

Abstract
Systemic juvenile idiopathic arthritis (sJIA) is a childhood rheumatic disease of unknown origin. Dysregulated innate immunity is implicated in disease pathology. We investigated if IL-1 inhibition affects circulating cytokines and monocyte gene expression. CD14+ monocytes from patients in the RAPPORT trial were analyzed by RT-PCR for expression of IL1B and transcription factors associated with monocyte activation. Serum IL-1ra decreased with treatment, and IL-18BP transiently increased. Serum levels of IL-1β, IL-6, IL-10 and IL-18 were unchanged. IRF5 and STAT6 were decreased, and PPARG was increased, independent of clinical response, and may represent a skew towards a PPARG-driven M2-like phenotype. IL1B expression was decreased in early clinical responders. A transient increase in STAT1, and a decrease in SOCS1 preceded the reduction in IL1B in early clinical responders. Changes in IL1B/STAT1/SOCS1 could be associated with crosstalk between IL-1 and IFN pathways in sJIA. These transcriptional changes might be useful as drug response biomarkers.
Keywords: Juvenile arthritis, Monocytes, Cytokine
1. Introduction
Systemic juvenile idiopathic arthritis (sJIA) is a chronic rheumatic condition of childhood, with strong similarity to adult onset Still’s disease; both are characterized by remitting fever, transient rash, and arthritis; sJIA etiology remains unknown [1]. Although the adaptive immune response may also contribute [2], several lines of evidence indicate that dysregulation of innate immunity, especially of the monocyte/macrophage lineage, plays a major role in sJIA pathology [1]. Activated monocytes constitute a significant component of the perivascular and synovial infiltrates observed during sJIA flares [3]. Several experimental studies of peripheral blood mononuclear cells show activation of innate immune cells, including upregulation of monocyte genes that cannot be explained solely by increase in monocyte number [4–8]. Exposure of monocytes from healthy donors to plasma or serum from sJIA patients induces expression of genes related to innate immunity and production of IL-1β and IL-6 [4, 9]. Additionally, the association of sJIA with macrophage activation syndrome (MAS) is a prominent, and potentially fatal, complication of the disease [10], although MAS also can occur in association with other conditions, including classical autoimmune diseases, such as SLE, and infections. Importantly, the exact nature of the immune dysfunction in sJIA is still unknown [1, 11].
The most compelling evidence for innate mediators, IL-1 and IL-6, as important drivers of the inflammation of sJIA, is the striking therapeutic responses to IL-1 or IL-6 blockade in patients [4, 12–16]. Treatment with anti-IL-1 medication is successful in treating a large percentage (close to 60%) of sJIA patients followed for up to 2 years [15, 17, 18]. It has been proposed that anti-IL-1 treatment may modify the course of the disease and reduce long term arthritis, if administered early in the disease [19–21].
We have observed a number of changes in circulating sJIA monocyte activation phenotype, with some features associated primarily with active disease [6, 7]. One aspect we have investigated is monocyte polarization. Polarization refers to distinct patterns of monocyte/macrophage activation, based on different types of stimulation. The extreme ends of the polarization spectrum have been named M1 and M2 [22, 23]. The M1/M2 paradigm has its shortcomings, given the complexity of monocyte/macrophage activation [24, 25], but it is a useful framework to compare monocyte phenotype across different situations. The classically activated M1 is more proinflammatory, whereas the alternatively activated M2 is associated with resolution of inflammation and tissue repair [23, 26]. Despite the inflammatory nature of sJIA, we and others have found increased expression of genes and cell markers associated with M2-like response in peripheral blood monocytes, in addition to markers associated with M1-like response, suggesting these M2-associated markers may reflect a compensatory response to active inflammation in tissues [6, 8, 27]. In addition to the polarizing cytokines, monocytes and macrophages are highly sensitive to other cues in their microenvironments [9, 23]. Indeed, based on transcriptional analysis, striking heterogeneity is the hallmark of the programs expressed by these myeloid cells [28, 29].
Do the sJIA circulating monocytes contribute to IL-1-driven pathogenic activity in sJIA or do their phenotypes reflect the influence of elevated IL-1 on these cells? The RAPPORT trial (Randomized Placebo Phase Study of Rilonacept in the Treatment of Systemic Juvenile Idiopathic Arthritis) offered a unique opportunity to investigate changes in circulating monocyte phenotypes in the context of changes in in vivo IL-1 modulated by IL-1 blockade. RAPPORT was a randomized, placebo-controlled trial of rilanocept, an IL-1 “trap” [30]. Rilonacept is an Fc region of the human IgG1 fused to the extracellular domains of the 2 components of the IL-1 receptor, IL-1R1 and the IL-1 receptor accessory protein (IL-1RAcP). As such, it can bind IL-1β, IL-1α and may also bind to the endogenous IL-1 inhibitor, IL-1ra. The RAPPORT clinical trial tested the short-term safety of rilonacept and its efficacy for articular and systemic manifestations of sJIA. The drug was well-tolerated and showed efficacy in treating active sJIA [30].
We hypothesized that IL-1 inhibition modifies the activation profile of circulating monocytes in sJIA. The activation profiles of monocytes/macrophages are shaped by transcription factors (TFs) that regulate downstream genes, determining the type of response manifest by the cell [28]. We examined a panel of key TFs associated with known monocyte activation profiles before and after treatment with rilonacept to determine the effect of IL-1 inhibition on these profiles. We also investigated whether specific changes in monocyte TF expression are associated with the clinical response to IL-1 blockade. We also investigated if circulating IL-1ra and other key cytokines previously implicated in sJIA [31] were altered by IL-1 inhibition.
2. Materials and Methods:
2.1. RAPPORT trial:
The Randomized Placebo Phase Study of Rilonacept in the Treatment of Systemic Juvenile Idiopathic (RAPPORT) was designed so that half the subjects were randomized to begin placebo for 4 weeks followed by active drug (“the placebo group”), while the other half received active drug without a placebo phase (“the rilonacept group”); the total duration of the trial was 24 weeks (6 months) [30]. Patients in the placebo group received placebo for 4 weeks and rilonacept for 20 weeks; patients in the rilonacept group received the drug for 24 weeks. A loading dose of the drug was administered at Day 0 (rilonacept) or Day 28 (Placebo), followed by weekly maintenance doses [30]. Patients continue to receive systemic corticosteroids during the trial. The trial enrolled 71 patients, below its planned enrollment of 100, with 57 completing the study. Inclusion/exclusion criteria and the baseline demographic and clinical characteristics of the study participants are described in Ilowite et al [30]. Time to response was the endpoint, based on the adapted American College of Rheumatology Pediatric 30 (ACR Pedi 30) criteria [32], together with absence of fever and tapering of the systemic steroids. Each subject was clinically evaluated every two weeks, starting from the beginning, for a total of 14 weeks, and then evaluated again at week 24. The median time to achieve the primary end point, defined by composite end point of an ACR Pedi 30 response, absence of fever and corticosteroid taper, was 4 weeks of treatment [30]. Based on this finding, for the purpose of this study we defined a patient as an early responder if the patient achieved ACR Pedi 30 (considered as a “Yes”) before 4 weeks or less of treatment, and continued to achieve ACR Pedi 30 or at least had more “Yes” at later visits (≥10 weeks in study) than earlier visits (<10 weeks in study). Patients were classified as late responders if it took more than 4 weeks of treatment to achieve ACR Pedi 30, or the patient did not achieve ACR Pedi 30 during the duration of the trial (24 weeks). We obtained samples from 35 patients (see below); 23 patients were classified as early responders and 12 as late responders.
2.2. Patients’ samples:
De-identified, coded frozen PBMC samples from patients participating in the RAPPORT trial were received from the Tissue Repository Unit at Cincinnati Children’s Hospital Medical Center (CCHMC, PI Susan Thompson). Total RNA isolated from CD14+ monocytes was received from the Baylor Institute for Immunology Research. Blood draws were performed at baseline (week 0), weeks 2, 4, 14 and 24 of the trial. PBMC samples or total RNA from a total of 35 individual patients were obtained; we did not obtain serial samples from all patients. Serum samples were obtained for ELISA assays from a total of 45 patients. Only samples from the RAPPORT study were analyzed, to avoid introduction of sample collection and processing bias. This study was approved by the Administrative Panels on Human Subjects Research from Stanford University.
2.3. ELISA for serum cytokines:
IL-1 receptor antagonist (IL-1ra) and IL-8 levels in serum were measured by kinetic sandwich ELISA (Peprotech, Rocky Hill, NJ, USA). Serum levels of IL-1β, IL-6 and IL-10 were measured using Cytometric Bead Array (CBA) (BD Biosciences, San Jose, CA). IL-18 serum levels were measured using Human IL-18 Platinum ELISA (Affymetrix/eBioscience/ThermoFisher Scientific); IL-18 Binding Protein (BP) was measured using IL-18BP ELISA from Abcam (Cambridge, MA).
