

Abstract
Objective
The triggering receptor expressed on myeloid cells 2 (TREM2) p.R47H substitution (rs75932628) is a risk factor for Alzheimer disease (AD) but has not been well studied in relation to the risk of multiple system atrophy (MSA); the aim of this study was to evaluate the association between the TREM2 p.R47H variant and the risk of MSA.
Methods
A total of 168 patients with pathologically confirmed MSA, 89 patients with clinically diagnosed MSA, and 1,695 controls were included. TREM2 p.R47H was genotyped and assessed for association with MSA. Positive results in the Taqman genotyping assay were confirmed by Sanger sequencing. The primary comparison involved patients with pathologically confirmed MSA and controls due to the definitive MSA diagnosis in the pathologically confirmed series.
Results
We identified TREM2 p.R47H in 3 patients with pathologically confirmed MSA (1.79%), 1 patient with clinically diagnosed MSA (1.12%), and 7 controls (0.41%). Minimal AD pathology was observed for the pathologically confirmed MSA p.R47H carriers. For the primary comparison of patients with pathologically confirmed MSA and controls, risk of disease was significantly higher for p.R47H carriers (odds ratio [OR]: 4.39, p = 0.033). When supplementing the 168 pathologically confirmed patients with the 89 clinically diagnosed and examining the combined MSA series, the association with TREM2 p.R47H remained significant (OR: 3.81, p = 0.034).
Conclusions
Our preliminary results suggest that the TREM2 p.R47H substitution may be a risk factor for MSA, implying a link to neuroinflammatory processes, especially microglial activation. Validation of this finding will be important, given our relatively small sample size; meta-analytic approaches will be needed to better define the role of this variant in MSA.
Multiple system atrophy (MSA) is a rare, rapidly progressing, adult-onset neurodegenerative disease that is clinically characterized by parkinsonism, autonomic failure, pyramidal symptoms, and cerebellar ataxia and pathologically defined by the presence of α-synuclein-positive glial cytoplasmic inclusions.1 Uncovering genetic risk factors for MSA has proven difficult because of the challenge of accumulating a reasonable sample size and potential for clinical misdiagnosis. Nonetheless, studies have nominated several genes that may be associated with the risk of MSA.1,2 The first genome-wide association study of MSA was recently completed, and although no genome-wide significant associations were identified, suggestive associations were identified involving variants in MAPT, FBXO47, ELOVL7, and EDN1.3 Clearly, much remains to be understood regarding the genetics of MSA, and replication of previously nominated MSA susceptibility loci remains crucial.
A relatively rare substitution p.R47H (rs75932628) in the triggering receptor expressed on myeloid cells 2 (TREM2) protein is now a well-established risk factor for Alzheimer disease (AD) and has been suggested to play a role in other neurodegenerative disorders such as frontotemporal dementia (FTD) and Parkinson disease (PD).4 Of interest, mutations in the TREM2 gene were originally reported as a genetic cause of Nasu-Hakola disease, a rare leukodystrophy characterized by progressive presenile dementia and bone cysts.4 Given the important role of microglia in neuroinflammation, we evaluated the association between the TREM2 p.R47H variant and the risk of MSA using a predominantly neuropathologically confirmed MSA series.
Methods
Study participants
This study included 168 patients with pathologically confirmed MSA, 89 patients with clinically diagnosed MSA, and 1,695 healthy controls. The pathologically confirmed MSA series consisted of all cases obtained from the Mayo Clinic brain bank for neurodegenerative disorders in Jacksonville, FL, between 1998 and 2017. Pathologically confirmed MSA was diagnosed by a single neuropathologist (D.W.D.) as previously described.5 Immunohistochemistry for α-synuclein (NACP, 1:3,000 rabbit polyclonal) was performed on sections of the basal forebrain, striatum, midbrain, pons, medulla, and cerebellum to establish a neuropathologic diagnosis. MSA cases were pathologically subclassified as MSA with predominant striatonigral involvement (MSA-P), MSA with predominant olivopontocerebellar involvement (MSA-C), and MSA with equally severe involvement of striatonigral and olivopontocerebellar systems (MSA-mixed).5 One case was not assigned pathologic subtype because only the pons was available for histology.
