

Abstract
The enormous diversity of proteoforms produces tremendous complexity within cellular proteomes, facilitates intricate networks of molecular interactions, and constitutes a formidable analytical challenge for biomedical researchers. Currently, quantitative whole-proteome profiling often relies on non-targeted liquid chromatography – mass spectrometry (LC-MS), which samples proteoforms broadly, but can suffer from lower accuracy, sensitivity, and reproducibility compared with targeted LC-MS. Recent advances in bottom-up proteomics using targeted LC-MS have enabled previously unachievable identification and quantification of target proteins and posttranslational modifications within complex samples. Consequently, targeted LC-MS is rapidly advancing biomedical research, especially systems biology research in diverse areas that include proteogenomics, interactomics, kinomics, and biological pathway modeling. With the recent development of targeted LC-MS assays for nearly the entire human proteome, targeted LC-MS is positioned to enable quantitative proteomic profiling of unprecedented quality and accessibility to support fundamental and clinical research. Here we review recent applications of bottom-up proteomics using targeted LC-MS for systems biology research.
Keywords: bottom-up proteomics, parallel reaction monitoring, quantification, selected reaction monitoring, systems biology, targeted mass spectrometry
Graphical abstract

1. Introduction
Bottom-up proteomics using high-performance liquid chromatography coupled to mass spectrometry (LC-MS) has developed into a powerful and highly versatile technology that is enabling rapid advances in diverse areas of biomedical research, clinical diagnostics, and biotechnology [1, 2]. This is especially true for the emerging scientific discipline of systems biology [3]. Beyond basic proteomic profiling, proteomics has recently evolved to enable identification and quantification of protein isoforms (e.g., splice isoforms, single amino acid polymorphisms, and other genetic variants), numerous posttranslational modifications (PTMs), protein turnover, protein conformations, protein-protein interactions, protein interactions with other molecules, and protein-protein subcellular proximity [1, 2]. Recent novel applications include the search for “missing proteins” (genes and transcripts that appear to encode proteins but direct experimental evidence is lacking), kinomics, and enzymatic activity assays related to protein modification (described below).
Bottom-up proteomics LC-MS workflows typically use data-dependent acquisition (DDA, often referred to as shotgun MS), data-independent acquisition (DIA), or targeted LC-MS. DDA involves real-time semi-stochastic intensity-based selection of analytes for fragmentation, and is often the method of choice for discovery-level proteomics. DIA is an emerging technology which involves nonstochastic multiplexed fragmentation using relatively wide precursor ion isolation windows [4, 5]. Compared to DDA, DIA spectra are more challenging to analyze, but DIA can identify more peptides with better tandem MS sampling reproducibility, and with better quantification accuracy and precision [6, 7]. As publicly available DDA spectral libraries and DIA software are further developed, the need for experimentalists to perform DDA prior to each DIA experiment will decrease. Importantly, this requires robust control of the false discovery rate of peptide, protein, and PTM identification.
In a targeted LC-MS experiment, a target list of analyte ion descriptors (e.g., precursor and fragment ion m/z, collision energy, LC elution time) is pre-designated to perform MS1 of the precursor ion and/or tandem MSn of one or more fragment ions [8, 9]. Targeted LC-MS assays are generally designed with the use of DDA LC-MS data, and therefore targeted LC-MS has greatly benefitted from large-scale proteome-wide DDA LC-MS studies [10], which include studies of the yeast proteome [11], the human proteome [12, 13], and the mouse proteome and phosphoproteome [14]. Bottom-up proteomics using targeted LC-MS is a rapidly developing technology, and numerous methods and protocols have been published [15-21]. Neither DDA nor DIA require pre-MS designation of target analytes; as such, neither is targeted MS per se. However, because data analysis of DIA spectra can involve a target list of analyte descriptors (DIA analyses typically require a library of precursor and fragment ion m/z values produced using DDA MS), DIA is often classified as a form of targeted proteomics.
Compared with targeted MS, DDA and DIA workflows typically require significantly less preparation and result in far broader proteomic coverage, but both technologies are currently less sensitive, accurate, precise, and robust [7, 22-25]. Consequently, DDA and DIA are often used for discovery-level experimentation, whereas targeted MS is often used for biomarker validation and absolute quantification. Clinical biomarker applications of bottom-up proteomics using targeted LC-MS have been recently described in two detailed reviews [26, 27]. Therefore, here we focus on applications of targeted LC-MS for bottom-up proteomics in systems biology research (Fig. 1).
Figure 1. Timeline of selected applications of targeted LC-MS for systems biology research.
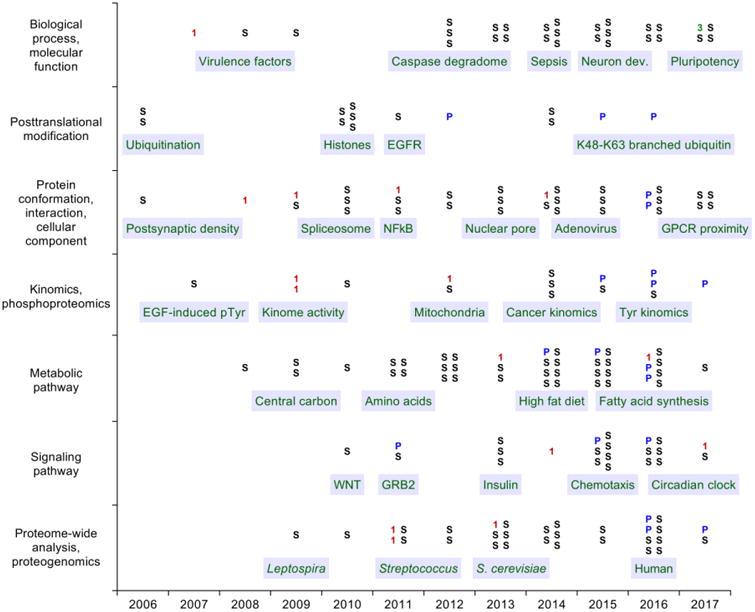
Research articles were partitioned into seven research categories and plotted by publication year. The symbols (1, 3, P, and S) indicate the principal MS scan type that was used for quantification (MS1, MS3, PRM, and SRM, respectively). Selected research topics are noted.
Quantitative immunoassays (e.g., densitometric western blots and enzyme-linked immunosorbent assays) are widespread, and can be of very high quality. Alongside quantitative immunoassays, targeted LC-MS has developed into a powerful alternative approach that can be much more selective, can have a much wider dynamic range, is often more amenable to multiplexing, and can otherwise be of roughly similar quality [28]. For particularly challenging targets, the two strategies can be integrated by immunoenriching target proteins or peptides, and subsequently performing targeted LC-MS.
The earliest LC-MS quantification procedure involved simply analyzing extracted ion chromatograms of precursor ions, and this technique still performs very well in some applications (described below). Currently, the most common targeted technique is selected reaction monitoring (SRM), which is also referred to as multiple reaction monitoring (MRM) [29-32]. SRM uses MS2 data for quantification, and is typically performed using a triple quadrupole MS. Due to the recent development of high resolution mass spectrometers (resolution ≥ ∼50,000) that have fast scan rates (scan frequency ≥ ∼10 Hz), wide dynamic ranges (range ≥ ∼1,000), and accurate and precise quantification, parallel reaction monitoring (PRM) has emerged as a powerful alternative to SRM [33]. Like SRM, PRM uses MS2 data for quantification, but whereas SRM uses fragment ion monitoring, PRM uses high resolution MS2 full scans to monitor the intensity of multiple fragment ions in parallel (low resolution MS2 full scans are rarely used; these are classified as PRM below). Occasionally, targeted LC-MS using MS3 (two stages of fragmentation) is used for qualitative reasons (e.g., identifying a phosphopeptide by fragmenting a neutral loss ion or distinguishing between two highly similar peptides) or for quantitative reasons (e.g., to avoid ratio compression resulting from quantification using isobaric tags). MS3 is highly selective and can be highly accurate, but it can suffer from lower sensitivity compared with MS1 and MS2.
Relative quantification of an analyte across multiple biological samples can be achieved by using the label-free approach (not labeling the analyte using stable isotopes, and performing LC-MS of each sample separately). Alternatively, relative quantification can be performed using a single LC-MS run by simultaneously analyzing a mixture of both unlabeled and stable isotope labeled (SIL) forms of the analyte. For absolute quantification, quantified SIL standards are used [34]. Metabolic labeling strategies include 13C labeling, 15N labeling, stable isotope labeling by amino acids in cell culture (SILAC), and stable isotope labeling of mammals (SILAM). Alternatively, chemical labels include 18O, SIL dimethylation, isobaric tags for relative and absolute quantitation (iTRAQ), mass differential tags for relative and absolute quantification (mTRAQ), tandem mass tags (TMT), isotope-coded affinity tags (ICAT), and isotope-coded protein labels (ICPL). Standards for qualitative and quantitative proteomics include peptides prepared using solid-phase peptide synthesis (SPPS), peptides prepared using recombinant expression of a quantification concatemer (QconCAT), and intact purified protein standards. The lattermost standards are typically used for protein standard absolute quantification (PSAQ) workflows.
Targeted MS experiments depend heavily on specialized software, and this has been reviewed by others [35-37]. Recently developed software programs for targeted MS assay development include MRMaid [38], PeptideClassifier [39] (notable for its ability to select isoform-specific peptides), PeptidePicker [40], PeptideManager [41] (notable for its support of multi-species experiments), PREGO [42], and Skyline [43]. Prediction of quantotypic peptides, which are peptides that can be assayed to accurately identify and quantify a specific proteoform or a specific set of proteoforms (e.g., a set of splice isoforms) remains challenging due to, for example, alternative splicing, PTMs, and chemical artefacts that result from MS sample preparation [44, 45].
Databases of DDA MS2 spectra of proteotypic peptides are often helpful during targeted MS assay development [46], and include The Global Proteome Machine [47], The NIST Libraries of Peptide Tandem Mass Spectra [48], and The ProteomeXchange consortium [49] (which integrates the iProX, jPost, MassIVE, PeptideAtlas, and PRIDE databases). In the absence of DDA data, proteotypic peptide prediction can be performed using CONSeQuence [50], ESP predictor [51], PeptideRank [52], PeptideSieve [53], and STEPP [54]. Unique ion signatures can be calculated to avoid MS signal interference using Sigpep [55] and SRMCollider [56]. Targeted MS software programs for peptide identification and quantification include Ariadne [57], Anubis [58], AuDIT [59], mProphet [60], MRMer [61], Pinnacle (Optys Tech Corp. Philadelphia, PA, http://www.optystech.com/), Skyline [43], and SpectroDive (Biognosys Inc., Schlieren, Switzerland, https://biognosys.com/). Downstream data analysis including statistical modeling can be performed using MSstats [62], Qualis-SIS [63], and SRMstats [64]. Online data management and sharing tools include the CPTAC assay portal [65], Panorama [66], and SRMAtlas [67].
2. Biological processes and molecular functions
Targeted proteomics has been used to study numerous biological functions and disorders including autism, cancer, metabolic syndrome, and neuron development (Table 1). For example, SRM was used to develop pluripotency assays of reprogrammed human fibroblasts [68]. In a second example, 188 biological processes in yeast (e.g., osmotic balance, glucose metabolism, autophagy, and DNA damage) were studied using a multiplexed LC-SRM assay [69]. This “sentinel fingerprint assay” was used to quantify 300 target peptides to assay the abundance of 156 proteins, 11 target peptides to assay degradation products from one protein, and 166 target phosphopeptides to assay 80 phosphoproteins.
Table 1. Selected studies of biological processes and molecular functions.
| Topic | Species (specimen) | MS scan | Proteins (peptides) | PTMs | Isotopic labeling (absolute quantification) | Ref. |
|---|---|---|---|---|---|---|
| Adipogenesis transcription factors | Mouse (3T3-L1 preadipocytes) | SRM | 10 (25) | None | PSAQ (copies/cell) | [150] |
| Antibiotic treatment | Leptospira interrogans | SRM | 39 (151) | None | None, SPPS (copies/cell) | [151] |
| Autism, schizophrenia | Mouse (frontal cortex, hippocampus) | SRM | 38 (86) | None | SPPS | [152] |
| Biomarkers of cellular processes | Saccharomyces cerevisiae | SRM | 182 (690) | None, phosphorylation | None, SPPS | [69] |
| Cancer invasion secretomics | Human (U87 glioblastoma cell line) | SRM | 65 (183) | None | SPPS | [153] |
| Cancer subtypes | Human (30 breast cancer cell lines) | SRM | 319 (645) | None | SPPS (pmol/mg protein) | [71] |
| Cancer-related proteins | Human (60 tumor cell lines) | MS3 | 68 (126) | None | SPPS-TMT | [70] |
| Caspase degradomics | Human (Jurkat cell line) | PRM, SRM | 275 (314) | None | None, SPPS | [154] |
| Caspase degradomics during apoptosis | Human (DB, Jurkat A3, MM.1S cells) | SRM | 533 (789) | None | None | [155] |
| Caspase degradomics during apoptosis | Human (Jurkat cell line) | SRM | 350 (431) | None | None | [76] |
| Chicory leaf infection | Dickeya dadantii | SRM | 445 (782) | None | None | [156] |
| Degradomics of wound healing | Sus scrofa (skin) | SRM | 9 (19) | None | iTRAQ | [157] |
| Estrogen receptor-mediated expression | Human (MCF-7 breast cancer cell line) | SRM | 62 (65) | None | SILAC | [72] |
| Heat shock response chaperones | Saccharomyces cerevisiae | SRM | 47 (166) | None | QconCAT (copies/cell) | [158] |
| Histone H1 | Human (3 myeloid cell types) | SRM | 8 (13) | None | None | [159] |
| Ketamine pharmacodynamics | Rat (cerebrum, hippocampus) | SRM | 49 (98) | None | SPPS | [160] |
| Metabolic syndrome | Mouse (liver) | SRM | 144 (312) | None | SILAC cells | [7] |
| Molecular chaperones | Saccharomyces cerevisiae | SRM | 51 (77) | None | QconCAT (copies/cell) | [73] |
| Muscle development | Gallus gallus (skeletal muscle) | MS1 | 20 (20) | None | QconCAT (pmol/mg protein) | [161] |
| Neuronal development | Human (3 neural stem cell lines) | SRM | 175 (279) | None | SPPS | [162] |
| Oncogenesis transcription factors | Human (8 lung cancer cell lines) | SRM | 28 (36) | None | SPPS (pmol/mg protein) | [163] |
| Parkinson's disease | Rat (PC12 cell line) | SRM | 2 (20) | None, nitration | None | [164] |
| PhoP-PhoQ virulence gene expression | Salmonella typhimurium | SRM | 92 (152) | None | SIL dimethylation | [165] |
| Pluripotency evaluation | Human (fibroblasts, stem cells, embryoi bodies) | d SRM | 15 (33) | None | SPPS (pmol/pmol GAPDH) | [68] |
| Sepsis | Human (neutrophils, plasma) | SRM | 49 (136) | None | None | [166] |
| Small GTPase activity assays | Human (platelets) | SRM | 12 (17) | None | SPPS | [75] |
| Transcription regulation | Saccharomyces cerevisiae | SRM | 209 (355) | None | SPPS (copies/cell), SILAC | [74] |
| Virulence factors | Streptococcus pyogenes | SRM | 21 (48) | None | ICPL | [167] |
n.i., not indicated
Cancer-related proteins have been studied using targeted LC-MS. Isobaric tagging combined with targeted LC-MS3 was used to perform high-throughput relative quantification of 69 cancer-related proteins across the National Cancer Institute NCI-60 panel of sixty cancer cell lines [70]. In this study, the samples as well as the peptide targets were multiplexed, and a correlation between BAZ1B abundance and doxorubicin sensitivity was discovered. Another study used targeted proteomics and transcriptomics to identify breast cancer subtypes [71]. SRM was also used to study estrogen receptor alpha-regulated protein expression in MCF-7 breast cancer cells [72].
Targeted LC-MS has also been used to study sets of proteins related by biochemical function (Table 1). The absolute abundance of 51 chaperone proteins in yeast cells was measured using LC-SRM, and the substrate flux of each chaperone was calculated [73]. Chromatin immunoprecipitation coupled with LC-SRM was used to characterize transcriptional regulation of the environmentally regulated FLO11 promoter in yeast [74]. Affinity purification coupled with LC-SRM was used to perform activity assays of twelve small GTPases within human platelets stimulated with thrombin and lysophosphatidic acid [75]. LC-SRM was also used to measure the kinetics of caspase-mediated proteolysis of 350 proteins in lysates and living cells [76].
3. Posttranslational modifications
Targeted LC-MS is highly amenable to the study of posttranslationally modified proteins (Table 2). In a recent report, 30 phosphorylation sites within epidermal growth factor receptor (EGFR) were profiled using both DDA and targeted MS of primary tumor explants and 31 lung cancer cell lines [77]. From this data, the authors were able to identify sites related to EGFR activation and erlotinib-mediated inhibition. A separate study found related results [78]. Notably, the authors of the later study successfully developed an LC-MS3 assay to distinguish between two extremely similar isobaric EGFR phosphopeptides, demonstrating the utility of targeted LC-MS to study extensively modified proteins.
Table 2. Selected studies of posttranslational modifications.
| Topic | Species (specimen) | MS scan | Proteins (peptides) | PTMs | Isotopic labeling (absolute quantification) | Ref. |
|---|---|---|---|---|---|---|
| Cell-division cycle | Human (Jurkat cell line) | SRM | 1 (4) | None, phosphorylation | SPPS (stoichiometry) | [168] |
| Cyclin-B1 ubiquitination | Human (recombinantly expressed) | SRM | 1 (11) | Ubiquitination | SPPS (stoichiometry) | [169] |
| EphA2 autophosphorylation | Human (recombinantly expressed) | SRM | 1 (23) | None, phosphorylation | None, SPPS (stoichiometry) | [170] |
| Epidermal growth factor receptor | Human (31 lung cancer cell lines) | SRM | 1 (7) | None, phosphorylation | SPPS | [77] |
| Epidermal growth factor receptor | Human (A431 carcinoma cell line) | MS3, PRM | 1 (15) | None, phosphorylation | None, SPPS (stoichiometry) | [78] |
| Heavy metal ubiquitinomics | Human, Saccharomyces cerevisiae | SRM | 1 (24) | None, phosphorylation, ubiquitination | SPPS (pmol/mg protein) | [171] |
| Histone H3 | Human, mouse (40 cell lines) | PRM | 1 (66) | Acetylation, methylation, dimethylation, trimethylation, phosphorylation, ubiquitination | SPPS (stoichiometry), SILAC | [79] |
| Histones | Human (U-937, HL-60 cell lines) | SRM | 4 (21) | Acetylation, methylation, dimethylation, trimethylation, ubiquitination | SPPS (stoichiometry) | [80] |
| HSP27 isobaric phosphopeptides | Human (breast cancer cells, tumor) | SRM | 1 (4) | None, phosphorylation | SPPS (stoichiometry) | [172] |
| Intracellular Cys oxidation | Human (MCF-7, HCA2 cell lines) | SRM | 2 (14) | Oxidation | SIL alkylation | [173] |
| K48-K63 branched ubiquitin | Human (U2OS, HEK293-F cell lines) | PRM | 1 (13) | Ubiquitination | SPPS (pmol/injection) | [81] |
| Lyn phosphorylation stoichiometry | Human (myeloma cells, xenograft tumor) | SRM | 1 (14) | None, phosphorylation | None | [174] |
| Mercury toxicology | Mouse (WEHI-231 B lymphoma cell line) | SRM | 1 (5) | None, phosphorylation | None | [175] |
Proteoforms containing multiple PTMs can be especially challenging to study. Histones, for example, can often be heavily modified. In one study, genes known to be active in epigenetic processes were knocked-down in 293T cells, and a novel targeted MS workflow was used to quantify modified histones [79]. In the same report, the authors used the workflow to profile knockdowns, knockouts, and drug treatments of murine stem cells. A similar study used LC-SRM and discovered that histone H2B ubiquitination inversely correlated with H3 methylation in the U937 human leukemia cell line [80].