2.4. Monocyte isolation:
Monocytes were isolated from frozen peripheral blood mononuclear cells (PBMCs) from RAPPORT subjects by positive selection with CD14 microbeads (Miltenyi Biotec, San Diego, CA, USA). Positive selection was chosen as the method of isolation that provided the highest yield and level of purity. Dead cells were removed prior to monocyte isolation using the Dead Cell Removal Kit (Miltenyi Biotec). Isolated monocytes were assessed for purity using flow cytometry. The mean percentage of CD14+ monocytes was 84% (range 60–99%) in post-sort samples.
2.5. Genes analyzed:
CD14+ monocyte expression of the following genes was analyzed: Interleukin-1beta (IL1B) and the ATP Purinergic Receptor P2X7 (P2RX7), involved in IL-1β secretion [33]. Transcription factors (TF) and related genes: M1-associated TF: Interferon Regulatory factor (IRF) family 5 (IRF5); Signal transducers and activators of transcription (STAT) family 1 (STAT1) and its negative regulator suppressor of cytokine signaling 1 (SOCS1); M2 associated genes: IRF4, STAT6, Kruppel-Like Factor 4 (KLF4), and peroxisome proliferator-activated receptor gamma (PPARG). Primer sequences and brief description of genes analyzed are on Table 1. Housekeeping genes: eukaryotic translation elongation factor 1 alpha1 (EEF1A1), protein phosphatase 1, catalytic subunit, gamma isoform (PPP1CC) [27], and Ribosomal Protein L12 (RPL12). Primer sequences for all genes were obtained from primerdepot.nci.nih.gov/ and synthesized at Stanford University Pan Facility.
Table 1:
Gene information and primer sequences.
| Gene symbol | Function(s) in monocyte/macrophages | Right primer sequence | Left primer sequence |
|---|---|---|---|
| IL1B | Cytokine produced by activated monocytes/macrophages | AAG CCC TTG CTG TAG TGG TG | GAA GCT GAT GGC CCT AAA CA |
| P2RX7 | Cell surface receptor for extracellular ATP; involved in IL-1β processing and release through activation of NLRP3 infammasome | GCT TGT CAC TCA CCA GAG CA | CTC GGA TCC AGA GCA TGA AT |
| M1-related genes | |||
| IRF5 | Induces expression of IL-12 p40 subunit; suppresses IL-10 expression with GM-CSF | GAT GGA CTG GTT CAT GGC AG | CAG AGC TCA GCT TGG TCC C |
| STAT1 | Induced by ΙFΝγ stimulation | TGA ATA TTC CCC GAC TGA GC | AGG AAG ACC CAA TCC AGA TGT |
| SOCS1 | Inhibitor of STAT1 signaling | CAC ATG GTT CCA GGC AAG TA | CTA CCT GAG CTC CTT CCC CT |
| M2-related genes | |||
| IRF4 | Promotes expression of several M2-related genes in a chitin model (helminth derived), or in response to M-CSF; negative regulator of TLR signaling; increased expression with IL-4 stimulation | GAG TCA CCT GGA ATC TTG GC | CCT GCA AGC TCT TTG ACA CA |
| KLF4 | Induced by IL-4 via STAT6; may inhibit expression of M1-induced genes | GTC AGT TCA TC TGA GCG GG | AGA GTT CCC ATC TCA AGG CA |
| PPARG | Induced by IL-4; PPARG activity amplified via STAT6 | GAG AGATCC ACG GAG CTG AT | AGG CCA TTT TGT CAA ACG AG |
| STAT6 | Activated by IL-4; essential role in M2-type polarization | ACT TTT TCT GGG GGC ATC TT | AGA AGA CAG CAG AGG GGT TG |
2.6. RT-PCR:
Total RNA from isolated monocytes was obtained using RNeasy Mini or Micro kit (Qiagen, Germantown, MD, USA) depending on monocyte number, and transcribed into cDNA using iScript Reverse Transcriptase (Bio-Rad, Hercules, CA, USA). cDNA (in duplicate) was amplified using SsoAdvanced™ SYBR® Green Supermix in a CFX384 qPCR machine (Bio-Rad). Relative amounts of transcripts for each gene were determined by the standard curve method, and adjusted for expression of housekeeping genes (combined expression of PPICC, EEF1A1, and RPL12). Each primer set was tested for assay efficiency, amplification of residual DNA and the profile of the melting curve. Additionally, cDNA from isolated monocytes from a single donor was used as a calibrator throughout the study.
2.7. Statistical analysis:
All statistical analyses were performed with IBM SPSS Statistics version 21. We obtained descriptive statistics from all data analyzed. We analyzed that data using a mixed effects regression model for repeated measures [34]. This approach allowed the missing data at some time points and accounted for partial pairing. To analyze if IL-1 inhibition, irrespective of clinical response, lead to change in any variable (cytokine or gene) tested, we analyzed for changes from baseline using time as a fixed effect. For analysis of effect of clinical response on the variables tested, we analyzed the early and late responder groups separately for changes during the duration of treatment. As this is an exploratory study, unpaired 2-group comparisons were also performed as described in the text, so as not to overlook any differences that may be small, but potentially biologically meaningful [35].
3. Results
3.1. Serum IL-1ra drops shortly after start of anti-IL-1 treatment
Levels of IL-1ra, the endogenous IL-1 receptor antagonist, positively correlate with IL-1 activity [36]. To assess whether treatment with rilonacept effectively reduces IL-1 activity, we measured the levels of circulating IL-1ra. In comparison to baseline, levels of IL-1ra dropped significantly in the rilonacept group, 2 weeks after initiation of treatment, which was the earliest time point measured (Fig. 1A, Table 2). The level of IL-1ra stayed significantly below baseline levels in the rilonacept group throughout the 24 weeks (Fig. 1A, Table 2). For the placebo group, levels changed little during the placebo phase (weeks 0 to 4), decreasing significantly with treatment by weeks 14 and 24 of the trial (weeks 10 and 20 of the treatment) (Fig. 1B, Table 2). We also assessed circulating IL-1β levels, which were mostly close to the detection level of the assay (4 pg/ml), with no significant change during the trial (Table 2). These results are in line with the literature [37, 38] and our own data on an unrelated sJIA cohort (Macaubas et al, unpublished), which found that the levels are close to the assay detection levels, even during sJIA disease activity.
Figure 1: Serum IL-1ra decreases after 2 weeks of rilonacept treatment.
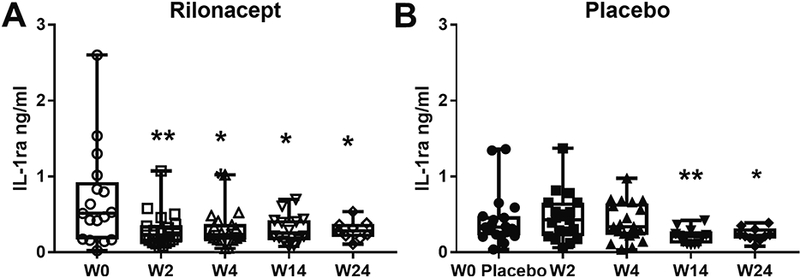
IL-1ra was detected in serum samples from patients in the RAPPORT trial as described in Material and Methods; (A), rilonacept group and (B), placebo group. Serum samples from a total of 45 patients were analyzed; samples were obtained at baseline, and week 2, 4, 14 and 24 of the trial; not all time points were available from all patients. Boxplots extend from the 25th to 75th percentiles, and the whiskers extend from the minimum to the maximum point; middle lane represents the median. Statistical analysis used mixed effects regression model. P values associated with type III tests of fixed effects. One symbol, p<0.05, two symbols, p≤0.01 in relation to baseline.