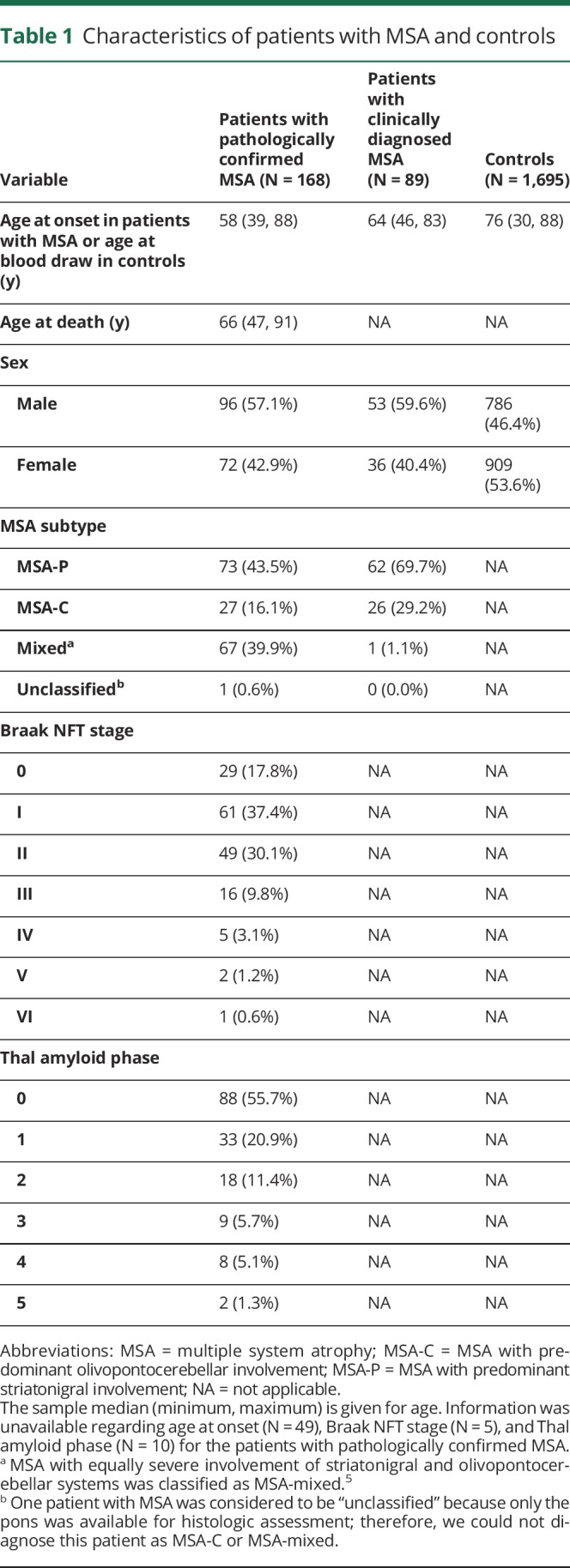
The clinically diagnosed patients (N = 81 probable MSA, N = 8 possible MSA) were seen at the Mayo Clinic in Rochester, MN (N = 35), or Jacksonville, FL (N = 54), where diagnosis was made according to the criteria of Gilman et al.6 Clinically diagnosed MSA cases were free of notable cognitive symptoms that might suggest the presence of an overlapping dementia or other neurodegenerative conditions. Pathologic assessment and clinical diagnoses were made without the knowledge of the TREM2 p.R47H genotype. Controls were also from the Mayo Clinic in Rochester, MN (N = 991), or Jacksonville, FL (N = 704). Controls were free of neurologic symptoms and without a family history of movement disorders. All participants were unrelated non-Hispanic Caucasians. The primary comparison of TREM2 p.R47H involved the patients with pathologically confirmed MSA and controls. In secondary analysis, we combined the patients with pathologically confirmed MSA and patients with clinically diagnosed MSA and compared this combined MSA series with the controls. Table 1 shows characteristics of patients with MSA and controls.
Table 1.
Characteristics of patients with MSA and controls
Standard protocol approvals, registrations, and patient consents
The Mayo Clinic IRB approved the use of human participants in this study.
Genetic analysis
Genomic DNA was extracted from brain tissue or peripheral blood monocytes using standard protocols. Genotyping of TREM2 exon 2 variant rs75932628 (NM_018965.3:c.140G>A, p.R47H) was performed using a custom TaqMan Allelic Discrimination Assay on an ABI 7900HT Fast Real-Time PCR system (Applied Bio-systems, Foster City, CA) according to the manufacturer's instructions (sequences available upon request). Genotype calls were made using SDS 2.2.2 software. Positive or ambiguous results in the TaqMan assay were also confirmed and resolved via Sanger sequencing. There was no evidence of a departure from Hardy-Weinberg equilibrium in controls (p = 0.93).