Among the most challenging PTMs to study are polymeric, branching PTMs such as glycosylation, polyubiquitination, and poly-ADP-ribosylation. Ohtake and colleagues used targeted MS to study polyubiquitin K48-K63 branched chains [81]. The authors found that, in response to interleukin-1β, the E3 ubiquitin ligase HUWE1 produces K48 branches on K63 chains of TRAF6. These K48-K63 branches protected TRAF6 from deubiquitination, resulting in amplification of nuclear factor κB signaling.
4. Protein conformation, protein-protein interaction, and cellular components
Targeted proteomics has been used to measure the stoichiometry of numerous protein complexes including the centrosome, the focal adhesion complex, the nuclear pore, the ribosome, and the spliceosome (Table 3). Shi and colleagues used LC-SRM to determine how ribosomal heterogeneity determines selectivity for subpools of transcripts [82]. Integration of LC-SRM with super-resolution microscopy and cryo-electron tomography was used to determine the structure of the human nuclear pore complex, and to discover that it varies across tissues, cancer cell types, and diseases [83, 84]. Ori and colleagues investigated spatiotemporal variation of human protein complex stoichiometry using numerous transcriptomic and proteomic technologies (including LC-SRM), and the nucleosome remodeling deacetylase (NuRD) complex was discovered to be an example of paralog switching within a moderately-variable protein complex [85].
Table 3. Selected studies of protein conformation, protein-protein interaction, and cellular components.
| Topic | Species (specimen) | MS scan | Proteins (peptides) | PTMs | Isotopic labeling (absolute quantification) | Ref. |
|---|---|---|---|---|---|---|
| 60S pre-ribosome | Saccharomyces cerevisiae | SRM | 51 (149) | None | None | [176] |
| Adenovirus | Adenovirus particles | SRM | 13 (33) | None | SPPS (stoichiometry) | [177] |
| ASK signalosome | Human (HEK293 cell line) | PRM | 99 (265) | None | None, SPPS (stoichiometry) | [178] |
| Blood-Streptococcus interactomics | Human (plasma) | SRM | 152 (406) | None | None | [179] |
| Blood-Streptococcus interactomics | Human (plasma), Streptococcus pyogenes | SRM | 56 (76) | None | SPPS (stoichiometry) | [89] |
| Centrosome | Human (5 cell lines) | SRM | 9 (18) | None | SPPS (copies/centrosome) | [180] |
| Cohesin complex | Human (MCF-7, HeLa cell lines) | MS1 | 11 (22) | None | QconCAT (stoichiometry) | [181] |
| Cullin-RING complex | Human (HEK293T cell line) | SRM | 25 (38) | None | SPPS (stoichiometry) | [182] |
| Focal adhesion complex | Human (breast cancer cells, tumor) | MS1, SRM | 17 (27) | None | None, SILAC | [183] |
| G protein subcellular localization | Mouse (4 brain tissues) | SRM | 12 (33) | None | None | [184] |
| GPCR proximity labeling | Human (HEK293 cell line) | SRM | 65 (187) | None | None | [90] |
| Hsp90-associated proteins | Human (HeLa cell line) | PRM | 4 (36) | None | SILAC | [185] |
| MP1-p14 complex | Mouse (recombinantly expressed) | MS1, SRM | 2 (10) | None | SPPS-mTRAQ (stoichiometry) | [186] |
| NFκB complex | Human (A549 lung epithelial cell line) | SRM | 3 (3) | None | SPPS (copies/cell) | [187] |
| NFκB complex | Human (SK-N-AS cell line) | SRM | 4 (11) | None | QconCAT (copies/cell) | [188] |
| Nuclear pore | Human (45 cells, tissues) | SRM | 47 (157) | None | SPPS (stoichiometry) | [83] |
| Nuclear pore | Human (HEK293 cell line) | SRM | 32 (76) | None | SPPS (stoichiometry) | [84] |
| Numb-associated endocytosis | Human (HEK293T cell line) | SRM | 14 (40) | None | None | [189] |
| NuRD complex | Human (HEK293, HeLa cell lines) | SRM | 10 (24) | None | SPPS | [85] |
| Podocyte slit diaphragm nephrin | Rat (renal glomeruli) | SRM | 1 (1) | None | SPPS (copies/cell) | [190] |
| Postsynaptic density | Rat (brain) | SRM | 112 (337) | None | None, SPPS | [191] |
| Postsynaptic density | Rat (brain) | SRM | 32 (32) | None | SPPS (pmol/mg protein) | [192] |
| Postsynaptic density | Rat (cerebral cortex) | SRM | 42 (89) | None | QconCAT (copies/postsynaptic density) | [193] |
| Protein complexes | Human (recombinantly expressed) | SRM | 6 (2) | None | SPPS (stoichiometry) | [194] |
| Protein conformation dynamics | Saccharomyces cerevisiae | SRM | 135 (697) | None | None | [86] |
| Protein phosphatase 2A complexes | Human (HEK293 cell line) | MS1 | 10 (n.i.) | None | SPPS (stoichiometry) | [195] |
| Ribosome | Escherichia coli | MS1 | 24 (41) | None | QconCAT (stoichiometry) | [196] |
| Ribosome | Mouse (ES-E14 embryonic stem cells) | SRM | 15 (28) | None | SPPS (stoichiometry) | [82] |
| RNA polymerase complex | Bacillus subtilis | SRM | 8 (16) | None | SPPS-mTRAQ (stoichiometry) | [197] |
| Rod photoreceptor outer segment | Rat (retinas) | SRM | 2 (2) | None | SPPS | [198] |
| Spliceosome | Human (HeLa cell line) | SRM | 5 (10) | None | SPPS (stoichiometry) | [199] |
| Transducin heterotrimeric G-protein | Bos taurus (retina) | MS1 | 3 (3) | None | QconCAT (stoichiometry) | [200] |
n.i., not indicated
Limited proteolysis (LiP) integrated with LC-SRM has been used to measure differences in protein conformation across experimental conditions [86, 87]. LiP-SRM was used to measure differences between two conformational states of the amyloid-forming protein α-synuclein (monomeric versus polymeric fibrillar, which are globally different structurally) and of myoglobin (unbound versus bound to heme, which are only structurally different at a single α-helical fold). LiP, DDA, and SRM were integrated to globally profile protein conformation differences between yeast cultured in glucose- versus ethanol-based media. Not surprisingly, conformational differences in the core carbon metabolism pathway were detected. Unexpectedly, the carboxy-terminal region of the 14-3-3 protein BMH1 was dramatically different during glucose- and ethanol-based metabolism. A BMH1-knockout strain displayed a growth defect in the ethanol-based medium, confirming that BMH1 has an as yet undetermined role in yeast ethanol metabolism. In a related study, DDA LC-MS was used to globally measure differences in protein conformation across cancer drug treatments (targeted MS was not used) [88]. Therefore, the use of LC-MS to discover changes in protein conformation across experimental conditions has developed into a novel and powerful methodology to discover changes in protein folding and/or protein-protein interaction.
In addition to studying protein conformation and protein-protein interaction, targeted MS has been used to study cellular components such as the adenovirus, the postsynaptic density, and the Gram-positive bacterial cell surface (Table 3). In the lattermost study, LC-SRM was used to produce a structural model of the Streptococcus pyogenes cell surface that included adhered human blood plasma proteins [89]. Targeted LC-MS has also been used to identify and quantify proteins proximal to G protein-coupled receptors (GPCRs) during signaling [90]. The β2 adrenergic receptor and the δ-opioid receptor were each coupled to an engineered ascorbic acid peroxidase (APEX). This enabled APEX catalyzed proximity labeling, discovery of proximal proteins using DDA MS, and quantification of proximal proteins using targeted MS to reveal spatiotemporal signaling by and trafficking of both GPCRs. WWP2 and TOM1 were identified as novel mediators of δ-opioid receptor degradation subsequent to prolonged activation (possibly via ubiquitination and trafficking to lysosomes).
5. Kinomics and phosphoproteomics
Targeted phosphoproteomic profiling has been used to investigate drug-induced phosphorylation, EGF-induced tyrosine phosphorylation, and mitochondrial phosphoproteomics (Table 4). It was first used to quantify EGF-induced tyrosine phosphorylation initially discovered using DDA LC-MS [91]. Seven time points following EGF treatment of 184A1 human mammary epithelial cells were analyzed using SRM, and 31 novel EGF-regulated tyrosine phosphorylation sites were discovered. In a second phosphoproteomic study, targeted MS enabled simplified profiling of drug-induced phosphorylation cascades using the P100 abridged set of target phosphopeptides [92]. In this investigation, clusters of correlated phosphosites were identified using DDA LC-MS, and each cluster was assayed using targeted MS of one or two representative phosphopeptide members. Hundreds of drug-treatment samples were rapidly profiled, and it was discovered that each drug produced a highly reproducible and distinct P100 phospho-signature. These two reports demonstrate the utility of targeted phosphoproteomics downstream of DDA LC-MS.
Table 4. Selected kinomics and phosphoproteomics studies.
| Topic | Species (specimen) | MS scan | Proteins (peptides) | PTMs | Isotopic labeling (absolute quantification) | Ref. |
|---|---|---|---|---|---|---|
| Breast cancer kinomics | Human (MCF-7 breast cancer cell lines) | SRM | 150 (179) | None | SIL chemical tag | [94] |
| Cancer Tyr kinomics | Human (12 cancer cell lines) | PRM | 86 (307) | None | None | [95] |
| Colorectal cancer kinomics | Human (HCT116 colorectal cancer cells) | PRM | 173 (822) | None | None | [96] |
| Drug-induced phosphorylation | Human (5 cell lines) | PRM | 86 (96) | Phosphorylation | SPPS | [92] |
| EGF-induced pTyr | Human (184A1 mammary epithelial cells) | SRM | 144 (226) | Phosphorylation | iTRAQ | [91] |
| Kinome activity during differentiation | Human (HeLa, THP-1 cell lines) | MS1 | 47 (60) | Phosphorylation | SPPS (pmol/(min*mg protein)) | [100] |
| Kinome activity in cancer | Human (14 cell lines, tumor) | MS1 | 73 (90) | Phosphorylation | SPPS (pmol/(min*mg protein)) | [99] |
| Kinome activity in cancer | Human (9 cell lines) | MS1 | 73 (90) | Phosphorylation | SPPS (pmol/(min*mg protein)) | [98] |
| Kinomics | Human (12 cell lines) | SRM | 83 (204) | None | None | [44] |
| Kinomics | Saccharomyces cerevisiae | SRM | 118 (214) | None | None | [201] |
| Kinomics of arsenic poisoning | Human (GM00637 skin fibroblast cell line) | SRM | 234 (245) | None | SILAC | [202] |
| Kinomics of diabetes | Human (HEK293T, GM00637 cell lines) | SRM | 328 (402) | None | SIL chemical tag | [203] |
| Lung cancer kinomics | Human (lung cancer tumor, cells) | PRM, SRM | 329 (789) | None, phosphorylation | None | [97] |
| Lung, skin cancer kinomics | Human (cancer cells, tumor) | SRM | 270 (301) | None | SIL chemical tag | [93] |
| Mitochondrial phosphoproteomics | Mouse (heart) | SRM | 7 (23) | None, phosphorylation | SPPS | [204] |
Targeted kinomics has been performed by coupling affinity enrichment of active protein kinases with targeted LC-MS, and this has been used to study breast, colorectal, lung, and skin cancer, as well as diabetes and arsenic poisoning (Table 4). Significant kinome reprogramming was discovered by comparing dasatinib-sensitive and insensitive melanoma cells, and also lung tumor and adjacent normal lung tissue [93]. A comparison of radiation therapy sensitive and resistant breast cancer cells revealed abundance alterations of kinases that control cell cycle progression and DNA repair [94]. Tyrosine kinase profiling was used to investigate EGF stimulation of skin cancer cells, APC mutation within colon cancer cells, ten colorectal cancer cell lines, and erlotinib-sensitive and insensitive lung cancer cells [95]. Colorectal cancer cell kinomics revealed compensatory activation of transforming growth factor beta (TGF-β) receptor superfamily members in response to treatment with three different mitogen-activated protein kinase (MAPK) inhibitors [96]. Fang and colleagues integrated kinomics and tyrosine phosphoproteomics to study lung cancer cell lines and tumors [97]. The activity of many kinases (measured using desthiobiotin-ATP labeling) correlated with their phosphorylation state. This study demonstrated the high value of integrating affinity enrichment of active protein kinases, phosphopeptide-enrichment, and targeted MS to study signaling cascades.
In addition to profiling the abundance and phosphorylation state of the kinome, targeted LC-MS has also been used to assay the enzymatic activity of the kinome using a method termed KAYAK (Kinase ActivitY Assay for Kinome profiling) (Table 4). In a KAYAK assay, the activation state of many kinases within a cell lysate is measured by incubating the lysate with a peptide library and subsequently performing targeted LC-MS of the resulting phosphopeptides. KAYAK was first used to profile the activity of the kinome upon mitogen stimulation, during the cell cycle, and across cancer cell lines [98, 99]. Fast protein liquid chromatography (FPLC) coupled with KAYAK was used to identify phosphorylation activity, the responsible kinase, and any associated protein complex members. A novel SRC-catalyzed tyrosine phosphorylation site on phosphatidylinositol 3-kinase (PI3K) regulatory subunits was discovered. In addition, the CDC2 – CCNB1 complex was identified as an activated kinase during mitosis. Therefore, KAYAK and FPLC-KAYAK have emerged as powerful methods for quantitative comparative kinome activity profiling and for the discovery of the responsible kinase(s). More recently, KAYAK was used to measure dose response curves of the PKC inhibitor Ro-31-8425 on kinases involved in monocyte differentiation into macrophages [100].
6. Metabolic pathways
A fundamental goal of systems biology is the comprehensive characterization of biological pathways to enable accurate pathway simulation at the molecular interaction level. These simulations are needed for the diagnosis of diseases, to design therapeutic interventions, and for pathway engineering. Numerous targeted proteomics investigations have been focused on the characterization of metabolic pathways (Table 5). Some of these projects used targeted proteomics to support Escherichia coli metabolic pathway engineering. These included optimizing the production of a sesquiterpene [101], engineering the mevalonate and tyrosine biosynthesis pathways [102], and increasing tyrosine production [103]. A novel principal component analysis was successfully applied to targeted proteomics and metabolomics data to direct engineering of the mevalonate pathway [104]. Fine-tuning the expression of a polyketide pathway protein was used to optimize the production of metabolites that could function as possible future biofuels [105].
Table 5. Selected studies of metabolic pathways.