Table 2:
Cytokine levels in serum of RAPPORT samples
| Cytokine | Placebo Group | Rilonacept Group | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Week 0 |
Week 2 |
Week 4 |
Week 14 |
Week 24 |
Week 0 |
Week 2 |
Week 4 |
Week 14 |
Week 24 |
|
| IL-1ra | 330 (222-477) | 429 (210-657) | 336 (228-647) | 207 (114.8-276.8)* | 222 (182-315.3)* | 514 (171-925) | 228 (137-357)* | 231 (165.8-378.8)* | 253.5 (160.5-424.5)* | 285 (201-376)* |
| IL-1β | 5.5 (4.7-17.9) | 5.2 (4.2-11.9) | 7.1 (5-18.7) | 5 (4.2-5.9) | 12.4 (4.8-39.4) | 5.2 (4.6-8.2) | 6.9 (4.4-9.4) | 9.1 (5.5-20.3 | 5.3 (5.3-12.2) | 6.3 (5-10) |
| IL-6 | 65.6 (23-359) | 48.4 (24-75.3) | 51.3 (26-178.9) | 30.5 (22.6-103.7) | 41.3 (26.1-120.1) | 85.9 (31-150.8) | 40.2 (24.2-121) | 46.4 (24.9-100.2) | 75.9 (27.2-84.4) | 67.4 (33.7-118.7) |
| IL-8 | 72 (33-312) | 165 (48-825) | 376.5 (43.5-753) | 141 (38.3-9229 | 540 (72-477.2) | 177 (58.5-1823) | 111 (33-462.5) | 120 (29.3-1092) | 148.5 (26.3-514.5) | 384 (120-7298) |
| IL-10 | 15.8 (12.4-24.3) | 13.3 (11.8-16.9) | 13.1 (12-18.9) | 13.1 (11.6-14.7) | 12.7 (11.9-15.4) | 13.2 (10.9-17.4) | 14.5 (13.1-18) | 14 (12.5-17.9) | 12.8 (11.9-16.7) | 13.6 (13-15) |
| IL-18 | 849.5 (63.8-2446) | 637.5 (92.5-3246) | 970 (254.5-2749) | 1599 (820.5-4947) | 1001 (56.3-2295) | 412 (41.5-8102) | 1241 (177-2210) | 2498 (66-7652) | 2381 (1322-5919) | 1789 (283-5640) |
| IL-18BP | 58.5 (24.8-155.7) | 56.9 (29.9-109.7) | 63.6 (0.7-75.4) | 64.6 (2-146.8) | 40.7 (19.1-123.6) | 31.2 (5.9-106.2) | 60.8 (1-135.1) | 165.9 (55.3-334.3)* | 234.7 (59.7-778.2) | 93.6 (0.4-205.4 |
Values expressed as median and interquartile range; unit: pg/ml
Statistical analysis used mixed effects regression model. P values associated with type III tests of fixed effects. One symbol,
p<0.05 in relation to baseline.
We also measured serum levels of IL-6, another cytokine involved in sJIA. We found no significant difference in circulating levels during the trial (Table 2). Another cytokine, IL-8, has been reported to respond to anti-IL-1 treatment [39], although in a short time frame. We did not observe a significant change in serum IL-8, suggesting that rilonacept treatment in sJIA does not alter this cytokine (Table 2). We also analyzed levels of IL-10, a regulatory cytokine often associated with suppression of immune response [40]; levels of serum IL-10 were unchanged throughout the trial (Table 2).
We also assessed levels of circulating IL-18 and IL-18 BP in a smaller set of samples (N=32). Elevated serum IL-18 is associated with sJIA [41], and treatment with IL-18BP, which binds IL-18 with high affinity and blocks its activity, is a potential treatment for sJIA [12]. Levels of serum IL-18 did not change with rilonacept treatment (Table 2). Levels of IL-18BP were increased at week 4 compared to baseline in the rilonacept group (Table 2); levels remains elevated in relation to baseline at week 14 but did not reach significance. By week 24, levels of IL-18BP were closer to baseline in the rilonacept group; no significant changes were observed in the placebo group (Table 2). It has been shown that IL-18 binding obscures measurement of IL-18BP [42], so the levels reported here are likely underestimated.
3.2. IL-1 inhibition decreases IL1B gene expression in CD14+ monocytes
We initially tested if IL-1 inhibition, irrespective of clinical response, leads to changes in gene expression in CD14+ monocytes. To perform this analysis samples were grouped based solely on duration (weeks) of treatment with rilonacept. The groups were weeks 0–4 (baseline), 10 (for placebo group) or 14 weeks (for the rilonacept group) and 20 (for placebo group) and 24 weeks (for the rilonacept group). There were no statistically significant difference s at baseline between the placebo and rilonacept groups for any of the parameters tested (p > 0.05).
We found that expression of IL1B in CD14+ monocytes was decreased significantly after 10–14 weeks and 20–24 weeks of rilonacept treatment (Fig. 2A), compared to baseline. We also measured expression of P2RX7, a purinergic receptor that recognizes ATP and mediates a range of functions, including inflammasome activation for IL-1 processing and secretion [33]. A significant decreased expression of P2RX7 compared to baseline was observed at weeks 20–24 of treatment (Fig. 2B).
Figure 2: Monocyte IL1B, STAT6 and IRF5 expression is reduced, and PPARG expression increased, after 20–24 weeks of rilonacept treatment.
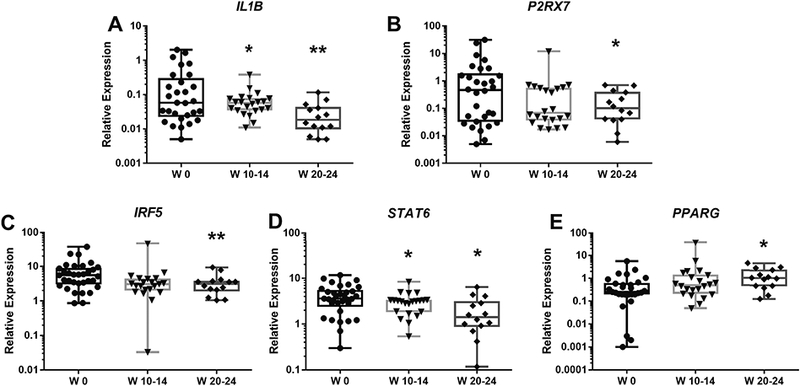
Total RNA was extracted from peripheral blood CD14+ monocytes; gene expression was determined using RT-PCR, as described in Material and Methods. X-Axis indicates the number of weeks (W) of treatment with rilonacept. Boxplots extend from the 25th to 75th percentiles, and the whiskers extend from the minimum to the maximum point; middle lane represents the median. Y-axis shows log10 scale. Statistical analysis used mixed effects regression model. P values associated with type III tests of fixed effects. One symbol, p<0.05; two symbols, p≤0.01 in relation to baseline.
3.3. IRF5 and STAT6 expression decreases and PPARG increases following IL-1 inhibition
We chose to analyze a panel of TFs, based on their known roles in regulating monocyte/macrophage functional phenotype (Table 1). Monocyte expression of IRF5, a M1-related TF, was significantly decreased after 20–24 weeks of treatment (Fig. 2C). STAT6 (a M2-related TF) expression by CD14+ monocytes was significantly decreased after 10–14 and 20–24 weeks of treatment (Fig. 2D). The only TF that showed a statistically significant increase with rilonacept treatment was PPARG. The elevation in PPARG expression achieved statistical significance after 20–24 weeks of treatment (Fig. 2E) in comparison to baseline.
3.4. Expression of other TFs analyzed did not change with IL-1 inhibition
We found that expression of the M1-related TF STAT1 and its negative regulator SOCS1 did not show significant changes from the baseline during the follow-up period (Fig. 3A, B). Similarly, the M2-related TFs IRF4 and KLF4 changed little from baseline throughout the treatment weeks (Fig. 3C, D)
Figure 3: Expression of STAT1, SOCS1, KLF4 and IRF4 does not change after rilonacept treatment.
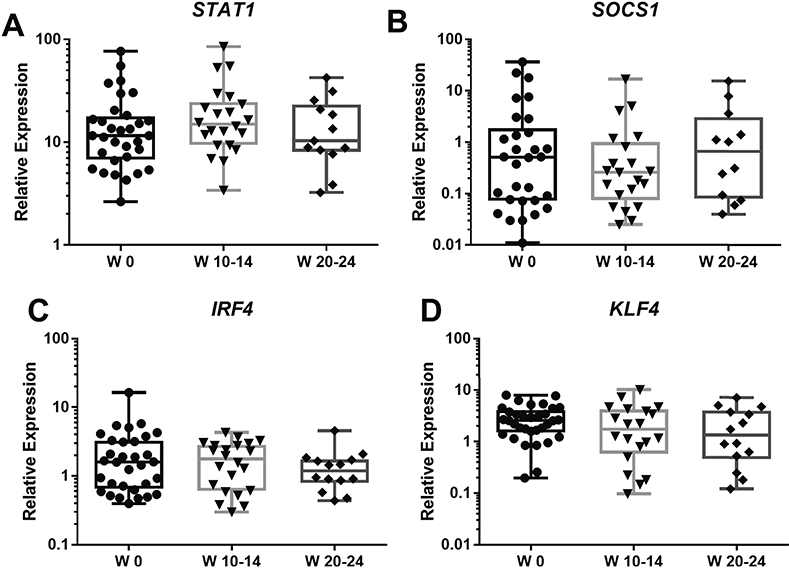
Total RNA was extracted from peripheral blood CD14+ monocytes; gene expression was determined using RT-PCR, as described in Material and Methods. X-Axis indicates the number of weeks (W) of treatment with rilonacept. Boxplots extend from the 25th to 75th percentiles, and the whiskers extend from the minimum to the maximum point; middle lane represents the median. Y-axis shows log10 scale. Statistical analysis used mixed effects regression model. All comparisons p>0.05, and deemed non-significant.
