, Alvarez M, Miao Z, Bhagat Y, Nikkola E, Cruz-Bautista I, Arellano-Campos O, Muñoz-Hernández LL, et al. Molecular Characterization of the Lipid Genome-Wide Association Study Signal on Chromosome 18q11.2 Implicates HNF4A-Mediated Regulation of the TMEM241 Gene. Arterioscler Thromb Vasc Biol. 2016 doi: 10.1161/ATVBAHA.116.307182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cannon ME, Duan Q, Wu Y, Zeynalzadeh M, Xu Z, Kangas AJ, Soininen P, Ala-Korpela M, Civelek M, Lusis AJ, et al. Trans-ancestry Fine Mapping and Molecular Assays Identify Regulatory Variants at the ANGPTL8 HDL-C GWAS Locus. G3 (Bethesda) 2017 doi: 10.1534/g3.117.300088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27*.Kessler T, Wobst J, Wolf B, Eckhold J, Vilne B, Hollstein R, Von Ameln S, Dang TA, Sager HB, Rumpf PM, et al. Functional Characterization of the GUCY1A3 Coronary Artery Disease Risk Locus. Circulation. 2017 doi: 10.1161/CIRCULATIONAHA.116.024152. This study used chromatin annotation data and transcriptional reporterassays to identify a regulatory variant at the GUCY1A3 locus. This studyalso highlighted direction of effect, showing that lower GUCY1A3expression is associated with increased atherosclerosis in humans andmice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28*.Reschen ME, Gaulton KJ, Lin D, Soilleux EJ, Morris AJ, Smyth SS, O'Callaghan CA. Lipid-Induced Epigenomic Changes in Human Macrophages Identify a Coronary Artery Disease-Associated Variant that RegulatesPPAP2B Expression through Altered C/EBP-Beta Binding. PLoS Genet. 2015 doi: 10.1371/journal.pgen.1005061. This study used chromatin annotation data in primary human macrophages before and after stimulation with oxidized low-density lipoprotein. Analysis of differential chromatin peaks between unstimulated and stimulated cells identified a specific enhancer region at thePPAP2B locus containing a variant that showed allelic effects on transcriptional activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khetarpal SA, Schjoldager KT, Christoffersen C, Raghavan A, Edmondson AC, Reutter HM, Ahmed B, Ouazzani R, Peloso GM, Vitali C, et al. Loss of Function of GALNT2 Lowers High-Density Lipoproteins in Humans, Nonhuman Primates, and Rodents. Cell Metab. 2016 doi: 10.1016/j.cmet.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patwardhan RP, Lee C, Litvin O, Young DL, Pe'Er D, Shendure J. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat Biotechnol. 2009 doi: 10.1038/nbt.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patwardhan RP, Hiatt JB, Witten DM, Kim MJ, Smith RP, May D, Lee C, Andrie JM, Lee SI, Cooper GM, et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 2012 doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, Rogov P, Feizi S, Gnirke A, Callan CG, Kinney JB, et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 2012 doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwasnieski JC, Mogno I, Myers CA, Corbo JC, Cohen BA. Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc Natl Acad Sci. 2012 doi: 10.1073/pnas.1210678109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arnold CD, Gerlach D, Stelzer C, Boryń ŁM, Rath M, Stark A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 2013;339:1074–7. doi: 10.1126/science.1232542. doi:10.1126/science. 1232542. [DOI] [PubMed] [Google Scholar]
- 35.Vockley CM, Guo C, Majoros WH, Nodzenski M, Scholtens DM, Hayes MG, Lowe WL, Reddy TE. Massively parallel quantification of the regulatory effects of noncoding genetic variation in a human cohort. Genome Res. 2015 doi: 10.1101/gr.190090.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36**.Tewhey R, Kotliar D, Park DS, Liu B, Winnicki S, Reilly SK, Andersen KG, Mikkelsen TS, Lander ES, Schaffner SF, et al. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell. 2016 doi: 10.1016/j.cell.2016.04.027. This study used MPRAs to test 32,373 variants for regulatory activity and identified 842 variants showing allelic differences in expression. Among these variants were 53 variants associated with diseases and traits, including variants associated with triglycerides and coronary artery disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37**.Pashos EE, Park YS, Wang X, Raghavan A, Yang W, Abbey D, Peters DT, Arbelaez J, Hernandez M, Kuperwasser N, et al. Large, Diverse Population Cohorts of hiPSCs and Derived Hepatocyte-like Cells Reveal Functional Genetic Variation at Blood Lipid-Associated Loci. Cell Stem Cell. 2017 doi: 10.1016/j.stem.2017.03.017. This study incorporated MPRAs, disruption of regulatory elements, and genome editing of specific variants to perform functional studies and identify molecular mechanisms at three GWAS loci for lipid traits. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science (80-) 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science (80-) 2013 doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science (80-) 2013 doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Danner E, Bashir S, Yumlu S, Wurst W, Wefers B, Kühn R. Control of gene editing by manipulation of DNA repair mechanisms. Mamm Genome. 2017 doi: 10.1007/s00335-017-9688-5. [DOI] [PubMed] [Google Scholar]
- 42*.Ye J, Tucker NR, Weng LC, Clauss S, Lubitz SA, Ellinor PT. A Functional Variant Associated with Atrial Fibrillation Regulates PITX2c Expression through TFAP2a. Am J Hum Genet. 2016 doi: 10.1016/j.ajhg.2016.10.001. This study identified a regulatory variant, rs2595104, at an association signal for atrial fibrillation using both chromatin annotation data and transcriptional reporter assays. CRISPR-Cas9 methods were used to delete the region spanning rs2595104 and to create an allelic substitution of rs2595104. Both methods showed a reduction of PITX2c gene expression in human stem-cell-derived cardiomyocytes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43*.Roman TS, Cannon ME, Vadlamudi S, Buchkovich ML, Wolford BN, Welch RP, Morken MA, Kwon GJ, Varshney A, Kursawe R, et al. A type 2 diabetes-associated functional regulatory variant in a pancreatic islet enhancer at the ADCY5 locus. Diabetes. 2017:2521–2530. doi: 10.2337/db17-0464. This study described a GWAS variant with allelic effects on transcriptional activity and transcription factor binding consistent with the direction of a colocalized association with gene expression. Homozygous CRISPR-mediated deletions of the surrounding enhancer element validated the effect on gene expression and demonstrated an effect on Adcy5 gene function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Musunuru K, Pirruccello JP, Do R, Peloso GM, Guiducci C, Sougnez C, Garimella KV, Fisher S, Abreu J, Barry AJ, et al. Exome Sequencing, ANGPTL3 Mutations, and Familial Combined Hypolipidemia. N Engl J Med. 2010 doi: 10.1056/NEJMoa1002926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu YX, Redon V, Yu H, Querbes W, Pirruccello J, Liebow A, Deik A, Trindade K, Wang X, Musunuru K, et al. Role of angiopoietin-like 3 (ANGPTL3) in regulating plasma level of low-density lipoprotein cholesterol. 2018 doi: 10.1016/j.atherosclerosis.2017.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46**.Gupta RM, Hadaya J, Trehan A, Zekavat SM, Roselli C, Klarin D, Emdin CA, Hilvering CRE, Bianchi V, Mueller C, et al. A Genetic Variant Associated with Five Vascular Diseases Is a Distal Regulator of Endothelin-1 Gene Expression. Cell. 2017 doi: 10.1016/j.cell.2017.06.049. These authors used multiple functional assays, including genomic editing of variant alleles, to show that rs9349379 is a regulatory variant affecting expression of endothelin-1 (EDN1) gene expression and protein levels at a GWAS signal for vascular disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Warren CR, O'Sullivan JF, Friesen M, Becker CE, Zhang X, Liu P, Wakabayashi Y, Morningstar JE, Shi X, Choi J, et al. Induced Pluripotent Stem Cell Differentiation Enables Functional Validation of GWAS Variants in Metabolic Disease. Cell Stem Cell. 2017 doi: 10.1016/j.stem.2017.01.010. [DOI] [PubMed] [Google Scholar]