| Topic | Species (specimen) | MS scan | Proteins (peptides) | PTMs | Isotopic labeling (absolute quantification) | Ref. |
|---|---|---|---|---|---|---|
| Acetyl-CoA biosynthesis | Escherichia coli, Saccharomyces cerevisiae | PRM | 177 (901) | None | None | [205] |
| Central carbon metabolism | Bacillus subtilis | SRM | 41 (85) | None | QconCAT | [206] |
| Central carbon metabolism | Corynebacterium glutamicum | SRM | 10 (30) | None | QconCAT (copies/cell) | [207] |
| Central carbon metabolism | Corynebacterium glutamicum | SRM | 19 (57) | None | 15N cells | [208] |
| Central carbon metabolism | Escherichia coli | SRM | 22 (99) | None | PSAQ (pmol/ml cytoplasm) | [209] |
| Central carbon metabolism | Human (colorectal cancer cells, tumors) | PRM, SRM | 75 (208) | None | None, SPPS (pmol/mg protein) | [210] |
| Central carbon metabolism | Human (MCF-7 breast cancer cell line) | SRM | 76 (134) | None | None | [211] |
| Central carbon metabolism | Saccharomyces cerevisiae | SRM | 137 (260) | None | SPPS (copies/cell) | [212] |
| Central carbon, amino acid metabolism | Saccharomyces cerevisiae | SRM | 135 (300) | None | 13C cells | [213] |
| Central carbon, amino acid metabolism | Saccharomyces cerevisiae | SRM | 137 (303) | None | 13C cells | [214] |
| Central carbon, amino acid metabolism | Saccharomyces cerevisiae | SRM | 228 (428) | None | 15N cells | [215] |
| Central carbon, amino acid metabolism | Saccharomyces cerevisiae | SRM | 4 (11) | None, phosphorylation | SPPS (pmol/ml lysate) | [106] |
| Central metabolic pathways | Escherichia coli | SRM | 392 (665) | None | QconCAT (pmol/injection) | [216] |
| Citric acid cycle | Mouse (liver) | SRM | 4 (58) | None | SILAM | [217] |
| Drug metabolism | Human (liver) | SRM | 25 (51) | None | QconCAT (pmol/mg protein) | [218] |
| Drug metabolism | Human (liver) | SRM | 14 (30) | None | SPPS (pmol/mg protein) | [219] |
| Drug metabolism | Human (liver) | SRM | 22 (38) | None | 18O-QconCAT (pmol/mg protein) | [220] |
| Drug metabolism | Human (liver, intestine) | SRM | 13 (13) | None | SPPS (pmol/mg protein) | [221] |
| Drug metabolism | Mouse (6 tissues) | SRM | 27 (27) | None | None, SPPS (pmol/mg protein) | [222] |
| Drug metabolism and transport | Human (intestine, liver, kidney) | PRM, SRM | 43 (44) | None | SPPS (pmol/mg protein) | [223] |
| Drug transport | Human (jejunum, ileum) | SRM | 10 (10) | None | SPPS (pmol/mg protein) | [224] |
| Drug transport | Human (jejunum, ileum) | SRM | 6 (6) | None | QconCAT (pmol/mg protein) | [225] |
| Drug transport | Human (renal cortex) | SRM | 17 (17) | None | SPPS (pmol/mg protein) | [226] |
| Drug transport | Mouse (brain, liver, kidney) | SRM | 36 (38) | None | SPPS (pmol/mg protein) | [227] |
| Eicosanoid synthesis pathway | Mouse (RAW 264.7 macrophage cell line) | SRM | 29 (41) | None | SILAC | [107] |
| Fatty acid synthesis pathway | Escherichia coli | SRM | 12 (22) | None | PSAQ (pmol/ml lysate) | [228] |
| Glycolytic pathway | Human (6 cell lines) | SRM | 24 (80) | None | SIL dimethylation | [229] |
| Glycolytic pathway | Saccharomyces cerevisiae | SRM | 27 (59) | None | QconCAT (copies/cell) | [230] |
| Glycolytic pathway | Saccharomyces cerevisiae | MS1 | 27 (59) | None | QconCAT (copies/cell) | [110] |
| High fat diet metabolic pathways | Mouse (liver) | SRM | 192 (309) | None | SILAC cells, SPPS | [231] |
| Metabolic and photosynthetic | Chlamydomonas reinhardtii | SRM | 88 (105) | None | SPPS (copies/cell) | [108] |
| pathways Mevalonate pathway | Escherichia coli | SRM | 17 (18) | None | QconCAT (pmol/ml cytoplasm) | [232] |
| Mevalonate pathway | Escherichia coli | SRM | 9 (n.i.) | None | None | [104] |
| Mevalonate, tyrosine pathways | Escherichia coli | SRM | 24 (48) | None | None | [102] |
| Mitochondrial metabolism | Human, mouse, rat (liver) | SRM | 57 (118) | None | QconCAT (pmol/mg protein) | [233] |
| Organohalide respiration | Dehalococcoides mccartyi | PRM, SRM | 10 (25) | None | SPPS (copies/cell) | [234] |
| Polyketide pathway | Escherichia coli | SRM | 2 (6) | None | None | [105] |
| Ribosome, glycolytic pathway | Saccharomyces cerevisiae | MS1 | 78 (102) | None | QconCAT (copies/cell) | [147] |
| Terpene pathway | Escherichia coli | SRM | 9 (9) | None | None | [101] |
| Terpene pathway | Picea abies (bark) | SRM | 16 (19) | None | SPPS | [109] |
| Tyr metabolic pathway | Escherichia coli | SRM | 11 (11) | None | None | [103] |
n.i., not indicated
In some investigations, targeted proteomics has been integrated with transcriptomics, metabolomics, and/or phosphoproteomics to study metabolic pathways. Oliveira and colleagues combined targeted proteomics with targeted phosphoproteomics to determine how protein abundance and phosphorylation affect enzymatic fluxes in yeast central metabolic pathways [106]. It was discovered that the absolute abundance of only the non-phosphorylated form of PDA1 correlated significantly with PDA1 enzymatic flux (total PDA1 abundance and phospho-Ser313 PDA1 abundance did not correlate with enzymatic flux). In another study, transcriptomics, targeted proteomics, and metabolomics were combined to produce a full picture of the macrophage prostaglandin biosynthetic pathway over a 24 hour time-course after stimulation with lipid A [107]. Using a similar approach, Wienkoop and colleagues used targeted proteomics and metabolomics to produce a detailed picture of metabolic and photosynthetic pathways within unicellular green algae [108]. In another multi-omic study, transcriptomics, targeted proteomics, and metabolomics were integrated to study the induction of terpene synthesis over a 32 day time-course in tree bark after treatment with an insect defense hormone [109].
In possibly the most extensive investigation of a metabolic pathway thus far, targeted proteomics, metabolomics, enzyme assays, and pathway modeling were integrated to construct and refine a model of the yeast glycolysis pathway [110]. Absolute abundance values of pathway proteins and metabolites were quantified using MS. Enzyme kinetics of purified proteins were assayed using in vitro conditions designed to mimic the in vivo environment. The protein abundance and kinetics data were input into an initial pathway model as parameters, pathway simulations were performed, and the resulting predicted metabolite abundances were compared to measured values. Eighteen iterations of model refinement were performed, partly to account for the effects of side reactions (e.g., the glycerol branch on the core glycolytic pathway), reducing the normalized root-mean-square deviation down to ∼30%.
7. Signaling pathways
Diverse signaling pathways have been studied using targeted LC-MS (Table 6). Quantification of circadian clock transcript and protein oscillations within wild type and knockout mice revealed the roles of clock proteins and enabled the development of a novel assay for circadian time [111]. Intriguingly, the delay between the circadian transcript and protein abundance peaks spanned from ∼0 to ∼8 hours via mechanisms that have yet to be discovered. In a recent report, we integrated DDA cellular proteomics, DDA secretomics, targeted secretomics, and transcriptomics to study pattern recognition receptor signaling [112]. This multi-omic approach enabled detailed comparisons of macrophages stimulated using individual pattern recognition receptor ligands (lipopolysaccharide, Pam3CSK4, and resiquimod) and whole bacteria (Pseudomonas aeruginosa, Staphylococcus aureus, and Burkholderia cenocepacia). Sabido and colleagues used targeted proteomics of the insulin signaling pathway merged with seven metabolic pathways to study metabolic syndrome resulting from a high-fat diet [113]. The metabolic pathways were: fatty acid biosynthesis, fatty acid β-oxidation, glycolysis and gluconeogenesis, pentose phosphate pathway, TCA cycle, ketogenesis, and glycogen metabolism. de Graaf and colleagues used targeted phosphoproteomics of the PI3K – mechanistic target of rapamycin (mTOR) – MAPK pathway to discover phosphorylation sites affected by oncogene-induced senescence and pharmacological intervention using BEZ235 (an inhibitor of both PI3K and mTOR) [114].
Table 6. Selected studies of signaling pathways.
| Topic | Species (specimen) | MS scan | Proteins (peptides) | PTMs | Isotopic labeling (absolute quantification) | Ref. |
|---|---|---|---|---|---|---|
| Chemotaxis signaling pathway | Mouse (RAW 264.7 macrophage cell line) | SRM | 41 (60) | None | SPPS (copies/cell) | [129] |
| Circadian clock | Mouse (liver) | SRM | 20 (124) | None, phosphorylation | QconCAT (pmol/mg protein), SIL dimethylation | [111] |
| DNA damage response network | Human (cells, tumor) | SRM | 26 (69) | None, phosphorylation | SPPS (pmol/mg protein) | [235] |
| DNA damage response network | Human (MCF-10A, blood cells) | SRM | 93 (107) | Phosphorylation | SPPS (pmol/mg protein) | [236] |
| EGF signaling pathway | Human (MCF-10A, tumor xenografts) | PRM, SRM | 10 (36) | Phosphorylation | SPPS-iTRAQ (copies/cell) | [237] |
| EGF-mediated Erk1 phosphorylation | Mouse (smooth muscle) | SRM | 1 (4) | None, phosphorylation | SPPS | [238] |
| EGFR-MAPK pathway | Human (8 cell lines) | SRM | 26 (53) | None, phosphorylation | SPPS (copies/cell) | [115] |
| ERK signaling pathway | Human (184A1 mammary epithelial cells) | SRM | 2 (8) | None, phosphorylation | SPPS (stoichiometry) | [239] |
| Galactose signaling pathway | Saccharomyces cerevisiae | SRM | 5 (11) | None | SPPS (copies/cell) | [128] |
| GRB2 signaling | Human (HEK293T cell line) | SRM | 90 (326) | None, phosphorylation | None | [240] |
| IGF-1 signaling pathway | Human (MCF-7 breast cancer cell line) | PRM | 75 (101) | Phosphorylation | None | [241] |
| Insulin, central metabolic pathways | Mouse (liver) | SRM | 144 (316) | None | SILAC cells | [113] |
| Neurotransmitter signaling | Mouse (6 brain tissues) | SRM | 260 (3501) | None | Recombinant protein | [242] |
| Pattern recognition receptor signaling | Arabidopsis thaliana (leaves) | SRM | 8 (13) | Phosphorylation | SPPS | [243] |
| Pattern recognition receptor signaling | Human (A549 lung epithelial cell line) | SRM | 10 (10) | None | SPPS | [244] |
| Pattern recognition receptor signaling | Mouse (RAW 264.7 macrophage cell line) | MS1 | 24 (178) | None | SILAC | [112] |
| Pattern recognition receptor signaling | Mouse (RAW 264.7 macrophage cell line) | PRM | 14 (14) | None | SPPS-mTRAQ (pmol/injection) | [245] |
| PI3K-mTOR-MAPK pathway | Human (2 lung cancer cell lines) | MS3, SRM | 30 (42) | Phosphorylation | SPPS (pmol/mg protein) | [246] |
| PI3K-mTOR-MAPK pathway | Human (TIG-3 fibroblast cell line) | SRM | 27 (51) | Phosphorylation | SPPS | [114] |
| RAF-MEK-ERK in vitro dynamics | Human, Xenopus laevis (recombinantly expressed) | MS1 | 2 (2) | Phosphorylation | SPPS | [117] |
| Synaptic glutamate signaling | Human (auditory cortex) | SRM | 155 (223) | None | SILAM mouse | [116] |
| WNT signaling pathway | Human (colon cancer cells, tissue) | SRM | 22 (85) | None | SPPS (copies/cell) | [247] |
Some quantitative LC-MS studies of signaling pathways have revealed patterns of conserved stoichiometry. Transcriptomics, targeted proteomics, and targeted phosphoproteomics were used to study the EGFR – MAPK pathway within a variety of normal and cancerous human cell types [115]. The stoichiometry of the pathway transcripts and proteins were found to be very similar across the cell types. The glutamatergic signaling pathway within the auditory cortex of schizophrenic and control subjects was quantitatively compared using LC-SRM, and pathway protein expression and co-expression were significantly correlated with the disease [116]. Dysregulation of co-expression strongly correlated with reduced dendritic spine density (a schizophrenia phenotype), demonstrating the high value of co-expression analysis of targeted proteomics data.
Quantification of pathway proteins and PTMs can be used to enable accurate pathway modeling. Targeted phosphoproteomics was used to study an in vitro minimal MAPK pathway consisting of only five proteins: a two stage phosphorylation cascade consisting of three proteins, and the two reverse reactions catalyzed by two phosphatases [117]. The experiments were designed to measure only quasi-steady-state behavior (reaction time = 30 min). Even in this simple system, perturbations caused by altering protein concentrations resulted in reequilibration of phosphorylation that required mass-action kinetics to correctly model (that is, simplistic inferences failed).
Determination of constants related to molecule-molecule interaction, molecular transformation (e.g., in protein conformation), and catalysis is necessary for the simulation of biological pathways at the molecular level. Numerous experimental methods have been developed to measure affinity constants in vitro (e.g., surface plasmon resonance) and in vivo (e.g., fluorescence cross-correlation spectrometry) [118-121]. In addition, structural modeling software has been developed that can be used to estimate affinity constants (PRODIGY, SDA, TransComp, and related tools) [122-127]. A novel strategy using targeted proteomics was used to measure in vivo dissociation constants of the yeast galactose signaling pathway consisting of galactose, four proteins (Gal1p, Gal3p, Gal4p, and Gal80p), and the genes transcriptionally activated by Gal4p (including those encoding Gal1p, Gal3p, and Gal80p) [128]. The abundance of the four proteins was systematically varied genetically and quantified using LC-SRM, and the pathway output (target gene transcription) was quantified. From these data, the protein-protein and protein-DNA dissociation constants were determined.
We used targeted proteomics to enable accurate pathway modeling of the mouse macrophage chemotaxis pathway [129]. RNA-seq was used to identify target protein splice isoforms and to estimate pathway protein absolute abundance values. LC-SRM was used to measure the absolute abundance of pathway proteins to accurately parameterize a pathway model. The Simmune software suite [130, 131] was used for rule-based pathway modeling, microscopy data were used for model training, and GTPase activation assay data were used for model accuracy testing. The model successfully simulated pathway behavior consistent with the GTPase data, which was not used for model training and was highly orthogonal to the microscopy data. In addition, 2,000 perturbed models were generated and used to demonstrate that the pathway model was robust. In this way, targeted MS and other state-of-the-art technologies are enabling the development of accurate and robust pathway models, which are critical to the advancement of systems biology, and which will aid the development of diagnostics, therapeutics, and personalized medicine.
8. Proteome-wide targeted MS and proteogenomics
Targeted MS has an important role in proteogenomics, especially coding sequence annotation (Table 7). Approximately 18% of the human proteome is classified as “missing” because there is not strong experimental evidence of the existence of these proteins [132]. To address this challenge, the Human Proteome Project is employing targeted proteomics and other technologies, and have confidently identified hundreds of formerly missing proteins. A typical strategy is to develop LC-SRM assays using synthesized peptide standards, and then to use these LC-SRM assays to analyze biological samples (selected because they express high levels of the corresponding transcript). Because of the excellent sensitivity and specificity of targeted MS, these efforts have often been very successful. For example, one study used DDA, PRM, and immunohistochemistry to confirm the expression of 206 previously missing proteins [133]. In another example, Omasits and colleagues combined a stringent re-analysis of proteomics and transcriptomics data with validation using LC-PRM to annotate coding sequences of Bartonella henselae [134]. Small coding sequences (∼50 residues or fewer) are especially challenging to annotate, and have recently been identified in numerous genomes including those of mammals [135, 136], mammalian mitochondria [137-139], and prokaryotes [140, 141]. For example, LC-MS1 and LC-SRM were used to identify and quantify small open reading frame-encoded polypeptides within human cancer cells [135, 136].
Table 7. Selected proteome-wide and proteogenomics studies.
| Topic | Species (specimen) | MS scan | Proteins (peptides) | PTMs | Isotopic labeling (absolute quantification) | Ref. |
|---|---|---|---|---|---|---|
| Coding sequence annotation | Bartonella henselae | PRM | 73 (107) | None | SPPS | [134] |
| Coding sequence annotation | Human (3 cancer cell lines) | SRM | 36 (62) | None | None | [135] |
| Coding sequence annotation | Human (glioblastoma, HepaRG cells) | SRM | 3 (6) | None | SPPS | [248] |
| Coding sequence annotation | Human (HeLa cell line) | SRM | 8 (9) | None | SPPS | [249] |
| Coding sequence annotation | Human (K-562 leukemia cell line) | MS1 | 8 (8) | None | SPPS (copies/cell) | [136] |
| Coding sequence annotation | Human (liver cells, tissue) | SRM | 81 (81) | None | SPPS (copies/cell) | [250] |
| Coding sequence annotation | Human (liver) | SRM | 57 (57) | None | None | [251] |
| Coding sequence annotation | Human (plasma) | SRM | 84 (84) | None | SPPS (pmol/ml plasma) | [252] |
| Coding sequence annotation | Human (plasma, liver) | SRM | 249 (516) | None | None | [253] |
| Coding sequence annotation | Human (plasma, liver) | SRM | 267 (267) | None | SPPS (copies/cell) | [254] |
| Coding sequence annotation | Human (plasma, liver, HepG2 cells) | SRM | 250 (693) | None | SPPS (copies/ml plasma, copies/cell) | [255] |
| Coding sequence annotation | Human (spermatozoa) | PRM | 31 (51) | None | SPPS | [133] |
| Glycoproteome-wide analysis | Human, mouse (plasma, serum) | SRM | 3360 (5568) | Glycosylation | SPPS (pmol/ml biofluid) | [256] |
| microRNA-mediated regulation | Caenorhabditis elegans | SRM | 215 (470) | None | 15N cells | [257] |
| microRNA-mediated regulation | Caenorhabditis elegans | SRM | 307 (591) | None | ICAT, 15N cells | [258] |
| Neurexin isoform profiling | Mouse (8 brain tissues) | SRM | 20 (44) | None | PSAQ (pmol/mg protein) | [259] |
| Proteome-wide abundance estimation | Bacillus subtilis | SRM | 7 (16) | None | QconCAT (copies/cell) | [260] |
| Proteome-wide abundance estimation | Bacillus subtilis, Staphylococcus aureus | SRM | 23 (55) | None | QconCAT (copies/cell) | [261] |
| Proteome-wide abundance estimation | Escherichia coli | SRM | 41 (41) | None | SPPS (copies/cell) | [262] |
| Proteome-wide abundance estimation | Human (9 cell lines, 11 tissues) | PRM | 55 (113) | None | QconCAT (copies/cell) | [146] |
| Proteome-wide abundance estimation | Human (U2OS cell line) | MS1 | 53 (71) | None | SPPS (copies/cell) | [144] |
| Proteome-wide abundance estimation | Leptospira interrogans | MS1 | 19 (29) | None | SPPS (copies/cell) | [263] |
| Proteome-wide abundance estimation | Leptospira interrogans | SRM | 19 (32) | None | SPPS (copies/cell) | [143] |
| Proteome-wide analysis | Human (5 cell lines) | SRM | 20225 (158015) | None | None | [148] |
| Proteome-wide analysis | Human (TIG-3 lung fibroblasts) | SRM | 16108 (138009) | None | PSAQ-mTRAQ (copies/cell), PSAQ (copies/cell), SPPS (copies/cell) | [149] |
| Proteome-wide analysis | Mycobacterium tuberculosis | SRM | 3894 (15679) | None | None, SPPS (pmol/mg protein) | [264] |
| Proteome-wide analysis | Saccharomyces cerevisiae | SRM | 1167 (1700) | None | QconCAT (copies/cell) | [265] |
| Proteome-wide analysis | Saccharomyces cerevisiae | SRM | 6399 (28216) | None | 15N cells | [266] |
| Proteome-wide analysis | Streptococcus pyogenes | SRM | 1332 (2594) | None | None | [267] |
| RNAi knockdown efficacy | Human (GM00639, HEK293T cells), Drosophila melanogaster | SRM | 6 (11) | None | None, SPPS | [268] |
| Single amino acid polymorphisms | Human (plasma) | SRM | 3 (6) | None | SPPS (pmol/ml plasma) | [269] |
Targeted proteomics coupled with other technologies has been used for proteome-wide absolute abundance estimation [142]. DDA and targeted LC-MS were combined to estimate protein abundances from precursor ion intensity values [143, 144]. Similarly, because transcript and protein abundance are sometimes significantly correlated [145], targeted proteomics coupled with transcriptomics has been used to estimate protein abundance from transcript abundance [129, 146]. It is important to note that transcript-protein absolute abundance correlations can vary dramatically across different target protein sets. For example, in yeast the transcript-protein correlation of the glycolysis pathway was strong (Spearman r = 0.97), whereas the correlation for ribosomal proteins was weak (Spearman r = ∼0) [147]. This disparity may have resulted from the very different ranges in protein abundance across the two target protein sets. The glycolysis proteins ranged from 40,000 – 1,500,000 copies per cell, whereas the ribosomal proteins only ranged from 250,000 – 400,000 copies per cell. Therefore, protein abundance estimation using targeted proteomics integrated with global transcriptomics requires that the target proteins range in abundance across many orders of magnitude.