3.5. Decreased expression of IL1B in CD14+ monocytes in early treatment responders
We next compared gene expression changes in patients who achieved an early or late clinical response to treatment, defined as achieving ACR Pedi 30 before (early) or after (late) 4 weeks of treatment (See ‘Rapport trial’ under Material and Methods). There were no statistically significant differences between early responders and late responders at baseline for any of the genes analyzed. After 20–24 weeks of treatment, IL1B expression decreased significantly in CD14+ monocytes of early responders, in comparison to baseline (Fig. 4A). In late responders, however, IL1B expression showed almost no change throughout the time that the samples were analyzed (Fig. 4A). Group to group comparison for week 20–24 time point show that IL1B expression was significantly lower in early responders in comparison to late responders (p<0.05). For P2RX7 expression, both groups showed a tendency to decreased expression without reaching statistical significance (Fig. 4B).
Figure 4: IL1B expression is decreased in rilonacept early responders, while STAT1 and SOCS1 are transiently affected with rilonacept treatment.
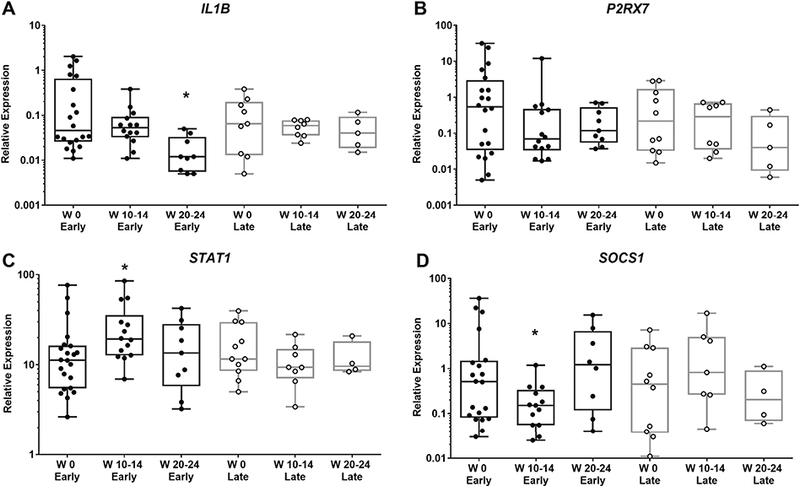
Total RNA was extracted from peripheral blood CD14+ monocytes; gene expression was determined using RT-PCR, as described in Material and Methods. X-Axis indicates the number of weeks (W) of treatment with rilonacept. Early (responders): achieved ACR Pedi 30 before 4 weeks or less of treatment; Late (responders): more than 4 weeks of treatment to achieve ACR Pedi 30. Boxplots extend from the 25th to 75th percentiles, and the whiskers extend from the minimum to the maximum point; middle lane represents the median. Y-axis shows log10 scale. Statistical analysis used mixed effects regression model. P values associated with type III tests of fixed effects. One symbol, p<0.05, in relation to baseline of specific responder group.
3.6. Transient changes in STAT1 and SOCS1 expression in early treatment responders
Analyzing samples based on clinical response to treatment showed that STAT1 was significantly increased in CD14+ monocytes of early responders at weeks 10–14, in comparison to baseline; there was no change for late responders. Group to group comparisons showed that STAT1 expression in early responders was significantly higher than in late responders at week 10–14 of treatment (p<0.05, Fig. 4C). The increase in STAT1 expression in early responders was transient, with values at week 20–24 closer to baseline. Concomitantly, SOCS1, the inhibitor of STAT1, was significantly lower in CD14+ monocytes of early responders at the same time point as elevated STAT1 (weeks 10–14 compared to baseline); no changes were observed for late responders (Fig. 4D). Using group to group comparisons, expression of SOCS1 at weeks 10–14 was lower in early responder in comparison to late responders (p<0.05). At weeks 20–24, there were no statistical differences between early and late responders.
3.7. IRF5, STAT6 and PPARG expression changes are independent of treatment response
Analysis of the samples based on treatment response showed a similar pattern for IRF5 and STAT6 expression in early and late responders, with a tendency toward decreased expression after 20–24 weeks (Fig. 5A, B). Similarly, for PPARG expression, there is a tendency for increased expression of PPARG in CD14+ monocytes in both early and late responders (Fig. 5C).
Figure 5: STAT6 and PPARG expression changes similarly in responders and nonresponders.
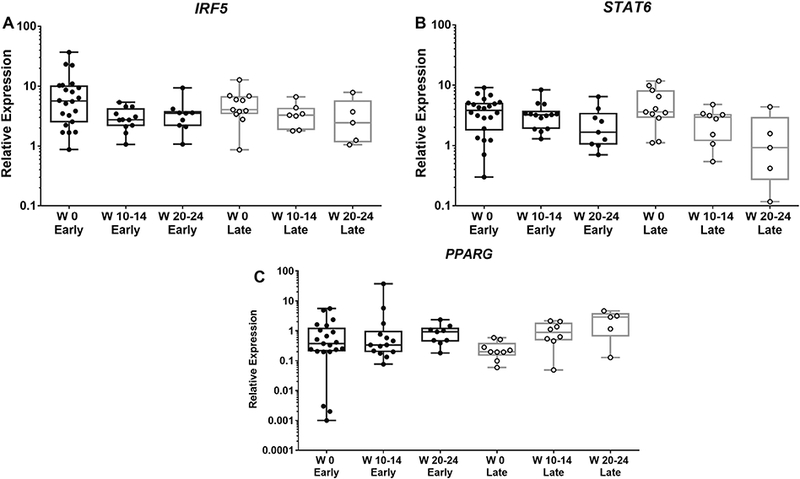
Total RNA was extracted from peripheral blood CD14+ monocytes; gene expression was determined using RT-PCR, as described in Material and Methods. X-Axis indicates the number of weeks (W) of treatment with rilonacept. Early (responders): achieved ACR Pedi 30 before 4 weeks or less of treatment; Late (responders): more than 4 weeks of treatment to achieve ACR Pedi 30. Boxplots extend from the 25th to 75th percentiles, and the whiskers extend from the minimum to the maximum point; middle lane represents the median. Y-axis shows log10 scale. Statistical analysis used mixed effects regression model. All comparisons p>0.05, and deemed non-significant.
3.8. Response to treatment did not affect expression of IRF4 or KLF4
Expression of the M2-related IRF4 and KLF4 in CD14+ monocytes did not show any noticeable changes associated with response to treatment (not shown)
4. Discussion
In this study, we find that systemic administration of the IL-1 trap rilonacept is associated with changes in the expression patterns of IL-1 and key transcription factors related to monocyte activation in circulating monocytes. Some gene expression changes are associated with clinical response to the treatment, whereas other changes are related only to exposure to rilonacept, arguing that distinct mechanisms may be involved in clinically related and clinically independent effects of IL-1 inhibition. As to some of its limitations, this study was performed with a limited number of samples, and changes between the early and late responders groups were statistically significant using only group to group comparisons. As such, further studies are necessary to confirm these observations.
We initially determined that IL-1 inhibition by rilonacept is associated with a significant drop in circulating IL-1ra, detected at the earliest time point assessed (2 weeks). This finding fits with the known role of IL-1 in the induction of IL-1RN [43], indicating that inhibition of IL-1 by rilonacept has a systemic effect on the IL-1 pathway. Effects of rilonacept on other cytokine tested were less noticeable; specifically, there was no significant effect on circulating levels of IL-6. IL-1 inhibition by anti-IL-1 antibody canakinumab was shown to decrease levels of serum IL-6 rapidly [44]. The failure of rilonacept to significantly decrease levels of serum IL-6 might be related to decreased efficiency of rilonacept versus canakinumab, although no direct comparison between these drugs has been performed [45]. Similarly, no decrease in serum levels of IL-18 were observed with rilonacept treatment, although a transient increase in serum IL-18BP was observed; the increase in IL-18BP may result in a temporary drop in active IL-18, even without changes in the total levels of IL-18. With canakinumab, IL-18 levels dropped significantly, although only after 8 weeks [44]. Elevated IL-18 levels have been shown to persist even during remission in sJIA [46]. Although IL-1ra levels dropped with rilonacept treatment, indicating systemic effect on IL-1 pathway, other pro-inflammatory pathways involved in IL-6, IL-8 and IL-18 induction may persist during treatment with rilonacept. For example, IL-18 has been shown to induce production of IL-6 and IL-8 independently of IL-1ß [47]; the failure of rilonacept to induce a significant drop of IL-18 levels may allow this cytokine to maintain production of IL-6 and IL-8, even when IL-1 pathways are inhibited. Rilonacept may also potentially bind to IL-1ra, and this may, paradoxically, decrease or delay its efficacy. Another possibility is that systemic induction of regulatory mechanisms, such as induction of IL-10, are not efficiently induced by rilonacept treatment.