Statistical analysis
We evaluated the association between TREM2 p.R47H and MSA using logistic regression models. Unadjusted models were first examined. Subsequently, although it is uncertain whether adjusting for any variable is reasonable, given the rare nature of this variant, we did also adjust our logistic regression models for age and sex in a sensitivity analysis. Odds ratios (ORs) and 95% confidence intervals were estimated. p ≤ 0.05 was considered statistically significant. All statistical analyses were performed using SAS (version 9.4; SAS Institute, Inc., Cary, NC).
Results
An evaluation of the association between TREM2 p.R47H and MSA is shown in table 2. The p.R47H variant was observed in 3 patients with pathologically confirmed MSA (1.79%). Of these 3 p.R47H carriers, neuropathologic examination was available for 2 and revealed MSA subtypes of MSA-P and MSA-C, Braak neurofibrillary tangle stages of I and III, and Thal amyloid phases of 0 and 0. For the 89 patients with clinically diagnosed MSA, there was 1 p.R47H carrier (1.12%, from Mayo Jacksonville), and this patient's MSA subtype was probable MSA-P. There were 7 carriers of p.R47H in the controls (0.41%, N = 4 Mayo Jacksonville, N = 3 Mayo Rochester), corresponding to a minor allele frequency of 0.21%, which is similar to the frequency reported in the ExAC database (Cambridge, MA [exac.broadinstitute.org]) for this population (0.26%).7 For the primary comparison of patients with pathologically confirmed MSA and controls, risk of MSA was significantly higher for p.R47H carriers (OR = 4.39, p = 0.033). When combining the pathologically confirmed and clinically diagnosed MSA series, the risk of MSA (vs controls) was also significantly higher (OR = 3.81, p = 0.034). In a sensitivity analysis adjusting for age and sex, the findings regarding the patients with pathologically confirmed MSA (OR = 3.55, p = 0.076) and the combined MSA series (OR = 3.17, p = 0.078) remained relatively consistent though weakened slightly and were no longer quite significant.
Table 2.
Association between TREM2 p.R47H and MSA

Discussion
TREM2 p.R47H is a well-established risk factor for AD4; however, results have been mixed for FTD and PD,4 while studies involving amyotrophic lateral sclerosis and dementia with Lewy bodies mostly point to a lack of association.4 Of interest, the results of our current study suggest that the TREM2 p.R47H variant may be associated with an increased risk of MSA. With a notably higher frequency in our combined MSA series (1.6%) compared with controls (0.4%), we observed a nearly 4-fold increased risk of MSA in p.R47H carriers. It is important that this finding was strongest in our pathologically confirmed MSA series. The absence of notable AD pathology in pathologically confirmed MSA p.R47H carriers indicates that the observed association was not driven by such pathology. The association was attenuated slightly when adjusting for age and sex; however, those results should not be overinterpreted because it is uncertain whether attempting any adjustment is reasonable, given the low frequency of p.R47H. These preliminary findings suggest that TREM2 p.R47H may play a role in susceptibility to MSA and that further study is warranted.
To the best of our knowledge, only 1 association study between TREM2 p.R47H and MSA has been conducted to date. In a study of 407 Chinese patients with clinically diagnosed MSA and 869 controls, the p.R47H substitution was observed in 1 MSA patient and no controls (p = 0.14).8 This negative result is not surprising because TREM2 p.R47H is extremely rare in East Asians.
How the TREM2 p.R47H variant could increase risk of MSA is an important topic for further study. MSA is regarded as an oligodendrogliopathy, but the mechanism of demyelination remains unclear. While demyelination depends primarily on oligodendrocytes, microglia also contribute to myelination and myelin homeostasis.9 In the CNS, TREM2 is predominantly expressed in microglia, and it has been shown to play critical roles in phagocytic clearance of apoptotic cells and disease-associated molecules and modulating microglial immune response.10 A recent study showed that TREM2 is required for myelin debris removal and remyelination after cuprizone-induced demyelination.11 We hypothesize that TREM2 p.R47H, a loss-of-function mutation, leads to impaired clearance of myelin debris and aberrant microglial activation, which result in increased risk of MSA.
Our results provide evidence that the TREM2 p.R47H substitution may be a genetic susceptibility factor for MSA. Our study is limited by the relatively small number of patients with pathologically confirmed MSA, and therefore, validation will be crucial. Recently, the clinical diagnostic accuracy of MSA was reported to be 62%, further underscoring the importance of using neuropathologically confirmed MSA cases in research studies.12 We hope that these initial promising results will form the basis for future examinations of TREM2 p.R47H in MSA.
Acknowledgment
The authors thank all those who have contributed to our research, particularly the patients and families who donated Brain and DNA samples for this work.