Recently, targeted LC-MS has developed into a technology capable of proteome-wide investigation, and currently the proteomes of four species have been analyzed almost in entirety: Mycobacterium tuberculosis, Streptococcus pyogenes, Saccharomyces cerevisiae, and Homo sapiens (Table 7). The first human proteome-wide investigation used SPPS to generate 166,174 peptide standards, which were used to successfully develop LC-SRM assays for 20,225 proteins (158,015 peptides) [148]. To demonstrate the utility of this resource, the authors investigated the effects of atorvastatin treatment on the cholesterol synthesis pathway in liver cells, and they also investigated a network of proteins associated with docetaxel inhibition of prostate cancer cell division. A second human proteome-wide investigation used 18,081 recombinant proteins to successfully develop LC-SRM assays for 16,108 proteins (138,009 peptides) [149]. The authors used this resource to quantify 634 enzymes to study the effects of oncogenesis on metabolic pathways. Because the development of a targeted LC-MS assay can be demanding and time-consuming, the proteome-wide development of human protein assays has greatly increased the accessibility of this powerful tool to scientists across a wide spectrum of biomedical research fields.
9. Conclusion
Proteomics has evolved far beyond basic proteomic profiling using DDA LC-MS. Targeted proteomics has been used to robustly quantify protein abundance, synthesis, degradation, PTMs, and other chemical modifications. Proteogenomic applications include identification of splice isoforms, identification of single amino acid polymorphisms and other genetic variants, identification of missing proteins, and quantification of DNA- and RNA-level regulation of protein expression (e.g., RNA interference). Functional proteomics applications include kinomics, enzymatic activity assays related to protein modification (e.g., protein phosphorylation, proteolysis), and measurement of protein conformation, protein-protein interaction, protein interaction with other molecules, and protein-protein subcellular proximity. These technologies are enabling biological pathway mapping, simulation, and engineering, and targeted proteomics integrated with other technologies (e.g., transcriptomics, metabolomics) has been especially productive. With the recent development of LC-SRM assays for nearly the entire human proteome, targeted proteomics has emerged as a powerful technology for biomedical research, clinical applications, and biotechnology research and development.
Significance.
This manuscript is a comprehensive review of the recent advances in bottom-up targeted proteomics research for cell signaling pathways and modeling.
Acknowledgments
The authors would like to thank Casey M. Daniels for helpful suggestions. This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Disclosures: The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537(7620):347–55. doi: 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- 2.Boersema PJ, Kahraman A, Picotti P. Proteomics beyond large-scale protein expression analysis. Curr Opin Biotechnol. 2015;34:162–70. doi: 10.1016/j.copbio.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 3.Germain RN, Meier-Schellersheim M, Nita-Lazar A, Fraser ID. Systems biology in immunology: a computational modeling perspective. Annu Rev Immunol. 2011;29:527–85. doi: 10.1146/annurev-immunol-030409-101317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sajic T, Liu Y, Aebersold R. Using data-independent, high-resolution mass spectrometry in protein biomarker research: perspectives and clinical applications. Proteomics Clin Appl. 2015;9(3-4):307–21. doi: 10.1002/prca.201400117. [DOI] [PubMed] [Google Scholar]
- 5.Bilbao A, Varesio E, Luban J, Strambio-De-Castillia C, Hopfgartner G, Muller M, Lisacek F. Processing strategies and software solutions for data-independent acquisition in mass spectrometry. Proteomics. 2015;15(5-6):964–80. doi: 10.1002/pmic.201400323. [DOI] [PubMed] [Google Scholar]
- 6.Collins BC, Hunter CL, Liu Y, Schilling B, Rosenberger G, Bader SL, Chan DW, Gibson BW, Gingras AC, Held JM, Hirayama-Kurogi M, Hou G, Krisp C, Larsen B, Lin L, Liu S, Molloy MP, Moritz RL, Ohtsuki S, Schlapbach R, Selevsek N, Thomas SN, Tzeng SC, Zhang H, Aebersold R. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat Commun. 2017;8(1):291. doi: 10.1038/s41467-017-00249-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Terfve C, Sabido E, Wu Y, Goncalves E, Choi M, Vaga S, Vitek O, Saez-Rodriguez J, Aebersold R. System-Wide Quantitative Proteomics of the Metabolic Syndrome in Mice: Genotypic and Dietary Effects. J Proteome Res. 2017;16(2):831–841. doi: 10.1021/acs.jproteome.6b00815. [DOI] [PubMed] [Google Scholar]
- 8.Ebhardt HA, Root A, Sander C, Aebersold R. Applications of targeted proteomics in systems biology and translational medicine. Proteomics. 2015;15(18):3193–208. doi: 10.1002/pmic.201500004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meyer JG, Schilling B. Clinical applications of quantitative proteomics using targeted and untargeted data-independent acquisition techniques. Expert Rev Proteomics. 2017;14(5):419–429. doi: 10.1080/14789450.2017.1322904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahrens CH, Brunner E, Qeli E, Basler K, Aebersold R. Generating and navigating proteome maps using mass spectrometry. Nat Rev Mol Cell Biol. 2010;11(11):789–801. doi: 10.1038/nrm2973. [DOI] [PubMed] [Google Scholar]
- 11.de Godoy LM, Olsen JV, Cox J, Nielsen ML, Hubner NC, Frohlich F, Walther TC, Mann M. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature. 2008;455(7217):1251–4. doi: 10.1038/nature07341. [DOI] [PubMed] [Google Scholar]
- 12.Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, Thomas JK, Muthusamy B, Leal-Rojas P, Kumar P, Sahasrabuddhe NA, Balakrishnan L, Advani J, George B, Renuse S, Selvan LD, Patil AH, Nanjappa V, Radhakrishnan A, Prasad S, Subbannayya T, Raju R, Kumar M, Sreenivasamurthy SK, Marimuthu A, Sathe GJ, Chavan S, Datta KK, Subbannayya Y, Sahu A, Yelamanchi SD, Jayaram S, Rajagopalan P, Sharma J, Murthy KR, Syed N, Goel R, Khan AA, Ahmad S, Dey G, Mudgal K, Chatterjee A, Huang TC, Zhong J, Wu X, Shaw PG, Freed D, Zahari MS, Mukherjee KK, Shankar S, Mahadevan A, Lam H, Mitchell CJ, Shankar SK, Satishchandra P, Schroeder JT, Sirdeshmukh R, Maitra A, Leach SD, Drake CG, Halushka MK, Prasad TS, Hruban RH, Kerr CL, Bader GD, Iacobuzio-Donahue CA, Gowda H, Pandey A. A draft map of the human proteome. Nature. 2014;509(7502):575–81. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilhelm M, Schlegl J, Hahne H, Gholami AM, Lieberenz M, Savitski MM, Ziegler E, Butzmann L, Gessulat S, Marx H, Mathieson T, Lemeer S, Schnatbaum K, Reimer U, Wenschuh H, Mollenhauer M, Slotta-Huspenina J, Boese JH, Bantscheff M, Gerstmair A, Faerber F, Kuster B. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509(7502):582–7. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 14.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143(7):1174–89. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bluemlein K, Ralser M. Monitoring protein expression in whole-cell extracts by targeted label- and standard-free LC-MS/MS. Nat Protoc. 2011;6(6):859–69. doi: 10.1038/nprot.2011.333. [DOI] [PubMed] [Google Scholar]
- 16.Stergachis AB, MacLean B, Lee K, Stamatoyannopoulos JA, MacCoss MJ. Rapid empirical discovery of optimal peptides for targeted proteomics. Nat Methods. 2011;8(12):1041–3. doi: 10.1038/nmeth.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manes NP, Mann JM, Nita-Lazar A. Selected Reaction Monitoring Mass Spectrometry for Absolute Protein Quantification. J Vis Exp. 2015;102:e52959. doi: 10.3791/52959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng Y, Picotti P. Selected Reaction Monitoring to Measure Proteins of Interest in Complex Samples: A Practical Guide. Methods Mol Biol. 2016;1394:43–56. doi: 10.1007/978-1-4939-3341-9_4. [DOI] [PubMed] [Google Scholar]
- 19.Faca VM. Selective Reaction Monitoring for Quantitation of Cellular Proteins. Methods Mol Biol. 2017;1546:213–221. doi: 10.1007/978-1-4939-6730-8_18. [DOI] [PubMed] [Google Scholar]
- 20.Russo P, Hood BL, Bateman NW, Conrads TP. Quantitative Mass Spectrometry by Isotope Dilution and Multiple Reaction Monitoring (MRM) Methods Mol Biol. 2017;1606:313–332. doi: 10.1007/978-1-4939-6990-6_20. [DOI] [PubMed] [Google Scholar]
- 21.Hoofnagle AN, Whiteaker JR, Carr SA, Kuhn E, Liu T, Massoni SA, Thomas SN, Townsend RR, Zimmerman LJ, Boja E, Chen J, Crimmins DL, Davies SR, Gao Y, Hiltke TR, Ketchum KA, Kinsinger CR, Mesri M, Meyer MR, Qian WJ, Schoenherr RM, Scott MG, Shi T, Whiteley GR, Wrobel JA, Wu C, Ackermann BL, Aebersold R, Barnidge DR, Bunk DM, Clarke N, Fishman JB, Grant RP, Kusebauch U, Kushnir MM, Lowenthal MS, Moritz RL, Neubert H, Patterson SD, Rockwood AL, Rogers J, Singh RJ, Van Eyk JE, Wong SH, Zhang S, Chan DW, Chen X, Ellis MJ, Liebler DC, Rodland KD, Rodriguez H, Smith RD, Zhang Z, Zhang H, Paulovich AG. Recommendations for the Generation, Quantification, Storage, and Handling of Peptides Used for Mass Spectrometry-Based Assays. Clin Chem. 2016;62(1):48–69. doi: 10.1373/clinchem.2015.250563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jsselstijn IL, Stoop MP, Stingl C, Sillevis Smitt PA, Luider TM, Dekker LJ. Comparative study of targeted and label-free mass spectrometry methods for protein quantification. J Proteome Res. 2013;12(4):2005–11. doi: 10.1021/pr301221f. [DOI] [PubMed] [Google Scholar]
- 23.Bauer M, Ahrne E, Baron AP, Glatter T, Fava LL, Santamaria A, Nigg EA, Schmidt A. Evaluation of data-dependent and -independent mass spectrometric workflows for sensitive quantification of proteins and phosphorylation sites. J Proteome Res. 2014;13(12):5973–88. doi: 10.1021/pr500860c. [DOI] [PubMed] [Google Scholar]
- 24.Faktor J, Sucha R, Paralova V, Liu Y, Bouchal P. Comparison of targeted proteomics approaches for detecting and quantifying proteins derived from human cancer tissues. Proteomics. 2017;17(5):1600323. doi: 10.1002/pmic.201600323. [DOI] [PubMed] [Google Scholar]
- 25.Kockmann T, Trachsel C, Panse C, Wahlander A, Selevsek N, Grossmann J, Wolski WE, Schlapbach R. Targeted proteomics coming of age - SRM, PRM and DIA performance evaluated from a core facility perspective. Proteomics. 2016;16(15-16):2183–92. doi: 10.1002/pmic.201500502. [DOI] [PubMed] [Google Scholar]
- 26.Sabbagh B, Mindt S, Neumaier M, Findeisen P. Clinical applications of MS-based protein quantification. Proteomics Clin Appl. 2016;10(4):323–45. doi: 10.1002/prca.201500116. [DOI] [PubMed] [Google Scholar]
- 27.Grebe SKG, Singh RJ. Clinical peptide and protein quantification by mass spectrometry (MS) Trends Analyt Chem. 2016;84:131–143. [Google Scholar]
- 28.Cross TG, Hornshaw MP. Can LC and LC-MS ever replace immunoassays? J Appl Bioanal. 2016;2(4):108–116. [Google Scholar]
- 29.Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat Methods. 2012;9(6):555–66. doi: 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- 30.Gianazza E, Tremoli E, Banfi C. The selected reaction monitoring/multiple reaction monitoring-based mass spectrometry approach for the accurate quantitation of proteins: clinical applications in the cardiovascular diseases. Expert Rev Proteomics. 2014;11(6):771–88. doi: 10.1586/14789450.2014.947966. [DOI] [PubMed] [Google Scholar]
- 31.Shi T, Song E, Nie S, Rodland KD, Liu T, Qian WJ, Smith RD. Advances in targeted proteomics and applications to biomedical research. Proteomics. 2016;16(15-16):2160–82. doi: 10.1002/pmic.201500449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vidova V, Spacil Z. A review on mass spectrometry-based quantitative proteomics: Targeted and data independent acquisition. Anal Chim Acta. 2017;964:7–23. doi: 10.1016/j.aca.2017.01.059. [DOI] [PubMed] [Google Scholar]
- 33.Lesur A, Domon B. Advances in high-resolution accurate mass spectrometry application to targeted proteomics. Proteomics. 2015;15(5-6):880–90. doi: 10.1002/pmic.201400450. [DOI] [PubMed] [Google Scholar]
- 34.Villanueva J, Carrascal M, Abian J. Isotope dilution mass spectrometry for absolute quantification in proteomics: concepts and strategies. J Proteomics. 2014;96:184–99. doi: 10.1016/j.jprot.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 35.Brusniak MY, Chu CS, Kusebauch U, Sartain MJ, Watts JD, Moritz RL. An assessment of current bioinformatic solutions for analyzing LC-MS data acquired by selected reaction monitoring technology. Proteomics. 2012;12(8):1176–84. doi: 10.1002/pmic.201100571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cham Mead JA, Bianco L, Bessant C. Free computational resources for designing selected reaction monitoring transitions. Proteomics. 2010;10(6):1106–26. doi: 10.1002/pmic.200900396. [DOI] [PubMed] [Google Scholar]
- 37.Colangelo CM, Chung L, Bruce C, Cheung KH. Review of software tools for design and analysis of large scale MRM proteomic datasets. Methods. 2013;61(3):287–98. doi: 10.1016/j.ymeth.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fan J, Mohareb F, Bond NJ, Lilley KS, Bessant C. MRMaid 2.0: mining PRIDE for evidence-based SRM transitions. OMICS. 2012;16(9):483–8. doi: 10.1089/omi.2011.0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qeli E, Ahrens CH. PeptideClassifier for protein inference and targeted quantitative proteomics. Nat Biotechnol. 2010;28(7):647–50. doi: 10.1038/nbt0710-647. [DOI] [PubMed] [Google Scholar]
- 40.Mohammed Y, Domanski D, Jackson AM, Smith DS, Deelder AM, Palmblad M, Borchers CH. PeptidePicker: a scientific workflow with web interface for selecting appropriate peptides for targeted proteomics experiments. J Proteomics. 2014;106:151–61. doi: 10.1016/j.jprot.2014.04.018. [DOI] [PubMed] [Google Scholar]
- 41.Demeure K, Duriez E, Domon B, Niclou SP. PeptideManager: a peptide selection tool for targeted proteomic studies involving mixed samples from different species. Front Genet. 2014;5:305. doi: 10.3389/fgene.2014.00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Searle BC, Egertson JD, Bollinger JG, Stergachis AB, MacCoss MJ. Using Data Independent Acquisition (DIA) to Model High-responding Peptides for Targeted Proteomics Experiments. Mol Cell Proteomics. 2015;14(9):2331–40. doi: 10.1074/mcp.M115.051300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pino LK, Searle BC, Bollinger JG, Nunn B, MacLean B, MacCoss MJ. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom Rev. 2017 doi: 10.1002/mas.21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Worboys JD, Sinclair J, Yuan Y, Jorgensen C. Systematic evaluation of quantotypic peptides for targeted analysis of the human kinome. Nat Methods. 2014;11(10):1041–4. doi: 10.1038/nmeth.3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nesvizhskii AI. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J Proteomics. 2010;73(11):2092–123. doi: 10.1016/j.jprot.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perez-Riverol Y, Alpi E, Wang R, Hermjakob H, Vizcaino JA. Making proteomics data accessible and reusable: current state of proteomics databases and repositories. Proteomics. 2015;15(5-6):930–49. doi: 10.1002/pmic.201400302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walsh GM, Lin S, Evans DM, Khosrovi-Eghbal A, Beavis RC, Kast J. Implementation of a data repository-driven approach for targeted proteomics experiments by multiple reaction monitoring. J Proteomics. 2009;72(5):838–52. doi: 10.1016/j.jprot.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lam H, Deutsch EW, Eddes JS, Eng JK, Stein SE, Aebersold R. Building consensus spectral libraries for peptide identification in proteomics. Nat Methods. 2008;5(10):873–5. doi: 10.1038/nmeth.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deutsch EW, Csordas A, Sun Z, Jarnuczak A, Perez-Riverol Y, Ternent T, Campbell DS, Bernal-Llinares M, Okuda S, Kawano S, Moritz RL, Carver JJ, Wang M, Ishihama Y, Bandeira N, Hermjakob H, Vizcaino JA. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017;45(D1):D1100–D1106. doi: 10.1093/nar/gkw936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eyers CE, Lawless C, Wedge DC, Lau KW, Gaskell SJ, Hubbard SJ. CONSeQuence: prediction of reference peptides for absolute quantitative proteomics using consensus machine learning approaches. Mol Cell Proteomics. 2011;10(11):M110 003384. doi: 10.1074/mcp.M110.003384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fusaro VA, Mani DR, Mesirov JP, Carr SA. Prediction of high-responding peptides for targeted protein assays by mass spectrometry. Nat Biotechnol. 2009;27(2):190–8. doi: 10.1038/nbt.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qeli E, Omasits U, Goetze S, Stekhoven DJ, Frey JE, Basler K, Wollscheid B, Brunner E, Ahrens CH. Improved prediction of peptide detectability for targeted proteomics using a rank-based algorithm and organism-specific data. J Proteomics. 2014;108:269–83. doi: 10.1016/j.jprot.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 53.Mallick P, Schirle M, Chen SS, Flory MR, Lee H, Martin D, Ranish J, Raught B, Schmitt R, Werner T, Kuster B, Aebersold R. Computational prediction of proteotypic peptides for quantitative proteomics. Nat Biotechnol. 2007;25(1):125–31. doi: 10.1038/nbt1275. [DOI] [PubMed] [Google Scholar]