We next examined if IL-1 inhibition by rilonacept affects expression of IL1B and transcription factors associated with monocyte activation in circulating CD14+ monocytes. When we analyzed samples based solely on duration (weeks) of rilonacept treatment, we found that IL1B expression in CD14+ monocytes is decreased compared to baseline following IL-1 blockade. This result agrees with the well-known property of IL-1 as an inducer of its own synthesis [48, 49]. Expression of P2RX7, which encodes a nucleotide receptor associated with inflammasome activation, IL-1 processing and release [33], also decreased, reaching significance after 20–24 weeks, arguing that blockade of IL-1 may also have moderate effect on P2RX7 expression in CD14+ monocytes. Other blood cells may be affected differently, as Quartier et al [15] found that expression of P2RX7 was increased following treatment with anakinra (recombinant IL-1ra), in analysis of whole blood, perhaps due to changes in neutrophils [50].
Among the TF associated with monocyte activation, we observed that after IL-1 inhibition, the M1-related TF IRF5 was decreased. We have previously observed increased in both M1/M2-associated markers during sJIA flare [6]; IRF5 has been found to be important for control of inflammation in macrophages [51], and the reduction of IRF5 expression may be related to reduction in the pro-inflammatory monocyte phenotype with IL-1 inhibition. We also found that STAT6 expression was decreased after rilonacept treatment. In the context of monocyte activation, STAT6 plays an essential role in M2 polarization mediated by IL-4 [26]. Given the previously observed mixed M1/M2-like skew of circulating sJIA monocytes [6], the decrease of STAT6 expression may represent a reduction in the M2-like phenotype of circulating monocytes in sJIA, perhaps indicating diminishing levels of inflammation in tissues. However, expression of other tested TFs associated with M2 development were less affected by IL-1 inhibition. KLF4 and IRF4 did not show a change in expression with IL-1 inhibition.
Another M2-related TF, PPARG [52], showed increased expression with IL-1 blockade. STAT6 has been shown to physically interact and amplify PPARG responses [53]. Nevertheless, blockade of IL-1 may play a direct role in regulating PPARG expression. In liver cells, treatment with IL-1, but not IL-6, reduces the mRNA expression of PPARG (and other PPARs) [54]. PPARG ligands also inhibit the secretion of IL-1 by stimulated monocytes, and induce production of IL-1ra [55]. Chen et al [56], in a study of monocytes from patients with atrial fibrillation, showed a significant negative correlation between serum levels of IL-1 and expression of PPARG. The inhibition of IL-1 may, directly or indirectly, contribute to increased expression of PPARG. Increased expression of PPARG, together with reduction in IRF5 and STAT6 expression, may skew monocytes in sJIA from a mixed M1/M2-driven to a PPARG-driven M2-like phenotype. Szanto et al [53] found that while the majority (85%) of genes induced by PPARG are STAT6-dependent, close to 30% of genes repressed by PPARG are STAT6 independent. PPARG activation has been shown to suppress IL-1β production [57]. These expression changes, if translated into active protein, could result in an increase of PPARG-repressive activity, independent of STAT6, following IL-1 blockade.
When we examined the transcription signatures in light of clinical responses to rilonacept (as determined by achieving ACR Pedi 30), we found that expression of IL1B was decreased significantly only in early responders after 20–24 weeks of treatment. At this point, the expression of IL1B was lower in early responders than late responders. Notably, the expression of IL1B in CD14+ from late responders was similar to baseline throughout the whole trial, although the number of late responders examined was small. This result indicates that, although rilonacept is generally associated with decreased IL1B, heterogeneity can be observed when clinical response is taken into account. In analysis of whole blood, the decrease in IL1B expression has also been observed to be more pronounced in sJIA responders to canakinumab, a human anti-IL-1β antibody [44]. Of note, IL1B mRNA expression is only one aspect of IL-1β biology. IL1B is translated into pro-IL-1β, which can be stored intracellularly; activation of IL-1β is dependent on inflammasome activation and cleavage by caspase-1, followed by secretion [58]. The significance of alterations in IL1B expression in sJIA monocytes following IL-1 inhibition needs to be confirmed by analysis of active protein.
We found that expression of STAT1 was transiently increased (during week 10–14) in early responders in comparison to baseline and in comparison to late responders. Concomitantly, the expression of SOCS1, suppressor of cytokine signaling, was decreased in early responders and lower in the early responder group in comparison to late responders over the same time period. We have previously observed that STAT1 phosphorylation in response to IFNγ and α was decreased in monocytes from untreated sJIA patients with active disease in comparison to controls, indicating that response to IFN is dysregulated in sJIA monocytes [27]. Additionally, an animal model based on administration of Freund’s complete adjuvant that reproduced several characteristic of sJIA, showed more robust and diverse sJIA-like symptoms in the absence of IFNγ [59], indicating a contribution from dysregulation in the IFNγ pathway in this model. We also found that IL-1 inhibition (with anakinra) or IL-6 inhibition (with tocilizumab) was associated with enhanced IFNγ-induced STAT1 phosphorylation compared to controls [27]. Furthermore, it has been shown that b lockade of IL-1 with anakinra revealed an IFN signature in whole blood from sJIA patients [15], and the levels of two IFN-inducible proteins (IP-10 and TRAIL) were elevated in serum following 6 months of anakinra treatment. These results suggest a relationship between IL-1 and IFN in sJIA. In other settings, reciprocal effects have been observed, including IFN inhibition of IL-1 activity [60, 61]. Work in host defense has indicated that production of IL-1 in vivo might be inhibited by IFN [62, 63], in a STAT1-dependent manner [62]. Guarda et al [61] showed that IFN signaling via STAT1 inhibits the activity of the NLRP1 and NLRP3 inflammasomes, consequently decreasing the production of mature IL-1β by caspase-1. In an animal model of tuberculosis, Mishra et al [64] found that the suppressive effect of IFNγ on IL-1 production was mediated by effects of nitric oxide (NO) on NLRP3 inflammasome assembly.
The clinical response to IL-1 blockade may involve, directly or indirectly, normalization of IFN signaling in monocytes, either via increased expression of STAT1 and/or decreased expression of SOCS1. Of note, SOCS1 can be induced by IL-21 [65]; investigations of the level of IL-21 in sJIA might be informative. An increase in IFN activity may follow IL-1 inhibition, as suggested by the results described in [15]. Although the STAT1/SOCS1 changes were transient, maybe as a result of appearance of other regulatory mechanisms, the increase in IFN signaling and possible effects on the NLRP3 inflammasome in early responders might further reinforce the IL-1 inhibition, resulting in further decrease of IL-1β expression. Although Quartier et al, 2011 [15] did not find any changes in expression of genes associated with the NLRP3 inflammasome in whole blood of anakinra-treated sJIA patients, analysis of isolated monocytes may be informative. Effects of IL-1 inhibition in other important pathways, such as NF-κB, which is activated by IL-1ß among other stimuli, may also occur. For example, there is evidence that IL-1 inhibition by anakinra may lead to decrease in nuclear translocation of NF-κB p65 [66].
The findings reported in this study refer only to gene expression. In addition to control of gene expression [67–69], multiple levels of post-transcriptional regulation exist, and it is unknown if IL-1 inhibition with rilonacept also alters the rate and/or stability of the transcripts, or other processes that control gene expression, such as epigenetic regulation [70]. An analysis of circulating CD14+ monocytes from RAPPORT subjects (Hay et al, unpublished) found that methylation changes in several genes could be detected when comparing monocytes at baseline with active disease to monocytes at week 24 with quiescent disease. Furthermore, it is unknown if the alterations in gene expression described here are reflected into translational changes of active proteins; additionally, post-translational modifications, such as phosphorylation, especially of STAT proteins, also influence molecule activity [71]. To understand the mechanism(s) of effects of IL-1 inhibition on sJIA monocytes, further studies analyzing protein levels and function will be needed.
Analysis of CD14+ monocyte gene expression following IL-1 inhibition with rilonacept shows that M1-associated IRF5 and M2-associated TF STAT6 are decreased, whereas PPARG expression increases, resulting in a monocyte phenotype of IRF5lo STAT6lo and PPARGhi, compared to baseline at active sJIA. The PPARG changes may be a direct effect of reduced IL-1. However, these changes are independent of therapeutic response, suggesting they may be necessary, but not sufficient, for clinical improvement. These changes may result in a persistent M2-like phenotype in circulating CD14+ monocytes (and perhaps tissue monocytes) even during sJIA quiescence, under influence of PPARG rather than STAT6. Our results also show that reductions in IL1B transcripts correlate most robustly with achieving less active sJIA on rilonacept. It will be of interest to determine whether diminished IL1B expression marks a distinct subset of blood monocytes compared to IRF5lo, STAT6lo, PPARGhi cells; single cell RNA sequencing will be required to answer this question. In addition to the association of reduced IL1B with clinical improvement, it is notable that approximately 30% of the early responders have higher baseline ILIB expression than the rest of all subjects studied. Thus, elevated initial monocyte IL1B transcripts may be a prognostic biomarker of responders to rilonacept and to IL-1 inhibition more generally [44]. In a recent study, alleles associated with higher expression of IL1RN (gene coding for IL1-ra) were associated with failure to respond to anakinra [72]. Early reduction of monocyte IL1B expression in rilonacept responders (and possibly reduction in monocyte P2XR7) strengthens the conclusion that IL-1 activity is a driver of sJIA in at least this subset of children, corroborating findings with other IL-1 inhibitors [15, 44]. An important outstanding goal is to immunophenotype incomplete and non-responders to identify candidate alternative therapeutic targets for these patients.
The other monocyte transcriptional changes associated with an early response to rilonacept (transient increase in STAT1 and decrease in SOCS-1 compared to late responders) suggest that a deficiency of interferon signaling may contribute to pathogenesis in IL-1 associated disease. Determining if the transcriptional changes are biomarkers of a therapeutic response to other medications that target IL-1 or to other interventions, such as IL-6 inhibition, will be useful.
Acknowledgments
We would like to thank Clarissa Klein, Paul Roever and Justin Lee for help with PCR experiments, Josephine Yoon for help with ELISA experiments, and Dr. Lu Tian, Department of Biomedical Data Science, Stanford, for guidance on statistical analyses. Funded by Arthritis Foundation 5906 and Training Program T32 Grant 2T32AR050942–06A1 (to YZ); NIH R01AR061297 and NIH 1R21AR062765 (to EDM); Department of Pediatrics at The Children’s Hospital at Montefiore (to ADH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5 : Rapport investigators:
Kristi Prather, Yuliya Lokhnygina (Duke Clinical Research Institute, Durham, NC)
Laura E. Schanberg (Duke University Medical Center, Durham, NC)
Melissa Elder (University of Florida, Gainesville, FL)
Diana Milojevic (Department of Pediatrics, University of California San Francisco; San Francisco, CA)
James W. Verbsky (Medical College of Wisconsin, Milwaukee, WI)
Steven J. Spalding (Cleveland Clinic, Cleveland, OH)
Yukiko Kimura (Hackensack University Medical Center, Hackensack, NJ)
Lisa F. Imundo (Columbia University Medical Center, New York, NY)
Marilynn G. Punaro (Texas Scottish Rite Hospital, Dallas, TX)
David D. Sherry (Children’s Hospital of Philadelphia, Philadelphia, PA)
Stacey E. Tarvin, Kathleen O’Neil (Riley Hospital for Children at Indiana University Health, IN) Lawrence S. Zemel (Connecticut Children’s Medical Center, Hartford, CT)
James D. Birmingham (Michigan State University, East Lansing, MI)
Beth S. Gottlieb (Steven and Alexandra Cohen Children’s Hospital, New Hyde Park, NY) Michael L. Miller (Ann and Robert H. Lurie Children’s Hospital, Chicago, IL)
Natasha M. Ruth (Medical University of South Carolina, Charleston, SC)
Carol A. Wallace (Seattle Children’s Hospital and Research Institute, Seattle, WA)
Nora G. Singer (Rainbow Babies and Children’s Hospital and Case Medical Center, Cleveland, OH)
Christy I. Sandborg (Stanford University School of Medicine, Stanford, CA)
References
- [1].Mellins ED, Macaubas C, Grom AA, Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions, Nature reviews. Rheumatology, 7 (2011) 416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ombrello MJ, Remmers EF, Tachmazidou I, Grom A, Foell D, Haas JP, Martini A, Gattorno M, Ozen S, Prahalad S, Zeft AS, Bohnsack JF, Mellins ED, Ilowite NT, Russo R, Len C, Hilario MO, Oliveira S, Yeung RS, Rosenberg A, Wedderburn LR, Anton J, Schwarz T, Hinks A, Bilginer Y, Park J, Cobb J, Satorius CL, Han B, Baskin E, Signa S, Duerr RH, Achkar JP, Kamboh MI, Kaufman KM, Kottyan LC, Pinto D, Scherer SW, Alarcon-Riquelme ME, Docampo E, Estivill X, Gul A, de Bakker PI, Raychaudhuri S, Langefeld CD, Thompson S, Zeggini E, Thomson W, Kastner DL, Woo P, HLA-DRB1*11 and variants of the MHC class II locus are strong risk factors for systemic juvenile idiopathic arthritis, Proceedings of the National Academy of Sciences of the United States of America, 112 (2015) 15970–15975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Frosch M, Metze D, Foell D, Vogl T, Sorg C, Sunderkotter C, Roth J, Early activation of cutaneous vessels and epithelial cells is characteristic of acute systemic onset juvenile idiopathic arthritis, Experimental dermatology, 14 (2005) 259–265. [DOI] [PubMed] [Google Scholar]
- [4].Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J, Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade, The Journal of experimental medicine, 201 (2005) 1479–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ogilvie EM, Khan A, Hubank M, Kellam P, Woo P, Specific gene expression profiles in systemic juvenile idiopathic arthritis, Arthritis and rheumatism, 56 (2007) 1954–1965. [DOI] [PubMed] [Google Scholar]
- [6].Macaubas C, Nguyen KD, Peck A, Buckingham J, Deshpande C, Wong E, Alexander HC, Chang SY, Begovich A, Sun Y, Park JL, Pan KH, Lin R, Lih CJ, Augustine EM, Phillips C, Hadjinicolaou AV, Lee T, Mellins ED, Alternative activation in systemic juvenile idiopathic arthritis monocytes, Clinical immunology (Orlando, Fla.), 142 (2012) 362–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Macaubas C, Nguyen K, Deshpande C, Phillips C, Peck A, Lee T, Park JL, Sandborg C, Mellins ED, Distribution of circulating cells in systemic juvenile idiopathic arthritis across disease activity states, Clinical immunology (Orlando, Fla.), 134 (2010) 206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fall N, Barnes M, Thornton S, Luyrink L, Olson J, Ilowite NT, Gottlieb BS, Griffin T, Sherry DD, Thompson S, Glass DN, Colbert RA, Grom AA, Gene expression profiling of peripheral blood from patients with untreated new-onset systemic juvenile idiopathic arthritis reveals molecular heterogeneity that may predict macrophage activation syndrome, Arthritis and rheumatism, 56 (2007) 3793–3804. [DOI] [PubMed] [Google Scholar]
- [9].Nguyen KD, Macaubas C, Truong P, Wang N, Hou T, Yoon T, Mellins ED, Serum amyloid A induces mitogenic signals in regulatory T cells via monocyte activation, Molecular immunology, 59 (2014) 172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ravelli A, Grom AA, Behrens EM, Cron RQ, Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment, Genes and immunity, 13 (2012) 289–298. [DOI] [PubMed] [Google Scholar]
- [11].Woo P, Systemic juvenile idiopathic arthritis: diagnosis, management, and outcome, Nature clinical practice. Rheumatology, 2 (2006) 28–34. [DOI] [PubMed] [Google Scholar]
- [12].Canny S, Mellins E, New frontiers in the treatment of systemic juvenile idiopathic arthritis, F1000Research, 6 (2017) 971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sandborg C, Mellins ED, A new era in the treatment of systemic juvenile idiopathic arthritis, The New England journal of medicine, 367 (2012) 2439–2440. [DOI] [PubMed] [Google Scholar]