- 54.Webb-Robertson BJ, Cannon WR, Oehmen CS, Shah AR, Gurumoorthi V, Lipton MS, Waters KM. A support vector machine model for the prediction of proteotypic peptides for accurate mass and time proteomics. Bioinformatics. 2008;24(13):1503–9. doi: 10.1093/bioinformatics/btn218. [DOI] [PubMed] [Google Scholar]
- 55.Helsens K, Mueller M, Hulstaert N, Martens L. Sigpep: calculating unique peptide signature transition sets in a complete proteome background. Proteomics. 2012;12(8):1142–6. doi: 10.1002/pmic.201100566. [DOI] [PubMed] [Google Scholar]
- 56.Rost H, Malmstrom L, Aebersold R. A computational tool to detect and avoid redundancy in selected reaction monitoring. Mol Cell Proteomics. 2012;11(8):540–9. doi: 10.1074/mcp.M111.013045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nasso S, Goetze S, Martens L. Ariadne's Thread: A Robust Software Solution Leading to Automated Absolute and Relative Quantification of SRM Data. J Proteome Res. 2015;14(9):3779–92. doi: 10.1021/pr500996s. [DOI] [PubMed] [Google Scholar]
- 58.Teleman J, Karlsson C, Waldemarson S, Hansson K, James P, Malmstrom J, Levander F. Automated selected reaction monitoring software for accurate label-free protein quantification. J Proteome Res. 2012;11(7):3766–73. doi: 10.1021/pr300256x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abbatiello SE, Mani DR, Keshishian H, Carr SA. Automated detection of inaccurate and imprecise transitions in peptide quantification by multiple reaction monitoring mass spectrometry. Clin Chem. 2010;56(2):291–305. doi: 10.1373/clinchem.2009.138420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reiter L, Rinner O, Picotti P, Huttenhain R, Beck M, Brusniak MY, Hengartner MO, Aebersold R. mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nat Methods. 2011;8(5):430–5. doi: 10.1038/nmeth.1584. [DOI] [PubMed] [Google Scholar]
- 61.Martin DB, Holzman T, May D, Peterson A, Eastham A, Eng J, McIntosh M. MRMer, an interactive open source and cross-platform system for data extraction and visualization of multiple reaction monitoring experiments. Mol Cell Proteomics. 2008;7(11):2270–8. doi: 10.1074/mcp.M700504-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choi M, Chang CY, Clough T, Broudy D, Killeen T, MacLean B, Vitek O. MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics. 2014;30(17):2524–6. doi: 10.1093/bioinformatics/btu305. [DOI] [PubMed] [Google Scholar]
- 63.Mohammed Y, Percy AJ, Chambers AG, Borchers CH. Qualis-SIS: automated standard curve generation and quality assessment for multiplexed targeted quantitative proteomic experiments with labeled standards. J Proteome Res. 2015;14(2):1137–46. doi: 10.1021/pr5010955. [DOI] [PubMed] [Google Scholar]
- 64.Chang CY, Picotti P, Huttenhain R, Heinzelmann-Schwarz V, Jovanovic M, Aebersold R, Vitek O. Protein significance analysis in selected reaction monitoring (SRM) measurements. Mol Cell Proteomics. 2012;11(4):M111 014662. doi: 10.1074/mcp.M111.014662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Whiteaker JR, Halusa GN, Hoofnagle AN, Sharma V, Mac Lean B, Yan P, Wrobel JA, Kennedy J, Mani DR, Zimmerman LJ, Meyer MR, Mesri M, Rodriguez H, C. Clinical Proteomic Tumor Analysis. Paulovich AG. CPTAC Assay Portal: a repository of targeted proteomic assays. Nat Methods. 2014;11(7):703–4. doi: 10.1038/nmeth.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sharma V, Eckels J, Taylor GK, Shulman NJ, Stergachis AB, Joyner SA, Yan P, Whiteaker JR, Halusa GN, Schilling B, Gibson BW, Colangelo CM, Paulovich AG, Carr SA, Jaffe JD, MacCoss MJ, MacLean B. Panorama: a targeted proteomics knowledge base. J Proteome Res. 2014;13(9):4205–10. doi: 10.1021/pr5006636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Farrah T, Deutsch EW, Kreisberg R, Sun Z, Campbell DS, Mendoza L, Kusebauch U, Brusniak MY, Huttenhain R, Schiess R, Selevsek N, Aebersold R, Moritz RL. PASSEL: the PeptideAtlas SRMexperiment library. Proteomics. 2012;12(8):1170–5. doi: 10.1002/pmic.201100515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baud A, Wessely F, Mazzacuva F, McCormick J, Camuzeaux S, Heywood WE, Little D, Vowles J, Tuefferd M, Mosaku O, Lako M, Armstrong L, Webber C, Cader MZ, Peeters P, Gissen P, Cowley SA, Mills K. Multiplex High-Throughput Targeted Proteomic Assay To Identify Induced Pluripotent Stem Cells. Anal Chem. 2017;89(4):2440–2448. doi: 10.1021/acs.analchem.6b04368. [DOI] [PubMed] [Google Scholar]
- 69.Soste M, Hrabakova R, Wanka S, Melnik A, Boersema P, Maiolica A, Wernas T, Tognetti M, von Mering C, Picotti P. A sentinel protein assay for simultaneously quantifying cellular processes. Nat Methods. 2014;11(10):1045–8. doi: 10.1038/nmeth.3101. [DOI] [PubMed] [Google Scholar]
- 70.Erickson BK, Rose CM, Braun CR, Erickson AR, Knott J, McAlister GC, Wuhr M, Paulo JA, Everley RA, Gygi SP. A Strategy to Combine Sample Multiplexing with Targeted Proteomics Assays for High-Throughput Protein Signature Characterization. Mol Cell. 2017;65(2):361–370. doi: 10.1016/j.molcel.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kennedy JJ, Abbatiello SE, Kim K, Yan P, Whiteaker JR, Lin C, Kim JS, Zhang Y, Wang X, Ivey RG, Zhao L, Min H, Lee Y, Yu MH, Yang EG, Lee C, Wang P, Rodriguez H, Kim Y, Carr SA, Paulovich AG. Demonstrating the feasibility of large-scale development of standardized assays to quantify human proteins. Nat Methods. 2014;11(2):149–55. doi: 10.1038/nmeth.2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Drabovich AP, Pavlou MP, Schiza C, Diamandis EP. Dynamics of Protein Expression Reveals Primary Targets and Secondary Messengers of Estrogen Receptor Alpha Signaling in MCF-7 Breast Cancer Cells. Mol Cell Proteomics. 2016;15(6):2093–107. doi: 10.1074/mcp.M115.057257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brownridge P, Lawless C, Payapilly AB, Lanthaler K, Holman SW, Harman VM, Grant CM, Beynon RJ, Hubbard SJ. Quantitative analysis of chaperone network throughput in budding yeast. Proteomics. 2013;13(8):1276–91. doi: 10.1002/pmic.201200412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mirzaei H, Knijnenburg TA, Kim B, Robinson M, Picotti P, Carter GW, Li S, Dilworth DJ, Eng JK, Aitchison JD, Shmulevich I, Galitski T, Aebersold R, Ranish J. Systematic measurement of transcription factor-DNA interactions by targeted mass spectrometry identifies candidate gene regulatory proteins. Proc Natl Acad Sci U S A. 2013;110(9):3645–50. doi: 10.1073/pnas.1216918110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang CC, Li R, Jiang H, Lin S, Rogalski JC, Liu K, Kast J. Development and application of a quantitative multiplexed small GTPase activity assay using targeted proteomics. J Proteome Res. 2015;14(2):967–76. doi: 10.1021/pr501010v. [DOI] [PubMed] [Google Scholar]
- 76.Agard NJ, Mahrus S, Trinidad JC, Lynn A, Burlingame AL, Wells JA. Global kinetic analysis of proteolysis via quantitative targeted proteomics. Proc Natl Acad Sci U S A. 2012;109(6):1913–8. doi: 10.1073/pnas.1117158109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang G, Fang B, Liu RZ, Lin H, Kinose F, Bai Y, Oguz U, Remily-Wood ER, Li J, Altiok S, Eschrich S, Koomen J, Haura EB. Mass spectrometry mapping of epidermal growth factor receptor phosphorylation related to oncogenic mutations and tyrosine kinase inhibitor sensitivity. J Proteome Res. 2011;10(1):305–19. doi: 10.1021/pr1006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sherrod SD, Myers MV, Li M, Myers JS, Carpenter KL, Maclean B, Maccoss MJ, Liebler DC, Ham AJ. Label-free quantitation of protein modifications by pseudo selected reaction monitoring with internal reference peptides. J Proteome Res. 2012;11(6):3467–79. doi: 10.1021/pr201240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Creech AL, Taylor JE, Maier VK, Wu X, Feeney CM, Udeshi ND, Peach SE, Boehm JS, Lee JT, Carr SA, Jaffe JD. Building the Connectivity Map of epigenetics: chromatin profiling by quantitative targeted mass spectrometry. Methods. 2015;72:57–64. doi: 10.1016/j.ymeth.2014.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Darwanto A, Curtis MP, Schrag M, Kirsch W, Liu P, Xu G, Neidigh JW, Zhang K. A modified “cross-talk” between histone H2B Lys-120 ubiquitination and H3 Lys-79 methylation. J Biol Chem. 2010;285(28):21868–76. doi: 10.1074/jbc.M110.126813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ohtake F, Saeki Y, Ishido S, Kanno J, Tanaka K. The K48-K63 Branched Ubiquitin Chain Regulates NF-kappaB Signaling. Mol Cell. 2016;64(2):251–266. doi: 10.1016/j.molcel.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 82.Shi Z, Fujii K, Kovary KM, Genuth NR, Rost HL, Teruel MN, Barna M. Heterogeneous Ribosomes Preferentially Translate Distinct Subpools of mRNAs Genome-wide. Mol Cell. 2017;67(1):71–83 e7. doi: 10.1016/j.molcel.2017.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ori A, Banterle N, Iskar M, Andres-Pons A, Escher C, Khanh Bui H, Sparks L, Solis-Mezarino V, Rinner O, Bork P, Lemke EA, Beck M. Cell type-specific nuclear pores: a case in point for context-dependent stoichiometry of molecular machines. Mol Syst Biol. 2013;9:648. doi: 10.1038/msb.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.von Appen A, Kosinski J, Sparks L, Ori A, DiGuilio AL, Vollmer B, Mackmull MT, Banterle N, Parca L, Kastritis P, Buczak K, Mosalaganti S, Hagen W, Andres-Pons A, Lemke EA, Bork P, Antonin W, Glavy JS, Bui KH, Beck M. In situ structural analysis of the human nuclear pore complex. Nature. 2015;526(7571):140–143. doi: 10.1038/nature15381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ori A, Iskar M, Buczak K, Kastritis P, Parca L, Andres-Pons A, Singer S, Bork P, Beck M. Spatiotemporal variation of mammalian protein complex stoichiometries. Genome Biol. 2016;17:47. doi: 10.1186/s13059-016-0912-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Feng Y, De Franceschi G, Kahraman A, Soste M, Melnik A, Boersema PJ, de Laureto PP, Nikolaev Y, Oliveira AP, Picotti P. Global analysis of protein structural changes in complex proteomes. Nat Biotechnol. 2014;32(10):1036–44. doi: 10.1038/nbt.2999. [DOI] [PubMed] [Google Scholar]
- 87.Schopper S, Kahraman A, Leuenberger P, Feng Y, Piazza I, Muller O, Boersema PJ, Picotti P. Measuring protein structural changes on a proteome-wide scale using limited proteolysis-coupled mass spectrometry. Nat Protoc. 2017;12(11):2391–2410. doi: 10.1038/nprot.2017.100. [DOI] [PubMed] [Google Scholar]
- 88.Savitski MM, Reinhard FB, Franken H, Werner T, Savitski MF, Eberhard D, Martinez Molina D, Jafari R, Dovega RB, Klaeger S, Kuster B, Nordlund P, Bantscheff M, Drewes G. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014;346(6205):1255784. doi: 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- 89.Sjoholm K, Kilsgard O, Teleman J, Happonen L, Malmstrom L, Malmstrom J. Targeted Proteomics and Absolute Protein Quantification for the Construction of a Stoichiometric Host-Pathogen Surface Density Model. Mol Cell Proteomics. 2017;16(4 suppl 1):S29–S41. doi: 10.1074/mcp.M116.063966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lobingier BT, Huttenhain R, Eichel K, Miller KB, Ting AY, von Zastrow M, Krogan NJ. An Approach to Spatiotemporally Resolve Protein Interaction Networks in Living Cells. Cell. 2017;169(2):350–360 e12. doi: 10.1016/j.cell.2017.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wolf-Yadlin A, Hautaniemi S, Lauffenburger DA, White FM. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc Natl Acad Sci U S A. 2007;104(14):5860–5. doi: 10.1073/pnas.0608638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Abelin JG, Patel J, Lu X, Feeney CM, Fagbami L, Creech AL, Hu R, Lam D, Davison D, Pino L, Qiao JW, Kuhn E, Officer A, Li J, Abbatiello S, Subramanian A, Sidman R, Snyder E, Carr SA, Jaffe JD. Reduced-representation Phosphosignatures Measured by Quantitative Targeted MS Capture Cellular States and Enable Large-scale Comparison of Drug-induced Phenotypes. Mol Cell Proteomics. 2016;15(5):1622–41. doi: 10.1074/mcp.M116.058354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xiao Y, Guo L, Wang Y. A targeted quantitative proteomics strategy for global kinome profiling of cancer cells and tissues. Mol Cell Proteomics. 2014;13(4):1065–75. doi: 10.1074/mcp.M113.036905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Guo L, Xiao Y, Fan M, Li JJ, Wang Y. Profiling global kinome signatures of the radioresistant MCF-7/C6 breast cancer cells using MRM-based targeted proteomics. J Proteome Res. 2015;14(1):193–201. doi: 10.1021/pr500919w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim HJ, Lin D, Lee HJ, Li M, Liebler DC. Quantitative Profiling of Protein Tyrosine Kinases in Human Cancer Cell Lines by Multiplexed Parallel Reaction Monitoring Assays. Mol Cell Proteomics. 2016;15(2):682–91. doi: 10.1074/mcp.O115.056713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Urisman A, Levin RS, Gordan JD, Webber JT, Hernandez H, Ishihama Y, Shokat KM, Burlingame AL. An Optimized Chromatographic Strategy for Multiplexing In Parallel Reaction Monitoring Mass Spectrometry: Insights from Quantitation of Activated Kinases. Mol Cell Proteomics. 2017;16(2):265–277. doi: 10.1074/mcp.M116.058172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fang B, Hoffman MA, Mirza AS, Mishall KM, Li J, Peterman SM, Smalley KS, Shain KH, Weinberger PM, Wu J, Rix U, Haura EB, Koomen JM. Evaluating kinase ATP uptake and tyrosine phosphorylation using multiplexed quantification of chemically labeled and post-translationally modified peptides. Methods. 2015;81:41–9. doi: 10.1016/j.ymeth.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yu Y, Anjum R, Kubota K, Rush J, Villen J, Gygi SP. A site-specific, multiplexed kinase activity assay using stable-isotope dilution and high-resolution mass spectrometry. Proc Natl Acad Sci U S A. 2009;106(28):11606–11. doi: 10.1073/pnas.0905165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kubota K, Anjum R, Yu Y, Kunz RC, Andersen JN, Kraus M, Keilhack H, Nagashima K, Krauss S, Paweletz C, Hendrickson RC, Feldman AS, Wu CL, Rush J, Villen J, Gygi SP. Sensitive multiplexed analysis of kinase activities and activity-based kinase identification. Nat Biotechnol. 2009;27(10):933–40. doi: 10.1038/nbt.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kunz RC, McAllister FE, Rush J, Gygi SP. A high-throughput, multiplexed kinase assay using a benchtop orbitrap mass spectrometer to investigate the effect of kinase inhibitors on kinase signaling pathways. Anal Chem. 2012;84(14):6233–9. doi: 10.1021/ac301116z. [DOI] [PubMed] [Google Scholar]
- 101.Redding-Johanson AM, Batth TS, Chan R, Krupa R, Szmidt HL, Adams PD, Keasling JD, Lee TS, Mukhopadhyay A, Petzold CJ. Targeted proteomics for metabolic pathway optimization: application to terpene production. Metab Eng. 2011;13(2):194–203. doi: 10.1016/j.ymben.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 102.Singh P, Batth TS, Juminaga D, Dahl RH, Keasling JD, Adams PD, Petzold CJ. Application of targeted proteomics to metabolically engineered Escherichia coli. Proteomics. 2012;12(8):1289–99. doi: 10.1002/pmic.201100482. [DOI] [PubMed] [Google Scholar]
- 103.Juminaga D, Baidoo EE, Redding-Johanson AM, Batth TS, Burd H, Mukhopadhyay A, Petzold CJ, Keasling JD. Modular engineering of L-tyrosine production in Escherichia coli. Appl Environ Microbiol. 2012;78(1):89–98. doi: 10.1128/AEM.06017-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Alonso-Gutierrez J, Kim EM, Batth TS, Cho N, Hu Q, Chan LJ, Petzold CJ, Hillson NJ, Adams PD, Keasling JD, Garcia Martin H, Lee TS. Principal component analysis of proteomics (PCAP) as a tool to direct metabolic engineering. Metab Eng. 2015;28:123–33. doi: 10.1016/j.ymben.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 105.Liu Q, Wu K, Cheng Y, Lu L, Xiao E, Zhang Y, Deng Z, Liu T. Engineering an iterative polyketide pathway in Escherichia coli results in single-form alkene and alkane overproduction. Metab Eng. 2015;28:82–90. doi: 10.1016/j.ymben.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 106.Oliveira AP, Ludwig C, Picotti P, Kogadeeva M, Aebersold R, Sauer U. Regulation of yeast central metabolism by enzyme phosphorylation. Mol Syst Biol. 2012;8:623. doi: 10.1038/msb.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sabido E, Quehenberger O, Shen Q, Chang CY, Shah I, Armando AM, Andreyev A, Vitek O, Dennis EA, Aebersold R. Targeted proteomics of the eicosanoid biosynthetic pathway completes an integrated genomics-proteomics-metabolomics picture of cellular metabolism. Mol Cell Proteomics. 2012;11(7):M111 014746. doi: 10.1074/mcp.M111.014746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wienkoop S, Weiss J, May P, Kempa S, Irgang S, Recuenco-Munoz L, Pietzke M, Schwemmer T, Rupprecht J, Egelhofer V, Weckwerth W. Targeted proteomics for Chlamydomonas reinhardtii combined with rapid subcellular protein fractionation, metabolomics and metabolic flux analyses. Mol Biosyst. 2010;6(6):1018–31. doi: 10.1039/b920913a. [DOI] [PubMed] [Google Scholar]
- 109.Zulak KG, Lippert DN, Kuzyk MA, Domanski D, Chou T, Borchers CH, Bohlmann J. Targeted proteomics using selected reaction monitoring reveals the induction of specific terpene synthases in a multi-level study of methyl jasmonate-treated Norway spruce (Picea abies) Plant J. 2009;60(6):1015–30. doi: 10.1111/j.1365-313X.2009.04020.x. [DOI] [PubMed] [Google Scholar]
- 110.Smallbone K, Messiha HL, Carroll KM, Winder CL, Malys N, Dunn WB, Murabito E, Swainston N, Dada JO, Khan F, Pir P, Simeonidis E, Spasic I, Wishart J, Weichart D, Hayes NW, Jameson D, Broomhead DS, Oliver SG, Gaskell SJ, McCarthy JE, Paton NW, Westerhoff HV, Kell DB, Mendes P. A model of yeast glycolysis based on a consistent kinetic characterisation of all its enzymes. FEBS Lett. 2013;587(17):2832–41. doi: 10.1016/j.febslet.2013.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Narumi R, Shimizu Y, Ukai-Tadenuma M, Ode KL, Kanda GN, Shinohara Y, Sato A, Matsumoto K, Ueda HR. Mass spectrometry-based absolute quantification reveals rhythmic variation of mouse circadian clock proteins. Proc Natl Acad Sci U S A. 2016;113(24):E3461–7. doi: 10.1073/pnas.1603799113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Koppenol-Raab M, Sjoelund V, Manes NP, Gottschalk RA, Dutta B, Benet ZL, Fraser ID, Nita-Lazar A. Proteome and Secretome Analysis Reveals Differential Post-transcriptional Regulation of Toll-like Receptor Responses. Mol Cell Proteomics. 2017;16(4 suppl 1):S172–S186. doi: 10.1074/mcp.M116.064261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sabido E, Wu Y, Bautista L, Porstmann T, Chang CY, Vitek O, Stoffel M, Aebersold R. Targeted proteomics reveals strain-specific changes in the mouse insulin and central metabolic pathways after a sustained high-fat diet. Mol Syst Biol. 2013;9:681. doi: 10.1038/msb.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.de Graaf EL, Kaplon J, Mohammed S, Vereijken LA, Duarte DP, Redondo Gallego L, Heck AJ, Peeper DS, Altelaar AF. Signal Transduction Reaction Monitoring Deciphers Site-Specific PI3K-mTOR/MAPK Pathway Dynamics in Oncogene-Induced Senescence. J Proteome Res. 2015;14(7):2906–14. doi: 10.1021/acs.jproteome.5b00236. [DOI] [PubMed] [Google Scholar]
- 115.Shi T, Niepel M, McDermott JE, Gao Y, Nicora CD, Chrisler WB, Markillie LM, Petyuk VA, Smith RD, Rodland KD, Sorger PK, Qian WJ, Wiley HS. Conservation of protein abundance patterns reveals the regulatory architecture of the EGFR-MAPK pathway. Sci Signal. 2016;9(436):rs6. doi: 10.1126/scisignal.aaf0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.MacDonald ML, Ding Y, Newman J, Hemby S, Penzes P, Lewis DA, Yates NA, Sweet RA. Altered glutamate protein co-expression network topology linked to spine loss in the auditory cortex of schizophrenia. Biol Psychiatry. 2015;77(11):959–68. doi: 10.1016/j.biopsych.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Prabakaran S, Gunawardena J, Sontag E. Paradoxical results in perturbation-based signaling network reconstruction. Biophys J. 2014;106(12):2720–8. doi: 10.1016/j.bpj.2014.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Piehler J. Spectroscopic techniques for monitoring protein interactions in living cells. Curr Opin Struct Biol. 2014;24:54–62. doi: 10.1016/j.sbi.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 119.Podobnik M, Krasevec N, Bedina Zavec A, Naneh O, Flasker A, Caserman S, Hodnik V, Anderluh G. How to Study Protein-protein Interactions. Acta Chim Slov. 2016;63(3):424–39. doi: 10.17344/acsi.2016.2419. [DOI] [PubMed] [Google Scholar]
- 120.Zhou M, Li Q, Wang R. Current Experimental Methods for Characterizing Protein-Protein Interactions. ChemMedChem. 2016;11(8):738–56. doi: 10.1002/cmdc.201500495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zheng X, Bi C, Li Z, Podariu M, Hage DS. Analytical methods for kinetic studies of biological interactions: A review. J Pharm Biomed Anal. 2015;113:163–80. doi: 10.1016/j.jpba.2015.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Qin S, Pang X, Zhou HX. Automated prediction of protein association rate constants. Structure. 2011;19(12):1744–51. doi: 10.1016/j.str.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Moal IH, Bates PA. Kinetic rate constant prediction supports the conformational selection mechanism of protein binding. PLoS Comput Biol. 2012;8(1):e1002351. doi: 10.1371/journal.pcbi.1002351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mosca R, Ceol A, Aloy P. Interactome3D: adding structural details to protein networks. Nat Methods. 2013;10(1):47–53. doi: 10.1038/nmeth.2289. [DOI] [PubMed] [Google Scholar]
- 125.Martinez M, Bruce NJ, Romanowska J, Kokh DB, Ozboyaci M, Yu X, Ozturk MA, Richter S, Wade RC. SDA 7: A modular and parallel implementation of the simulation of diffusional association software. J Comput Chem. 2015;36(21):1631–45. doi: 10.1002/jcc.23971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Xue LC, Rodrigues JP, Kastritis PL, Bonvin AM, Vangone A. PRODIGY: a web server for predicting the binding affinity of protein-protein complexes. Bioinformatics. 2016;32(23):3676–3678. doi: 10.1093/bioinformatics/btw514. [DOI] [PubMed] [Google Scholar]
- 127.Bienert S, Waterhouse A, de Beer TA, Tauriello G, Studer G, Bordoli L, Schwede T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017;45(D1):D313–D319. doi: 10.1093/nar/gkw1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gencoglu M, Schmidt A, Becskei A. Measurement of In Vivo Protein Binding Affinities in a Signaling Network with Mass Spectrometry. ACS Synth Biol. 2017;6(7):1305–1314. doi: 10.1021/acssynbio.6b00282. [DOI] [PubMed] [Google Scholar]
- 129.Manes NP, Angermann BR, Koppenol-Raab M, An E, Sjoelund VH, Sun J, Ishii M, Germain RN, Meier-Schellersheim M, Nita-Lazar A. Targeted Proteomics-Driven Computational Modeling of Macrophage S1P Chemosensing. Mol Cell Proteomics. 2015;14(10):2661–81. doi: 10.1074/mcp.M115.048918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Angermann BR, Klauschen F, Garcia AD, Prustel T, Zhang F, Germain RN, Meier-Schellersheim M. Computational modeling of cellular signaling processes embedded into dynamic spatial contexts. Nat Methods. 2012;9(3):283–9. doi: 10.1038/nmeth.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhang F, Angermann BR, Meier-Schellersheim M. The Simmune Modeler visual interface for creating signaling networks based on bi-molecular interactions. Bioinformatics. 2013;29(9):1229–30. doi: 10.1093/bioinformatics/btt134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Baker MS, Ahn SB, Mohamedali A, Islam MT, Cantor D, Verhaert PD, Fanayan S, Sharma S, Nice EC, Connor M, Ranganathan S. Accelerating the search for the missing proteins in the human proteome. Nat Commun. 2017;8:14271. doi: 10.1038/ncomms14271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vandenbrouck Y, Lane L, Carapito C, Duek P, Rondel K, Bruley C, Macron C, Gonzalez de Peredo A, Coute Y, Chaoui K, Com E, Gateau A, Hesse AM, Marcellin M, Mear L, Mouton-Barbosa E, Robin T, Burlet-Schiltz O, Cianferani S, Ferro M, Freour T, Lindskog C, Garin J, Pineau C. Looking for Missing Proteins in the Proteome of Human Spermatozoa: An Update. J Proteome Res. 2016;15(11):3998–4019. doi: 10.1021/acs.jproteome.6b00400. [DOI] [PubMed] [Google Scholar]
- 134.Omasits U, Varadarajan AR, Schmid M, Goetze S, Melidis D, Bourqui M, Nikolayeva O, Quebatte M, Patrignani A, Dehio C, Frey JE, Robinson MD, Wollscheid B, Ahrens CH. An integrative strategy to identify the entire protein coding potential of prokaryotic genomes by proteogenomics. Genome Res. 2017;27(12):2083–2095. doi: 10.1101/gr.218255.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ma J, Ward CC, Jungreis I, Slavoff SA, Schwaid AG, Neveu J, Budnik BA, Kellis M, Saghatelian A. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 2014;13(3):1757–65. doi: 10.1021/pr401280w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Slavoff SA, Mitchell AJ, Schwaid AG, Cabili MN, Ma J, Levin JZ, Karger AD, Budnik BA, Rinn JL, Saghatelian A. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat Chem Biol. 2013;9(1):59–64. doi: 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cobb LJ, Lee C, Xiao J, Yen K, Wong RG, Nakamura HK, Mehta HH, Gao Q, Ashur C, Huffman DM, Wan J, Muzumdar R, Barzilai N, Cohen P. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging (Albany NY) 2016;8(4):796–809. doi: 10.18632/aging.100943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee C, Kim KH, Cohen P. MOTS-c: A novel mitochondrial-derived peptide regulating muscle and fat metabolism. Free Radic Biol Med. 2016;100:182–187. doi: 10.1016/j.freeradbiomed.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yen K, Lee C, Mehta H, Cohen P. The emerging role of the mitochondrial-derived peptide humanin in stress resistance. J Mol Endocrinol. 2013;50(1):R11–9. doi: 10.1530/JME-12-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hobbs EC, Fontaine F, Yin X, Storz G. An expanding universe of small proteins. Curr Opin Microbiol. 2011;14(2):167–73. doi: 10.1016/j.mib.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Storz G, Wolf YI, Ramamurthi KS. Small proteins can no longer be ignored. Annu Rev Biochem. 2014;83:753–77. doi: 10.1146/annurev-biochem-070611-102400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Maass S, Becher D. Methods and applications of absolute protein quantification in microbial systems. J Proteomics. 2016;136:222–33. doi: 10.1016/j.jprot.2016.01.015. [DOI] [PubMed] [Google Scholar]
- 143.Malmstrom J, Beck M, Schmidt A, Lange V, Deutsch EW, Aebersold R. Proteome-wide cellular protein concentrations of the human pathogen Leptospira interrogans. Nature. 2009;460(7256):762–5. doi: 10.1038/nature08184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Beck M, Schmidt A, Malmstroem J, Claassen M, Ori A, Szymborska A, Herzog F, Rinner O, Ellenberg J, Aebersold R. The quantitative proteome of a human cell line. Mol Syst Biol. 2011;7:549. doi: 10.1038/msb.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang X, Liu Q, Zhang B. Leveraging the complementary nature of RNA-Seq and shotgun proteomics data. Proteomics. 2014;14(23-24):2676–87. doi: 10.1002/pmic.201400184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Edfors F, Danielsson F, Hallstrom BM, Kall L, Lundberg E, Ponten F, Forsstrom B, Uhlen M. Gene-specific correlation of RNA and protein levels in human cells and tissues. Mol Syst Biol. 2016;12(10):883. doi: 10.15252/msb.20167144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kito K, Okada M, Ishibashi Y, Okada S, Ito T. A strategy for absolute proteome quantification with mass spectrometry by hierarchical use of peptide-concatenated standards. Proteomics. 2016;16(10):1457–73. doi: 10.1002/pmic.201500414. [DOI] [PubMed] [Google Scholar]
- 148.Kusebauch U, Campbell DS, Deutsch EW, Chu CS, Spicer DA, Brusniak MY, Slagel J, Sun Z, Stevens J, Grimes B, Shteynberg D, Hoopmann MR, Blattmann P, Ratushny AV, Rinner O, Picotti P, Carapito C, Huang CY, Kapousouz M, Lam H, Tran T, Demir E, Aitchison JD, Sander C, Hood L, Aebersold R, Moritz RL. Human SRMAtlas: A Resource of Targeted Assays to Quantify the Complete Human Proteome. Cell. 2016;166(3):766–778. doi: 10.1016/j.cell.2016.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Matsumoto M, Matsuzaki F, Oshikawa K, Goshima N, Mori M, Kawamura Y, Ogawa K, Fukuda E, Nakatsumi H, Natsume T, Fukui K, Horimoto K, Nagashima T, Funayama R, Nakayama K, Nakayama KI. A large-scale targeted proteomics assay resource based on an in vitro human proteome. Nat Methods. 2017;14(3):251–258. doi: 10.1038/nmeth.4116. [DOI] [PubMed] [Google Scholar]
- 150.Simicevic J, Schmid AW, Gilardoni PA, Zoller B, Raghav SK, Krier I, Gubelmann C, Lisacek F, Naef F, Moniatte M, Deplancke B. Absolute quantification of transcription factors during cellular differentiation using multiplexed targeted proteomics. Nat Methods. 2013;10(6):570–6. doi: 10.1038/nmeth.2441. [DOI] [PubMed] [Google Scholar]
- 151.Ludwig C, Claassen M, Schmidt A, Aebersold R. Estimation of absolute protein quantities of unlabeled samples by selected reaction monitoring mass spectrometry. Mol Cell Proteomics. 2012;11(3):M111 013987. doi: 10.1074/mcp.M111.013987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wesseling H, Guest PC, Lee CM, Wong EH, Rahmoune H, Bahn S. Integrative proteomic analysis of the NMDA NR1 knockdown mouse model reveals effects on central and peripheral pathways associated with schizophrenia and autism spectrum disorders. Mol Autism. 2014;5:38. doi: 10.1186/2040-2392-5-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sangar V, Funk CC, Kusebauch U, Campbell DS, Moritz RL, Price ND. Quantitative proteomic analysis reveals effects of epidermal growth factor receptor (EGFR) on invasion-promoting proteins secreted by glioblastoma cells. Mol Cell Proteomics. 2014;13(10):2618–31. doi: 10.1074/mcp.M114.040428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Julien O, Zhuang M, Wiita AP, O'Donoghue AJ, Knudsen GM, Craik CS, Wells JA. Quantitative MS-based enzymology of caspases reveals distinct protein substrate specificities, hierarchies, and cellular roles. Proc Natl Acad Sci U S A. 2016;113(14):E2001–10. doi: 10.1073/pnas.1524900113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Shimbo K, Hsu GW, Nguyen H, Mahrus S, Trinidad JC, Burlingame AL, Wells JA. Quantitative profiling of caspase-cleaved substrates reveals different drug-induced and cell-type patterns in apoptosis. Proc Natl Acad Sci U S A. 2012;109(31):12432–7. doi: 10.1073/pnas.1208616109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rougemont B, Bontemps Gallo S, Ayciriex S, Carriere R, Hondermarck H, Lacroix JM, Le Blanc JC, Lemoine J. Scout-MRM: Multiplexed Targeted Mass Spectrometry-Based Assay without Retention Time Scheduling Exemplified by Dickeya dadantii Proteomic Analysis during Plant Infection. Anal Chem. 2017;89(3):1421–1426. doi: 10.1021/acs.analchem.6b03201. [DOI] [PubMed] [Google Scholar]
- 157.Sabino F, Hermes O, Egli FE, Kockmann T, Schlage P, Croizat P, Kizhakkedathu JN, Smola H, auf dem Keller U. In vivo assessment of protease dynamics in cutaneous wound healing by degradomics analysis of porcine wound exudates. Mol Cell Proteomics. 2015;14(2):354–70. doi: 10.1074/mcp.M114.043414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mackenzie RJ, Lawless C, Holman SW, Lanthaler K, Beynon RJ, Grant CM, Hubbard SJ, Eyers CE. Absolute protein quantification of the yeast chaperome under conditions of heat shock. Proteomics. 2016;16(15-16):2128–40. doi: 10.1002/pmic.201500503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nicholas D, Tang H, Zhang Q, Rudra J, Xu F, Langridge W, Zhang K. Quantitative proteomics reveals a role for epigenetic reprogramming during human monocyte differentiation. Mol Cell Proteomics. 2015;14(1):15–29. doi: 10.1074/mcp.M113.035089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wesseling H, Rahmoune H, Tricklebank M, Guest PC, Bahn S. A targeted multiplexed proteomic investigation identifies ketamine-induced changes in immune markers in rat serum and expression changes in protein kinases/phosphatases in rat brain. J Proteome Res. 2015;14(1):411–21. doi: 10.1021/pr5009493. [DOI] [PubMed] [Google Scholar]
- 161.Rivers J, Simpson DM, Robertson DH, Gaskell SJ, Beynon RJ. Absolute multiplexed quantitative analysis of protein expression during muscle development using QconCAT. Mol Cell Proteomics. 2007;6(8):1416–27. doi: 10.1074/mcp.M600456-MCP200. [DOI] [PubMed] [Google Scholar]
- 162.Dunkley T, Costa V, Friedlein A, Lugert S, Aigner S, Ebeling M, Miller MT, Patsch C, Piraino P, Cutler P, Jagasia R. Characterization of a human pluripotent stem cell-derived model of neuronal development using multiplexed targeted proteomics. Proteomics Clin Appl. 2015;9(7-8):684–94. doi: 10.1002/prca.201400150. [DOI] [PubMed] [Google Scholar]
- 163.Kim JS, Lee Y, Lee MY, Shin J, Han JM, Yang EG, Yu MH, Kim S, Hwang D, Lee C. Multiple reaction monitoring of multiple low-abundance transcription factors in whole lung cancer cell lysates. J Proteome Res. 2013;12(6):2582–96. doi: 10.1021/pr3011414. [DOI] [PubMed] [Google Scholar]