- [14].Giancane G, Minoia F, Davi S, Bracciolini G, Consolaro A, Ravelli A, IL-1 Inhibition in Systemic Juvenile Idiopathic Arthritis, Frontiers in pharmacology, 7 (2016) 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Quartier P, Allantaz F, Cimaz R, Pillet P, Messiaen C, Bardin C, Bossuyt X, Boutten A, Bienvenu J, Duquesne A, Richer O, Chaussabel D, Mogenet A, Banchereau J, Treluyer JM, Landais P, Pascual V, A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial), Annals of the rheumatic diseases, 70 (2011) 747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Verbsky JW, White AJ, Effective use of the recombinant interleukin 1 receptor antagonist anakinra in therapy resistant systemic onset juvenile rheumatoid arthritis, The Journal of rheumatology, 31 (2004) 2071–2075. [PubMed] [Google Scholar]
- [17].Ruperto N, Brunner HI, Quartier P, Constantin T, Wulffraat N, Horneff G, Brik R, McCann L, Kasapcopur O, Rutkowska-Sak L, Schneider R, Berkun Y, Calvo I, Erguven M, Goffin L, Hofer M, Kallinich T, Oliveira SK, Uziel Y, Viola S, Nistala K, Wouters C, Cimaz R, Ferrandiz MA, Flato B, Gamir ML, Kone-Paut I, Grom A, Magnusson B, Ozen S, Sztajnbok F, Lheritier K, Abrams K, Kim D, Martini A, Lovell DJ, Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis, The New England journal of medicine, 367 (2012) 2396–2406. [DOI] [PubMed] [Google Scholar]
- [18].Gattorno M, Piccini A, Lasiglie D, Tassi S, Brisca G, Carta S, Delfino L, Ferlito F, Pelagatti MA, Caroli F, Buoncompagni A, Viola S, Loy A, Sironi M, Vecchi A, Ravelli A, Martini A, Rubartelli A, The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis, Arthritis and rheumatism, 58 (2008) 1505–1515. [DOI] [PubMed] [Google Scholar]
- [19].Nigrovic PA, Mannion M, Prince FH, Zeft A, Rabinovich CE, van Rossum MA, Cortis E, Pardeo M, Miettunen PM, Janow G, Birmingham J, Eggebeen A, Janssen E, Shulman AI, Son MB, Hong S, Jones K, Ilowite NT, Cron RQ, Higgins GC, Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series, Arthritis and rheumatism, 63 (2011) 545–555. [DOI] [PubMed] [Google Scholar]
- [20].Vastert SJ, de Jager W, Noordman BJ, Holzinger D, Kuis W, Prakken BJ, Wulffraat NM, Effectiveness of first-line treatment with recombinant interleukin-1 receptor antagonist in steroid-naive patients with new-onset systemic juvenile idiopathic arthritis: results of a prospective cohort study, Arthritis & rheumatology (Hoboken, N.J.), 66 (2014) 1034–1043. [DOI] [PubMed] [Google Scholar]
- [21].Hedrich CM, Bruck N, Fiebig B, Gahr M, Anakinra: a safe and effective first-line treatment in systemic onset juvenile idiopathic arthritis (SoJIA), Rheumatology international, 32 (2012) 3525–3530. [DOI] [PubMed] [Google Scholar]
- [22].Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM, M-1/M-2 macrophages and the Th1/Th2 paradigm, Journal of immunology (Baltimore, Md. : 1950), 164 (2000) 6166–6173. [DOI] [PubMed] [Google Scholar]
- [23].Martinez FO, Sica A, Mantovani A, Locati M, Macrophage activation and polarization, Frontiers in bioscience : a journal and virtual library, 13 (2008) 453–461. [DOI] [PubMed] [Google Scholar]
- [24].Martinez FO, Gordon S, The M1 and M2 paradigm of macrophage activation: time for reassessment, F1000prime reports, 6 (2014) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, Wynn TA, Macrophage activation and polarization: nomenclature and experimental guidelines, Immunity, 41 (2014) 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gordon S, Martinez FO, Alternative activation of macrophages: mechanism and functions, Immunity, 32 (2010) 593–604. [DOI] [PubMed] [Google Scholar]
- [27].Macaubas C, Wong E, Zhang Y, Nguyen KD, Lee J, Milojevic D, Shenoi S, Stevens AM, Ilowite N, Saper V, Lee T, Mellins ED, Altered signaling in systemic juvenile idiopathic arthritis monocytes, Clinical immunology (Orlando, Fla.), 163 (2016) 66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lawrence T, Natoli G, Transcriptional regulation of macrophage polarization: enabling diversity with identity, Nature reviews. Immunology, 11 (2011) 750–761. [DOI] [PubMed] [Google Scholar]
- [29].Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, Jardine L, Dixon D, Stephenson E, Nilsson E, Grundberg I, McDonald D, Filby A, Li W, De Jager PL, Rozenblatt-Rosen O, Lane AA, Haniffa M, Regev A, Hacohen N, Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors, Science (New York, N.Y.), 356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ilowite NT, Prather K, Lokhnygina Y, Schanberg LE, Elder M, Milojevic D, Verbsky JW, Spalding SJ, Kimura Y, Imundo LF, Punaro MG, Sherry DD, Tarvin SE, Zemel LS, Birmingham JD, Gottlieb BS, Miller ML, O’Neil K, Ruth NM, Wallace CA, Singer NG, Sandborg CI, Randomized, double-blind, placebo-controlled trial of the efficacy and safety of rilonacept in the treatment of systemic juvenile idiopathic arthritis, Arthritis & rheumatology (Hoboken, N.J.), 66 (2014) 2570–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Avau A, Put K, Wouters CH, Matthys P, Cytokine balance and cytokine-driven natural killer cell dysfunction in systemic juvenile idiopathic arthritis, Cytokine & growth factor reviews, 26 (2015) 35–45. [DOI] [PubMed] [Google Scholar]
- [32].Giannini EH, Ruperto N, Ravelli A, Lovell DJ, Felson DT, Martini A, Preliminary definition of improvement in juvenile arthritis, Arthritis and rheumatism, 40 (1997) 1202–1209. [DOI] [PubMed] [Google Scholar]
- [33].Giuliani AL, Sarti AC, Falzoni S, Di Virgilio F, The P2X7 Receptor-Interleukin-1 Liaison, Frontiers in pharmacology, 8 (2017) 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Van Dongen HP, Olofsen E, Dinges DF, Maislin G, Mixed-model regression analysis and dealing with interindividual differences, Methods in enzymology, 384 (2004) 139–171. [DOI] [PubMed] [Google Scholar]
- [35].Althouse AD, Adjust for Multiple Comparisons? It’s Not That Simple, The Annals of thoracic surgery, 101 (2016) 1644–1645. [DOI] [PubMed] [Google Scholar]
- [36].Granowitz EV, Santos AA, Poutsiaka DD, Cannon JG, Wilmore DW, Wolff SM, Dinarello CA, Production of interleukin-1-receptor antagonist during experimental endotoxaemia, Lancet (London, England), 338 (1991) 1423–1424. [DOI] [PubMed] [Google Scholar]
- [37].Dinarello CA, Blocking IL-1 in systemic inflammation, The Journal of experimental medicine, 201 (2005) 1355–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].De Benedetti F, Pignatti P, Massa M, Sartirana P, Ravelli A, Martini A, Circulating levels of interleukin 1 beta and of interleukin 1 receptor antagonist in systemic juvenile chronic arthritis, Clinical and experimental rheumatology, 13 (1995) 779–784. [PubMed] [Google Scholar]
- [39].Sanda S, Bollyky J, Standifer N, Nepom G, Hamerman JA, Greenbaum C, Short-term IL-1beta blockade reduces monocyte CD11b integrin expression in an IL-8 dependent fashion in patients with type 1 diabetes, Clinical immunology (Orlando, Fla.), 136 (2010) 170–173. [DOI] [PubMed] [Google Scholar]
- [40].Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG, Regulation and functions of the IL-10 family of cytokines in inflammation and disease, Annual review of immunology, 29 (2011) 71–109. [DOI] [PubMed] [Google Scholar]
- [41].Kaplanski G, Interleukin-18: Biological properties and role in disease pathogenesis, Immunological reviews, 281 (2018) 138–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Weiss ES, Girard-Guyonvarc’h C, Holzinger D, de Jesus AA, Tariq Z, Picarsic J, Schiffrin EJ, Foell D, Grom AA, Ammann S, Ehl S, Hoshino T, Goldbach-Mansky R, Gabay C, Canna SW, Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome, Blood, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hurme M, Santtila S, IL-1 receptor antagonist (IL-1Ra) plasma levels are co-ordinately regulated by both IL-1Ra and IL-1beta genes, European journal of immunology, 28 (1998) 2598–2602. [DOI] [PubMed] [Google Scholar]