- 164.Danielson SR, Held JM, Schilling B, Oo M, Gibson BW, Andersen JK. Preferentially increased nitration of alpha-synuclein at tyrosine-39 in a cellular oxidative model of Parkinson's disease. Anal Chem. 2009;81(18):7823–8. doi: 10.1021/ac901176t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hu LZ, Zhang WP, Zhou MT, Han QQ, Gao XL, Zeng HL, Guo L. Analysis of Salmonella PhoP/PhoQ regulation by dimethyl-SRM-based quantitative proteomics. Biochim Biophys Acta. 2016;1864(1):20–8. doi: 10.1016/j.bbapap.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 166.Malmstrom E, Davidova A, Morgelin M, Linder A, Larsen M, Qvortrup K, Nordenfelt P, Shannon O, Dzupova O, Holub M, Malmstrom J, Herwald H. Targeted mass spectrometry analysis of neutrophil-derived proteins released during sepsis progression. Thromb Haemost. 2014;112(6):1230–43. doi: 10.1160/TH14-04-0312. [DOI] [PubMed] [Google Scholar]
- 167.Lange V, Malmstrom JA, Didion J, King NL, Johansson BP, Schafer J, Rameseder J, Wong CH, Deutsch EW, Brusniak MY, Buhlmann P, Bjorck L, Domon B, Aebersold R. Targeted quantitative analysis of Streptococcus pyogenes virulence factors by multiple reaction monitoring. Mol Cell Proteomics. 2008;7(8):1489–500. doi: 10.1074/mcp.M800032-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mayya V, Rezual K, Wu L, Fong MB, Han DK. Absolute quantification of multisite phosphorylation by selective reaction monitoring mass spectrometry: determination of inhibitory phosphorylation status of cyclin-dependent kinases. Mol Cell Proteomics. 2006;5(6):1146–57. doi: 10.1074/mcp.T500029-MCP200. [DOI] [PubMed] [Google Scholar]
- 169.Kirkpatrick DS, Hathaway NA, Hanna J, Elsasser S, Rush J, Finley D, King RW, Gygi SP. Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat Cell Biol. 2006;8(7):700–10. doi: 10.1038/ncb1436. [DOI] [PubMed] [Google Scholar]
- 170.Balasubramaniam D, Eissler CL, Stauffacher CV, Hall MC. Use of selected reaction monitoring data for label-free quantification of protein modification stoichiometry. Proteomics. 2010;10(23):4301–5. doi: 10.1002/pmic.201000232. [DOI] [PubMed] [Google Scholar]
- 171.Mirzaei H, Rogers RS, Grimes B, Eng J, Aderem A, Aebersold R. Characterizing the connectivity of poly-ubiquitin chains by selected reaction monitoring mass spectrometry. Mol Biosyst. 2010;6(10):2004–14. doi: 10.1039/c005242f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Xu F, Yang T, Sheng Y, Zhong T, Yang M, Chen Y. Simultaneous quantification of protein phosphorylation sites using liquid chromatography-tandem mass spectrometry-based targeted proteomics: a linear algebra approach for isobaric phosphopeptides. J Proteome Res. 2014;13(12):5452–60. doi: 10.1021/pr500339u. [DOI] [PubMed] [Google Scholar]
- 173.Held JM, Danielson SR, Behring JB, Atsriku C, Britton DJ, Puckett RL, Schilling B, Campisi J, Benz CC, Gibson BW. Targeted quantitation of site-specific cysteine oxidation in endogenous proteins using a differential alkylation and multiple reaction monitoring mass spectrometry approach. Mol Cell Proteomics. 2010;9(7):1400–10. doi: 10.1074/mcp.M900643-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jin LL, Tong J, Prakash A, Peterman SM, St-Germain JR, Taylor P, Trudel S, Moran MF. Measurement of protein phosphorylation stoichiometry by selected reaction monitoring mass spectrometry. J Proteome Res. 2010;9(5):2752–61. doi: 10.1021/pr100024a. [DOI] [PubMed] [Google Scholar]
- 175.Caruso JA, Stemmer PM, Dombkowski A, Caruthers NJ, Gill R, Rosenspire AJ. A systems toxicology approach identifies Lyn as a key signaling phosphoprotein modulated by mercury in a B lymphocyte cell model. Toxicol Appl Pharmacol. 2014;276(1):47–54. doi: 10.1016/j.taap.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Altvater M, Chang Y, Melnik A, Occhipinti L, Schutz S, Rothenbusch U, Picotti P, Panse VG. Targeted proteomics reveals compositional dynamics of 60S pre-ribosomes after nuclear export. Mol Syst Biol. 2012;8:628. doi: 10.1038/msb.2012.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Benevento M, Di Palma S, Snijder J, Moyer CL, Reddy VS, Nemerow GR, Heck AJ. Adenovirus composition, proteolysis, and disassembly studied by in-depth qualitative and quantitative proteomics. J Biol Chem. 2014;289(16):11421–30. doi: 10.1074/jbc.M113.537498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Federspiel JD, Codreanu SG, Palubinsky AM, Winland AJ, Betanzos CM, McLaughlin B, Liebler DC. Assembly Dynamics and Stoichiometry of the Apoptosis Signal-regulating Kinase (ASK) Signalosome in Response to Electrophile Stress. Mol Cell Proteomics. 2016;15(6):1947–61. doi: 10.1074/mcp.M115.057364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sjoholm K, Karlsson C, Linder A, Malmstrom J. A comprehensive analysis of the Streptococcus pyogenes and human plasma protein interaction network. Mol Biosyst. 2014;10(7):1698–708. doi: 10.1039/c3mb70555b. [DOI] [PubMed] [Google Scholar]
- 180.Bauer M, Cubizolles F, Schmidt A, Nigg EA. Quantitative analysis of human centrosome architecture by targeted proteomics and fluorescence imaging. EMBO J. 2016;35(19):2152–2166. doi: 10.15252/embj.201694462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ding C, Li Y, Kim BJ, Malovannaya A, Jung SY, Wang Y, Qin J. Quantitative analysis of cohesin complex stoichiometry and SMC3 modification-dependent protein interactions. J Proteome Res. 2011;10(8):3652–9. doi: 10.1021/pr2002758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Bennett EJ, Rush J, Gygi SP, Harper JW. Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics. Cell. 2010;143(6):951–65. doi: 10.1016/j.cell.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Liu NQ, Dekker LJ, Stingl C, Guzel C, De Marchi T, Martens JW, Foekens JA, Luider TM, Umar A. Quantitative proteomic analysis of microdissected breast cancer tissues: comparison of label-free and SILAC-based quantification with shotgun, directed, and targeted MS approaches. J Proteome Res. 2013;12(10):4627–41. doi: 10.1021/pr4005794. [DOI] [PubMed] [Google Scholar]
- 184.Betke KM, Rose KL, Friedman DB, Baucum AJ, 2nd, Hyde K, Schey KL, Hamm HE. Differential localization of G protein betagamma subunits. Biochemistry. 2014;53(14):2329–43. doi: 10.1021/bi500091p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chavez JD, Eng JK, Schweppe DK, Cilia M, Rivera K, Zhong X, Wu X, Allen T, Khurgel M, Kumar A, Lampropoulos A, Larsson M, Maity S, Morozov Y, Pathmasiri W, Perez-Neut M, Pineyro-Ruiz C, Polina E, Post S, Rider M, Tokmina-Roszyk D, Tyson K, Vieira Parrine Sant'Ana D, Bruce JE. A General Method for Targeted Quantitative Cross-Linking Mass Spectrometry. PLoS One. 2016;11(12):e0167547. doi: 10.1371/journal.pone.0167547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Holzmann J, Pichler P, Madalinski M, Kurzbauer R, Mechtler K. Stoichiometry determination of the MP1-p14 complex using a novel and cost-efficient method to produce an equimolar mixture of standard peptides. Anal Chem. 2009;81(24):10254–61. doi: 10.1021/ac902286m. [DOI] [PubMed] [Google Scholar]
- 187.Zhao Y, Widen SG, Jamaluddin M, Tian B, Wood TG, Edeh CB, Brasier AR. Quantification of activated NF-kappaB/RelA complexes using ssDNA aptamer affinity-stable isotope dilution-selected reaction monitoring-mass spectrometry. Mol Cell Proteomics. 2011;10(6):M111 008771. doi: 10.1074/mcp.M111.008771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bennett RJ, Simpson DM, Holman SW, Ryan S, Brownridge P, Eyers CE, Colyer J, Beynon RJ. DOSCATs: Double standards for protein quantification. Sci Rep. 2017;7:45570. doi: 10.1038/srep45570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Krieger JR, Taylor P, Gajadhar AS, Guha A, Moran MF, McGlade CJ. Identification and selected reaction monitoring (SRM) quantification of endocytosis factors associated with Numb. Mol Cell Proteomics. 2013;12(2):499–514. doi: 10.1074/mcp.M112.020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kawakami H, Kamiie J, Yasuno K, Kobayashi R, Aihara N, Shirota K. Dynamics of absolute amount of nephrin in a single podocyte in puromycin aminonucleoside nephrosis rats calculated by quantitative glomerular proteomics approach with selected reaction monitoring mode. Nephrol Dial Transplant. 2012;27(4):1324–30. doi: 10.1093/ndt/gfr492. [DOI] [PubMed] [Google Scholar]
- 191.Colangelo CM, Ivosev G, Chung L, Abbott T, Shifman M, Sakaue F, Cox D, Kitchen RR, Burton L, Tate SA, Gulcicek E, Bonner R, Rinehart J, Nairn AC, Williams KR. Development of a highly automated and multiplexed targeted proteome pipeline and assay for 112 rat brain synaptic proteins. Proteomics. 2015;15(7):1202–14. doi: 10.1002/pmic.201400353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cheng D, Hoogenraad CC, Rush J, Ramm E, Schlager MA, Duong DM, Xu P, Wijayawardana SR, Hanfelt J, Nakagawa T, Sheng M, Peng J. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol Cell Proteomics. 2006;5(6):1158–70. doi: 10.1074/mcp.D500009-MCP200. [DOI] [PubMed] [Google Scholar]
- 193.Lowenthal MS, Markey SP, Dosemeci A. Quantitative mass spectrometry measurements reveal stoichiometry of principal postsynaptic density proteins. J Proteome Res. 2015;14(6):2528–38. doi: 10.1021/acs.jproteome.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Vandemoortele G, Staes A, Gonnelli G, Samyn N, De Sutter D, Vandermarliere E, Timmerman E, Gevaert K, Martens L, Eyckerman S. An extra dimension in protein tagging by quantifying universal proteotypic peptides using targeted proteomics. Sci Rep. 2016;6:27220. doi: 10.1038/srep27220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wepf A, Glatter T, Schmidt A, Aebersold R, Gstaiger M. Quantitative interaction proteomics using mass spectrometry. Nat Methods. 2009;6(3):203–5. doi: 10.1038/nmeth.1302. [DOI] [PubMed] [Google Scholar]
- 196.Al-Majdoub ZM, Carroll KM, Gaskell SJ, Barber J. Quantification of the proteins of the bacterial ribosome using QconCAT technology. J Proteome Res. 2014;13(3):1211–22. doi: 10.1021/pr400667h. [DOI] [PubMed] [Google Scholar]
- 197.Delumeau O, Lecointe F, Muntel J, Guillot A, Guedon E, Monnet V, Hecker M, Becher D, Polard P, Noirot P. The dynamic protein partnership of RNA polymerase in Bacillus subtilis. Proteomics. 2011;11(15):2992–3001. doi: 10.1002/pmic.201000790. [DOI] [PubMed] [Google Scholar]
- 198.Hajkova D, Imanishi Y, Palamalai V, Rao KC, Yuan C, Sheng Q, Tang H, Zeng R, Darrow RM, Organisciak DT, Miyagi M. Proteomic changes in the photoreceptor outer segment upon intense light exposure. J Proteome Res. 2010;9(2):1173–81. doi: 10.1021/pr900819k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Schmidt C, Lenz C, Grote M, Luhrmann R, Urlaub H. Determination of protein stoichiometry within protein complexes using absolute quantification and multiple reaction monitoring. Anal Chem. 2010;82(7):2784–96. doi: 10.1021/ac902710k. [DOI] [PubMed] [Google Scholar]
- 200.Nanavati D, Gucek M, Milne JL, Subramaniam S, Markey SP. Stoichiometry and absolute quantification of proteins with mass spectrometry using fluorescent and isotope-labeled concatenated peptide standards. Mol Cell Proteomics. 2008;7(2):442–7. doi: 10.1074/mcp.M700345-MCP200. [DOI] [PubMed] [Google Scholar]
- 201.Picotti P, Rinner O, Stallmach R, Dautel F, Farrah T, Domon B, Wenschuh H, Aebersold R. High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat Methods. 2010;7(1):43–6. doi: 10.1038/nmeth.1408. [DOI] [PubMed] [Google Scholar]
- 202.Guo L, Xiao Y, Wang Y. Application of adenosine triphosphate affinity probe and scheduled multiple-reaction monitoring analysis for profiling global kinome in human cells in response to arsenite treatment. Anal Chem. 2014;86(21):10700–7. doi: 10.1021/ac502592d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Miao W, Xiao Y, Guo L, Jiang X, Huang M, Wang Y. A High-Throughput Targeted Proteomic Approach for Comprehensive Profiling of Methylglyoxal-Induced Perturbations of the Human Kinome. Anal Chem. 2016;88(19):9773–9779. doi: 10.1021/acs.analchem.6b02816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lam MP, Scruggs SB, Kim TY, Zong C, Lau E, Wang D, Ryan CM, Faull KF, Ping P. An MRM-based workflow for quantifying cardiac mitochondrial protein phosphorylation in murine and human tissue. J Proteomics. 2012;75(15):4602–9. doi: 10.1016/j.jprot.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Schilling B, MacLean B, Held JM, Sahu AK, Rardin MJ, Sorensen DJ, Peters T, Wolfe AJ, Hunter CL, MacCoss MJ, Gibson BW. Multiplexed, Scheduled, High-Resolution Parallel Reaction Monitoring on a Full Scan QqTOF Instrument with Integrated Data-Dependent and Targeted Mass Spectrometric Workflows. Anal Chem. 2015;87(20):10222–9. doi: 10.1021/acs.analchem.5b02983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kohlstedt M, Sappa PK, Meyer H, Maass S, Zaprasis A, Hoffmann T, Becker J, Steil L, Hecker M, van Dijl JM, Lalk M, Mader U, Stulke J, Bremer E, Volker U, Wittmann C. Adaptation of Bacillus subtilis carbon core metabolism to simultaneous nutrient limitation and osmotic challenge: a multi-omics perspective. Environ Microbiol. 2014;16(6):1898–917. doi: 10.1111/1462-2920.12438. [DOI] [PubMed] [Google Scholar]
- 207.Voges R, Corsten S, Wiechert W, Noack S. Absolute quantification of Corynebacterium glutamicum glycolytic and anaplerotic enzymes by QconCAT. J Proteomics. 2015;113:366–77. doi: 10.1016/j.jprot.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 208.Voges R, Noack S. Quantification of proteome dynamics in Corynebacterium glutamicum by (15)N-labeling and selected reaction monitoring. J Proteomics. 2012;75(9):2660–9. doi: 10.1016/j.jprot.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 209.Trauchessec M, Jaquinod M, Bonvalot A, Brun V, Bruley C, Ropers D, de Jong H, Garin J, Bestel-Corre G, Ferro M. Mass spectrometry-based workflow for accurate quantification of Escherichia coli enzymes: how proteomics can play a key role in metabolic engineering. Mol Cell Proteomics. 2014;13(4):954–68. doi: 10.1074/mcp.M113.032672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Hutton JE, Wang X, Zimmerman LJ, Slebos RJ, Trenary IA, Young JD, Li M, Liebler DC. Oncogenic KRAS and BRAF Drive Metabolic Reprogramming in Colorectal Cancer. Mol Cell Proteomics. 2016;15(9):2924–38. doi: 10.1074/mcp.M116.058925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Drabovich AP, Pavlou MP, Dimitromanolakis A, Diamandis EP. Quantitative analysis of energy metabolic pathways in MCF-7 breast cancer cells by selected reaction monitoring assay. Mol Cell Proteomics. 2012;11(8):422–34. doi: 10.1074/mcp.M111.015214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Picotti P, Bodenmiller B, Mueller LN, Domon B, Aebersold R. Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell. 2009;138(4):795–806. doi: 10.1016/j.cell.2009.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Matsuda F, Kinoshita S, Nishino S, Tomita A, Shimizu H. Targeted proteome analysis of single-gene deletion strains of Saccharomyces cerevisiae lacking enzymes in the central carbon metabolism. PLoS One. 2017;12(2):e0172742. doi: 10.1371/journal.pone.0172742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Matsuda F, Ogura T, Tomita A, Hirano I, Shimizu H. Nano-scale liquid chromatography coupled to tandem mass spectrometry using the multiple reaction monitoring mode based quantitative platform for analyzing multiple enzymes associated with central metabolic pathways of Saccharomyces cerevisiae using ultra fast mass spectrometry. J Biosci Bioeng. 2015;119(1):117–20. doi: 10.1016/j.jbiosc.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 215.Costenoble R, Picotti P, Reiter L, Stallmach R, Heinemann M, Sauer U, Aebersold R. Comprehensive quantitative analysis of central carbon and amino-acid metabolism in Saccharomyces cerevisiae under multiple conditions by targeted proteomics. Mol Syst Biol. 2011;7:464. doi: 10.1038/msb.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Batth TS, Singh P, Ramakrishnan VR, Sousa MML, Chan LJG, Tran HM, Luning EG, Pan EHY, Vuu KM, Keasling JD, Adams PD, Petzold CJ. A targeted proteomics toolkit for high-throughput absolute quantification of Escherichia coli proteins. Metab Eng. 2014;26:48–56. doi: 10.1016/j.ymben.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 217.Holman SW, Hammond DE, Simpson DM, Waters J, Hurst JL, Beynon RJ. Protein turnover measurement using selected reaction monitoring-mass spectrometry (SRM-MS) Philos Trans A Math Phys Eng Sci. 2016;374(2079):20150362. doi: 10.1098/rsta.2015.0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Achour B, Russell MR, Barber J, Rostami-Hodjegan A. Simultaneous quantification of the abundance of several cytochrome P450 and uridine 5′-diphospho-glucuronosyltransferase enzymes in human liver microsomes using multiplexed targeted proteomics. Drug Metab Dispos. 2014;42(4):500–10. doi: 10.1124/dmd.113.055632. [DOI] [PubMed] [Google Scholar]
- 219.Fallon JK, Neubert H, Hyland R, Goosen TC, Smith PC. Targeted quantitative proteomics for the analysis of 14 UGT1As and -2Bs in human liver using NanoUPLC-MS/MS with selected reaction monitoring. J Proteome Res. 2013;12(10):4402–13. doi: 10.1021/pr4004213. [DOI] [PubMed] [Google Scholar]
- 220.Li J, Zhou L, Wang H, Yan H, Li N, Zhai R, Jiao F, Hao F, Jin Z, Tian F, Peng B, Zhang Y, Qian X. A new sample preparation method for the absolute quantitation of a target proteome using (18)O labeling combined with multiple reaction monitoring mass spectrometry. Analyst. 2015;140(4):1281–90. doi: 10.1039/c4an02092h. [DOI] [PubMed] [Google Scholar]
- 221.Groer C, Busch D, Patrzyk M, Beyer K, Busemann A, Heidecke CD, Drozdzik M, Siegmund W, Oswald S. Absolute protein quantification of clinically relevant cytochrome P450 enzymes and UDP-glucuronosyltransferases by mass spectrometry-based targeted proteomics. J Pharm Biomed Anal. 2014;100:393–401. doi: 10.1016/j.jpba.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 222.Hersman EM, Bumpus NN. A targeted proteomics approach for profiling murine cytochrome P450 expression. J Pharmacol Exp Ther. 2014;349(2):221–8. doi: 10.1124/jpet.113.212456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Nakamura K, Hirayama-Kurogi M, Ito S, Kuno T, Yoneyama T, Obuchi W, Terasaki T, Ohtsuki S. Large-scale multiplex absolute protein quantification of drug-metabolizing enzymes and transporters in human intestine, liver, and kidney microsomes by SWATH-MS: Comparison with MRM/SRM and HR-MRM/PRM. Proteomics. 2016;16(15-16):2106–17. doi: 10.1002/pmic.201500433. [DOI] [PubMed] [Google Scholar]
- 224.Groer C, Bruck S, Lai Y, Paulick A, Busemann A, Heidecke CD, Siegmund W, Oswald S. LC-MS/MS-based quantification of clinically relevant intestinal uptake and efflux transporter proteins. J Pharm Biomed Anal. 2013;85:253–61. doi: 10.1016/j.jpba.2013.07.031. [DOI] [PubMed] [Google Scholar]
- 225.Harwood MD, Achour B, Russell MR, Carlson GL, Warhurst G, Rostami-Hodjegan A. Application of an LC-MS/MS method for the simultaneous quantification of human intestinal transporter proteins absolute abundance using a QconCAT technique. J Pharm Biomed Anal. 2015;110:27–33. doi: 10.1016/j.jpba.2015.02.043. [DOI] [PubMed] [Google Scholar]
- 226.Prasad B, Johnson K, Billington S, Lee C, Chung GW, Brown CD, Kelly EJ, Himmelfarb J, Unadkat JD. Abundance of Drug Transporters in the Human Kidney Cortex as Quantified by Quantitative Targeted Proteomics. Drug Metab Dispos. 2016;44(12):1920–1924. doi: 10.1124/dmd.116.072066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Kamiie J, Ohtsuki S, Iwase R, Ohmine K, Katsukura Y, Yanai K, Sekine Y, Uchida Y, Ito S, Terasaki T. Quantitative atlas of membrane transporter proteins: development and application of a highly sensitive simultaneous LC/MS/MS method combined with novel in-silico peptide selection criteria. Pharm Res. 2008;25(6):1469–83. doi: 10.1007/s11095-008-9532-4. [DOI] [PubMed] [Google Scholar]
- 228.Tao H, Zhang Y, Cao X, Deng Z, Liu T. Absolute quantification of proteins in the fatty acid biosynthetic pathway using protein standard absolute quantification. Synth Syst Biotechnol. 2016;1:150–7. doi: 10.1016/j.synbio.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Murphy JP, Pinto DM. Targeted proteomic analysis of glycolysis in cancer cells. J Proteome Res. 2011;10(2):604–13. doi: 10.1021/pr100774f. [DOI] [PubMed] [Google Scholar]
- 230.Carroll KM, Simpson DM, Eyers CE, Knight CG, Brownridge P, Dunn WB, Winder CL, Lanthaler K, Pir P, Malys N, Kell DB, Oliver SG, Gaskell SJ, Beynon RJ. Absolute quantification of the glycolytic pathway in yeast: deployment of a complete QconCAT approach. Mol Cell Proteomics. 2011;10(12):M111 007633. doi: 10.1074/mcp.M111.007633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Wu Y, Williams EG, Dubuis S, Mottis A, Jovaisaite V, Houten SM, Argmann CA, Faridi P, Wolski W, Kutalik Z, Zamboni N, Auwerx J, Aebersold R. Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell. 2014;158(6):1415–1430. doi: 10.1016/j.cell.2014.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Weaver LJ, Sousa MM, Wang G, Baidoo E, Petzold CJ, Keasling JD. A kinetic-based approach to understanding heterologous mevalonate pathway function in E. coli. Biotechnol Bioeng. 2015;112(1):111–9. doi: 10.1002/bit.25323. [DOI] [PubMed] [Google Scholar]
- 233.Wolters JC, Ciapaite J, van Eunen K, Niezen-Koning KE, Matton A, Porte RJ, Horvatovich P, Bakker BM, Bischoff R, Permentier HP. Translational Targeted Proteomics Profiling of Mitochondrial Energy Metabolic Pathways in Mouse and Human Samples. J Proteome Res. 2016;15(9):3204–13. doi: 10.1021/acs.jproteome.6b00419. [DOI] [PubMed] [Google Scholar]
- 234.Schiffmann C, Hansen R, Baumann S, Kublik A, Nielsen PH, Adrian L, von Bergen M, Jehmlich N, Seifert J. Comparison of targeted peptide quantification assays for reductive dehalogenases by selective reaction monitoring (SRM) and precursor reaction monitoring (PRM) Anal Bioanal Chem. 2014;406(1):283–91. doi: 10.1007/s00216-013-7451-7. [DOI] [PubMed] [Google Scholar]
- 235.Whiteaker JR, Zhao L, Yan P, Ivey RG, Voytovich UJ, Moore HD, Lin C, Paulovich AG. Peptide Immunoaffinity Enrichment and Targeted Mass Spectrometry Enables Multiplex, Quantitative Pharmacodynamic Studies of Phospho-Signaling. Mol Cell Proteomics. 2015;14(8):2261–73. doi: 10.1074/mcp.O115.050351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kennedy JJ, Yan P, Zhao L, Ivey RG, Voytovich UJ, Moore HD, Lin C, Pogosova-Agadjanyan EL, Stirewalt DL, Reding KW, Whiteaker JR, Paulovich AG. Immobilized Metal Affinity Chromatography Coupled to Multiple Reaction Monitoring Enables Reproducible Quantification of Phospho-signaling. Mol Cell Proteomics. 2016;15(2):726–39. doi: 10.1074/mcp.O115.054940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Curran TG, Zhang Y, Ma DJ, Sarkaria JN, White FM. MARQUIS: a multiplex method for absolute quantification of peptides and posttranslational modifications. Nat Commun. 2015;6:5924. doi: 10.1038/ncomms6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Liu X, Jin Z, O'Brien R, Bathon J, Dietz HC, Grote E, Van Eyk JE. Constrained selected reaction monitoring: quantification of selected post-translational modifications and protein isoforms. Methods. 2013;61(3):304–12. doi: 10.1016/j.ymeth.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Shi T, Gao Y, Gaffrey MJ, Nicora CD, Fillmore TL, Chrisler WB, Gritsenko MA, Wu C, He J, Bloodsworth KJ, Zhao R, Camp DG, 2nd, Liu T, Rodland KD, Smith RD, Wiley HS, Qian WJ. Sensitive targeted quantification of ERK phosphorylation dynamics and stoichiometry in human cells without affinity enrichment. Anal Chem. 2015;87(2):1103–10. doi: 10.1021/ac503797x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Bisson N, James DA, Ivosev G, Tate SA, Bonner R, Taylor L, Pawson T. Selected reaction monitoring mass spectrometry reveals the dynamics of signaling through the GRB2 adaptor. Nat Biotechnol. 2011;29(7):653–8. doi: 10.1038/nbt.1905. [DOI] [PubMed] [Google Scholar]
- 241.Lawrence RT, Searle BC, Llovet A, Villen J. Plug-and-play analysis of the human phosphoproteome by targeted high-resolution mass spectrometry. Nat Methods. 2016;13(5):431–4. doi: 10.1038/nmeth.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Takemori N, Takemori A, Matsuoka K, Morishita R, Matsushita N, Aoshima M, Takeda H, Sawasaki T, Endo Y, Higashiyama S. High-throughput synthesis of stable isotope-labeled transmembrane proteins for targeted transmembrane proteomics using a wheat germ cell-free protein synthesis system. Mol Biosyst. 2015;11(2):361–5. doi: 10.1039/c4mb00556b. [DOI] [PubMed] [Google Scholar]
- 243.Mithoe SC, Ludwig C, Pel MJ, Cucinotta M, Casartelli A, Mbengue M, Sklenar J, Derbyshire P, Robatzek S, Pieterse CM, Aebersold R, Menke FL. Attenuation of pattern recognition receptor signaling is mediated by a MAP kinase kinase kinase. EMBO Rep. 2016;17(3):441–54. doi: 10.15252/embr.201540806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Zhao Y, Tian B, Edeh CB, Brasier AR. Quantitation of the dynamic profiles of the innate immune response using multiplex selected reaction monitoring-mass spectrometry. Mol Cell Proteomics. 2013;12(6):1513–29. doi: 10.1074/mcp.M112.023465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Yan W, Luo J, Robinson M, Eng J, Aebersold R, Ranish J. Index-ion triggered MS2 ion quantification: a novel proteomics approach for reproducible detection and quantification of targeted proteins in complex mixtures. Mol Cell Proteomics. 2011;10(3):M110 005611. doi: 10.1074/mcp.M110.005611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Schmidlin T, Garrigues L, Lane CS, Mulder TC, van Doorn S, Post H, de Graaf EL, Lemeer S, Heck AJ, Altelaar AF. Assessment of SRM, MRM(3), and DIA for the targeted analysis of phosphorylation dynamics in non-small cell lung cancer. Proteomics. 2016;16(15-16):2193–205. doi: 10.1002/pmic.201500453. [DOI] [PubMed] [Google Scholar]
- 247.Chen Y, Gruidl M, Remily-Wood E, Liu RZ, Eschrich S, Lloyd M, Nasir A, Bui MM, Huang E, Shibata D, Yeatman T, Koomen JM. Quantification of beta-catenin signaling components in colon cancer cell lines, tissue sections, and microdissected tumor cells using reaction monitoring mass spectrometry. J Proteome Res. 2010;9(8):4215–27. doi: 10.1021/pr1005197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Carapito C, Lane L, Benama M, Opsomer A, Mouton-Barbosa E, Garrigues L, Gonzalez de Peredo A, Burel A, Bruley C, Gateau A, Bouyssie D, Jaquinod M, Cianferani S, Burlet-Schiltz O, Van Dorsselaer A, Garin J, Vandenbrouck Y. Computational and Mass-Spectrometry-Based Workflow for the Discovery and Validation of Missing Human Proteins: Application to Chromosomes 2 and 14. J Proteome Res. 2015;14(9):3621–34. doi: 10.1021/pr5010345. [DOI] [PubMed] [Google Scholar]
- 249.Sun H, Chen C, Shi M, Wang D, Liu M, Li D, Yang P, Li Y, Xie L. Integration of mass spectrometry and RNA-Seq data to confirm human ab initio predicted genes and lncRNAs. Proteomics. 2014;14(23-24):2760–8. doi: 10.1002/pmic.201400174. [DOI] [PubMed] [Google Scholar]
- 250.Poverennaya EV, Kopylov AT, Ponomarenko EA, Ilgisonis EV, Zgoda VG, Tikhonova OV, Novikova SE, Farafonova TE, Kiseleva YY, Radko SP, Vakhrushev IV, Yarygin KN, Moshkovskii SA, Kiseleva OI, Lisitsa AV, Sokolov AS, Mazur AM, Prokhortchouk EB, Skryabin KG, Kostrjukova ES, Tyakht AV, Gorbachev AY, Ilina EN, Govorun VM, Archakov AI. State of the Art of Chromosome 18-Centric HPP in 2016: Transcriptome and Proteome Profiling of Liver Tissue and HepG2 Cells. J Proteome Res. 2016;15(11):4030–4038. doi: 10.1021/acs.jproteome.6b00380. [DOI] [PubMed] [Google Scholar]
- 251.Chen C, Liu X, Zheng W, Zhang L, Yao J, Yang P. Screening of missing proteins in the human liver proteome by improved MRM-approach-based targeted proteomics. J Proteome Res. 2014;13(4):1969–78. doi: 10.1021/pr4010986. [DOI] [PubMed] [Google Scholar]
- 252.Kopylov AT, Ilgisonis EV, Moysa AA, Tikhonova OV, Zavialova MG, Novikova SE, Lisitsa AV, Ponomarenko EA, Moshkovskii SA, Markin AA, Grigoriev AI, Zgoda VG, Archakov AI. Targeted Quantitative Screening of Chromosome 18 Encoded Proteome in Plasma Samples of Astronaut Candidates. J Proteome Res. 2016;15(11):4039–4046. doi: 10.1021/acs.jproteome.6b00384. [DOI] [PubMed] [Google Scholar]
- 253.Ponomarenko EA, Kopylov AT, Lisitsa AV, Radko SP, Kiseleva YY, Kurbatov LK, Ptitsyn KG, Tikhonova OV, Moisa AA, Novikova SE, Poverennaya EV, Ilgisonis EV, Filimonov AD, Bogolubova NA, Averchuk VV, Karalkin PA, Vakhrushev IV, Yarygin KN, Moshkovskii SA, Zgoda VG, Sokolov AS, Mazur AM, Prokhortchouck EB, Skryabin KG, Ilina EN, Kostrjukova ES, Alexeev DG, Tyakht AV, Gorbachev AY, Govorun VM, Archakov AI. Chromosome 18 transcriptoproteome of liver tissue and HepG2 cells and targeted proteome mapping in depleted plasma: update 2013. J Proteome Res. 2014;13(1):183–90. doi: 10.1021/pr400883x. [DOI] [PubMed] [Google Scholar]
- 254.Ilgisonis EV, Kopylov AT, Zgoda VG. Dataset of target mass spectromic proteome profiling for human chromosome 18. Data Brief. 2016;8:1365–9. doi: 10.1016/j.dib.2016.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Zgoda VG, Kopylov AT, Tikhonova OV, Moisa AA, Pyndyk NV, Farafonova TE, Novikova SE, Lisitsa AV, Ponomarenko EA, Poverennaya EV, Radko SP, Khmeleva SA, Kurbatov LK, Filimonov AD, Bogolyubova NA, Ilgisonis EV, Chernobrovkin AL, Ivanov AS, Medvedev AE, Mezentsev YV, Moshkovskii SA, Naryzhny SN, Ilina EN, Kostrjukova ES, Alexeev DG, Tyakht AV, Govorun VM, Archakov AI. Chromosome 18 transcriptome profiling and targeted proteome mapping in depleted plasma, liver tissue and HepG2 cells. J Proteome Res. 2013;12(1):123–34. doi: 10.1021/pr300821n. [DOI] [PubMed] [Google Scholar]
- 256.Huttenhain R, Surinova S, Ossola R, Sun Z, Campbell D, Cerciello F, Schiess R, Bausch-Fluck D, Rosenberger G, Chen J, Rinner O, Kusebauch U, Hajduch M, Moritz RL, Wollscheid B, Aebersold R. N-glycoprotein SRMAtlas: a resource of mass spectrometric assays for N-glycosites enabling consistent and multiplexed protein quantification for clinical applications. Mol Cell Proteomics. 2013;12(4):1005–16. doi: 10.1074/mcp.O112.026617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Jovanovic M, Reiter L, Clark A, Weiss M, Picotti P, Rehrauer H, Frei A, Neukomm LJ, Kaufman E, Wollscheid B, Simard MJ, Miska EA, Aebersold R, Gerber AP, Hengartner MO. RIP-chip-SRM--a new combinatorial large-scale approach identifies a set of translationally regulated bantam/miR-58 targets in C. elegans. Genome Res. 2012;22(7):1360–71. doi: 10.1101/gr.133330.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Jovanovic M, Reiter L, Picotti P, Lange V, Bogan E, Hurschler BA, Blenkiron C, Lehrbach NJ, Ding XC, Weiss M, Schrimpf SP, Miska EA, Grosshans H, Aebersold R, Hengartner MO. A quantitative targeted proteomics approach to validate predicted microRNA targets in C. elegans. Nat Methods. 2010;7(10):837–42. doi: 10.1038/nmeth.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Schreiner D, Simicevic J, Ahrne E, Schmidt A, Scheiffele P. Quantitative isoform-profiling of highly diversified recognition molecules. Elife. 2015;4:e07794. doi: 10.7554/eLife.07794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Maabeta S, Wachlin G, Bernhardt J, Eymann C, Fromion V, Riedel K, Becher D, Hecker M. Highly precise quantification of protein molecules per cell during stress and starvation responses in Bacillus subtilis. Mol Cell Proteomics. 2014;13(9):2260–76. doi: 10.1074/mcp.M113.035741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Maass S, Sievers S, Zuhlke D, Kuzinski J, Sappa PK, Muntel J, Hessling B, Bernhardt J, Sietmann R, Volker U, Hecker M, Becher D. Efficient, global-scale quantification of absolute protein amounts by integration of targeted mass spectrometry and two-dimensional gel-based proteomics. Anal Chem. 2011;83(7):2677–84. doi: 10.1021/ac1031836. [DOI] [PubMed] [Google Scholar]
- 262.Schmidt A, Kochanowski K, Vedelaar S, Ahrne E, Volkmer B, Callipo L, Knoops K, Bauer M, Aebersold R, Heinemann M. The quantitative and condition-dependent Escherichia coli proteome. Nat Biotechnol. 2016;34(1):104–10. doi: 10.1038/nbt.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Schmidt A, Beck M, Malmstrom J, Lam H, Claassen M, Campbell D, Aebersold R. Absolute quantification of microbial proteomes at different states by directed mass spectrometry. Mol Syst Biol. 2011;7:510. doi: 10.1038/msb.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Schubert OT, Mouritsen J, Ludwig C, Rost HL, Rosenberger G, Arthur PK, Claassen M, Campbell DS, Sun Z, Farrah T, Gengenbacher M, Maiolica A, Kaufmann SH, Moritz RL, Aebersold R. The Mtb proteome library: a resource of assays to quantify the complete proteome of Mycobacterium tuberculosis. Cell Host Microbe. 2013;13(5):602–12. doi: 10.1016/j.chom.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Lawless C, Holman SW, Brownridge P, Lanthaler K, Harman VM, Watkins R, Hammond DE, Miller RL, Sims PF, Grant CM, Eyers CE, Beynon RJ, Hubbard SJ. Direct and Absolute Quantification of over 1800 Yeast Proteins via Selected Reaction Monitoring. Mol Cell Proteomics. 2016;15(4):1309–22. doi: 10.1074/mcp.M115.054288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Picotti P, Clement-Ziza M, Lam H, Campbell DS, Schmidt A, Deutsch EW, Rost H, Sun Z, Rinner O, Reiter L, Shen Q, Michaelson JJ, Frei A, Alberti S, Kusebauch U, Wollscheid B, Moritz RL, Beyer A, Aebersold R. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature. 2013;494(7436):266–70. doi: 10.1038/nature11835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Karlsson C, Malmstrom L, Aebersold R, Malmstrom J. Proteome-wide selected reaction monitoring assays for the human pathogen Streptococcus pyogenes. Nat Commun. 2012;3:1301. doi: 10.1038/ncomms2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Glukhova VA, Tomazela DM, Findlay GD, Monnat RJ, Jr, MacCoss MJ. Rapid assessment of RNAi-mediated protein depletion by selected reaction monitoring mass spectrometry. J Proteome Res. 2013;12(7):3246–54. doi: 10.1021/pr400067k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Su ZD, Sun L, Yu DX, Li RX, Li HX, Yu ZJ, Sheng QH, Lin X, Zeng R, Wu JR. Quantitative detection of single amino acid polymorphisms by targeted proteomics. J Mol Cell Biol. 2011;3(5):309–15. doi: 10.1093/jmcb/mjr024. [DOI] [PubMed] [Google Scholar]