- [44].Brachat AH, Grom AA, Wulffraat N, Brunner HI, Quartier P, Brik R, McCann L, Ozdogan H, Rutkowska-Sak L, Schneider R, Gerloni V, Harel L, Terreri M, Houghton K, Joos R, Kingsbury D, Lopez-Benitez JM, Bek S, Schumacher M, Valentin MA, Gram H, Abrams K, Martini A, Lovell DJ, Nirmala NR, Ruperto N, Early changes in gene expression and inflammatory proteins in systemic juvenile idiopathic arthritis patients on canakinumab therapy, Arthritis research & therapy, 19 (2017) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tarp S, Amarilyo G, Foeldvari I, Christensen R, Woo JM, Cohen N, Pope TD, Furst DE, Efficacy and safety of biological agents for systemic juvenile idiopathic arthritis: a systematic review and meta-analysis of randomized trials, Rheumatology (Oxford, England), 55 (2016) 669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Shimizu M, Yokoyama T, Yamada K, Kaneda H, Wada H, Wada T, Toma T, Ohta K, Kasahara Y, Yachie A, Distinct cytokine profiles of systemic-onset juvenile idiopathic arthritis associated macrophage activation syndrome with particular emphasis on the role of interleukin 18 in its pathogenesis, Rheumatology (Oxford, England), 49 (2010) 1645–1653. [DOI] [PubMed] [Google Scholar]
- [47].Lee JK, Kim SH, Lewis EC, Azam T, Reznikov LL, Dinarello CA, Differences in signaling pathways by IL-1beta and IL-18, Proceedings of the National Academy of Sciences of the United States of America, 101 (2004) 8815–8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Dinarello CA, Ikejima T, Warner SJ, Orencole SF, Lonnemann G, Cannon JG, Libby P, Interleukin 1 induces interleukin 1. I. Induction of circulating interleukin 1 in rabbits in vivo and in human mononuclear cells in vitro, Journal of immunology (Baltimore, Md. : 1950), 139 (1987) 1902–1910. [PubMed] [Google Scholar]
- [49].Manson JC, Symons JA, Di Giovine FS, Poole S, Duff GW, Autoregulation of interleukin 1 production, European journal of immunology, 19 (1989) 261–265. [DOI] [PubMed] [Google Scholar]
- [50].Karmakar M, Katsnelson MA, Dubyak GR, Pearlman E, Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1beta secretion in response to ATP, Nature communications, 7 (2016) 10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Weiss M, Blazek K, Byrne AJ, Perocheau DP, Udalova IA, IRF5 is a specific marker of inflammatory macrophages in vivo, Mediators of inflammation, 2013 (2013) 245804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Croasdell A, Duffney PF, Kim N, Lacy SH, Sime PJ, Phipps RP, PPARgamma and the Innate Immune System Mediate the Resolution of Inflammation, PPAR research, 2015 (2015) 549691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, Szeles L, Poliska S, Oros M, Evans RM, Barak Y, Schwabe J, Nagy L, STAT6 transcription factor is a facilitator of the nuclear receptor PPARgamma-regulated gene expression in macrophages and dendritic cells, Immunity, 33 (2010) 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kim MS, Sweeney TR, Shigenaga JK, Chui LG, Moser A, Grunfeld C, Feingold KR, Tumor necrosis factor and interleukin 1 decrease RXRalpha, PPARalpha, PPARgamma, LXRalpha, and the coactivators SRC-1, PGC-1alpha, and PGC-1beta in liver cells, Metabolism: clinical and experimental, 56 (2007) 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Meier CA, Chicheportiche R, Juge-Aubry CE, Dreyer MG, Dayer JM, Regulation of the interleukin-1 receptor antagonist in THP-1 cells by ligands of the peroxisome proliferator-activated receptor gamma, Cytokine, 18 (2002) 320–328. [DOI] [PubMed] [Google Scholar]
- [56].Chen X, Bing Z, He J, Jiang L, Luo X, Su Y, Kan B, Huang D, Wei Y, Downregulation of peroxisome proliferator-activated receptor-gamma expression in hypertensive atrial fibrillation, Clinical cardiology, 32 (2009) 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Glatz T, Stock I, Nguyen-Ngoc M, Gohlke P, Herdegen T, Culman J, Zhao Y, Peroxisome-proliferator-activated receptors gamma and peroxisome-proliferator-activated receptors beta/delta and the regulation of interleukin 1 receptor antagonist expression by pioglitazone in ischaemic brain, Journal of hypertension, 28 (2010) 1488–1497. [DOI] [PubMed] [Google Scholar]
- [58].Dinarello CA, IL-1: discoveries, controversies and future directions, European journal of immunology, 40 (2010) 599–606. [DOI] [PubMed] [Google Scholar]
- [59].Avau A, Mitera T, Put S, Put K, Brisse E, Filtjens J, Uyttenhove C, Van Snick J, Liston A, Leclercq G, Billiau AD, Wouters CH, Matthys P, Systemic juvenile idiopathic arthritis-like syndrome in mice following stimulation of the immune system with Freund’s complete adjuvant: regulation by interferon-gamma, Arthritis & rheumatology (Hoboken, N.J.), 66 (2014) 1340–1351. [DOI] [PubMed] [Google Scholar]
- [60].Hu X, Ho HH, Lou O, Hidaka C, Ivashkiv LB, Homeostatic role of interferons conferred by inhibition of IL-1-mediated inflammation and tissue destruction, Journal of immunology (Baltimore, Md. : 1950), 175 (2005) 131–138. [DOI] [PubMed] [Google Scholar]
- [61].Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P, Tschopp J, Type I interferon inhibits interleukin-1 production and inflammasome activation, Immunity, 34 (2011) 213–223. [DOI] [PubMed] [Google Scholar]
- [62].De Boer ML, Hu J, Kalvakolanu DV, Hasday JD, Cross AS, IFN-gamma inhibits lipopolysaccharide-induced interleukin-1 beta in primary murine macrophages via a Stat1-dependent pathway, Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research, 21 (2001) 485–494. [DOI] [PubMed] [Google Scholar]
- [63].Joshi VD, Kalvakolanu DV, Chen W, Zhang L, Kang TJ, Thomas KE, Vogel SN, Cross AS, A role for Stat1 in the regulation of lipopolysaccharide-induced interleukin-1beta expression, Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research, 26 (2006) 739–747. [DOI] [PubMed] [Google Scholar]
- [64].Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, Sassetti CM, Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta, Nature immunology, 14 (2013) 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Strengell M, Lehtonen A, Matikainen S, Julkunen I, IL-21 enhances SOCS gene expression and inhibits LPS-induced cytokine production in human monocyte-derived dendritic cells, Journal of leukocyte biology, 79 (2006) 1279–1285. [DOI] [PubMed] [Google Scholar]
- [66].Goncalves NP, Vieira P, Saraiva MJ, Interleukin-1 signaling pathway as a therapeutic target in transthyretin amyloidosis, Amyloid : the international journal of experimental and clinical investigation : the official journal of the International Society of Amyloidosis, 21 (2014) 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wong LH, Sim H, Chatterjee-Kishore M, Hatzinisiriou I, Devenish RJ, Stark G, Ralph SJ, Isolation and characterization of a human STAT1 gene regulatory element. Inducibility by interferon (IFN) types I and II and role of IFN regulatory factor-1, The Journal of biological chemistry, 277 (2002) 19408–19417. [DOI] [PubMed] [Google Scholar]
- [68].Bai L, Merchant JL, Transcription factor ZBP-89 is required for STAT1 constitutive expression, Nucleic acids research, 31 (2003) 7264–7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yuasa K, Hijikata T, Distal regulatory element of the STAT1 gene potentially mediates positive feedback control of STAT1 expression, Genes to cells : devoted to molecular & cellular mechanisms, 21 (2016) 25–40. [DOI] [PubMed] [Google Scholar]
- [70].Kim EG, Shin HJ, Lee CG, Park HY, Kim YK, Park HW, Cho SH, Min KU, Cho ML, Park SH, Lee CW, DNA methylation and not allelic variation regulates STAT6 expression in human T cells, Clinical and experimental medicine, 10 (2010) 143–152. [DOI] [PubMed] [Google Scholar]
- [71].Levy DE, Darnell JE Jr., Stats: transcriptional control and biological impact, Nature reviews. Molecular cell biology, 3 (2002) 651–662. [DOI] [PubMed] [Google Scholar]
- [72].Arthur VL, Shuldiner E, Remmers EF, Hinks A, Grom AA, Foell D, Martini A, Gattorno M, Ozen S, Prahalad S, Zeft AS, Bohnsack JF, Ilowite NT, Mellins ED, Russo R, Len C, Oliveira S, Yeung RSM, Rosenberg AM, Wedderburn LR, Anton J, Haas JP, Rosen-Wolff A, Minden K, Szymanski AM, Thomson W, Kastner DL, Woo P, Ombrello MJ, IL1RN Variation Influences both Disease Susceptibility and Response to Human Recombinant IL-1RA Therapy in Systemic Juvenile Idiopathic Arthritis, Arthritis & rheumatology (Hoboken, N.J.), (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]