Glossary
- AD
Alzheimer disease
- FTD
frontotemporal dementia
- MSA
multiple system atrophy
- MSA-C
MSA with predominant olivopontocerebellar involvement
- MSA-P
MSA with predominant striatonigral involvement
- OR
odds ratio
- PD
Parkinson disease
- TREM2
triggering receptor expressed on myeloid cells 2
Author contributions
K. Ogaki: drafting/revising the manuscript; study concept and design; and interpretation of data. M. G. Heckman and S. Koga: drafting/revising the manuscript and interpretation of data. Y.A. Martens: drafting/revising the manuscript. C. Labbé and O. Lorenzo-Betancor: revising the manuscript. R.L. Walton, A.I. Soto, and E.R. Vargas: revising the manuscript and interpretation of data. S. Fujioka: revising the manuscript. R.J. Uitti, J. Van Gerpen, and W.P. Cheshire: revising the manuscript and providing material for this study. S.G. Younkin: revising the manuscript. Z.K. Wszolek: revising the manuscript, providing material and funding for this study, and IRB committee approval. P.A. Low and W. Singer: revising the manuscript and providing material for this study. G. Bu: revising the manuscript. D.W. Dickson: drafting/revising the manuscript, providing material and funding for this study, and IRB committee approval. O.A. Ross: drafting/revising the manuscript; study concept and design; and interpretation of data.
Study funding
K. Ogaki is supported by a research grant from the NAITO Foundation, JSPS KAKENHI Grant Number JP17K14966 and a grant from Institute for Environmental and Gender-Specific Medicine, Juntendo University. R.J. Uitti, O.A. Ross, Z.K. Wszolek, and D.W. Dickson are partially supported by the NIH/NINDS P50 NS072187. O.A. Ross is supported by the NINDS R01# NS078086, U54 NS100693, DOD W81XWH-17-1-0249, and in part by the Michael J. Fox Foundation for Parkinson's Research. C. Labbé is the recipient of a F.R.S.Q. fellowship. Z.K. Wszolek receives research support from NIH/NIA (primary), and NIH/NINDS (secondary) 1U01AG045390-01A1, Mayo Clinic Center for Regenerative Medicine, Mayo Clinic Neuroscience Focused Research Team (Cecilia and Dan Carmichael Family Foundation, and the James C. and Sarah K. Kennedy Fund for Neurodegenerative Disease Research at Mayo Clinic in Florida), and is also partially supported by a gift from Carl Edward Bolch, Jr, and Susan Bass Bolch, The Sol Goldman Charitable Trust, and Donald G. and Jodi P. Heeringa. P.A. Low receives research support from the Cure MSA Foundation. Samples included in this study were clinical patients or brain donors to the brain bank at Mayo Clinic in Jacksonville, which is supported by the CurePSP Society for Progressive Supranuclear Palsy and Udall Center for Excellence in Parkinson Research (P50 NS072187), APDA Center for Advanced Research, an Alzheimer's disease Research Center grant (NIA P50 AG16574), and NINDS R01 NS078086.
Disclosure
K. Ogaki receives JSPS KAKENHI Grant Number JP17K14966 and a grant from Institute for Environmental & Gender-Specific Medicine, Juntendo University. M.G. Heckman serves as the editorial board member of Parkinsonism & Related Disorders. S. Koga and Y.A. Martens report no disclosures. C. Labbé receives a Fonds de recherche du Quebec-Sante postdoctoral fellowship. O. Lorenzo-Betancor, R.L. Walton, A.I. Soto, and E. Vargas report no disclosures. S. Fujioka receives JSPS KAKENHI Grant Number JP15K19501 and is an editorial board member of Parkinsonism & Related Disorders. R.J. Uitti serves as an associate editor of Neurology. J.Van Gerpen report no disclosures. W.P. Cheshire serves in the editorial boards of Parkinsonism & Related Disorders and Autonomic Neuroscience and as an associate editor of Clinical Autonomic Research. S.G. Younkin reports no disclosures. Z.K. Wszolek receives research support from P50 NS072187, NIH/NIA (primary), and NIH/NINDS (secondary) 1U01AG045390-01A1, Mayo Clinic Center for Regenerative Medicine, Mayo Clinic Neuroscience Focused Research Team (Cecilia and Dan Carmichael Family Foundation, and the James C. and Sarah K. Kennedy Fund for Neurodegenerative Disease Research at Mayo Clinic in Florida), the gift from Carl Edward Bolch, Jr., and Susan Bass Bolch, The Sol Goldman Charitable Trust, and Donald G. and Jodi P. Heeringa; serves as coeditor-in-chief of Parkinsonism & Related Disorders and an associate editor of the European Journal of Neurology; is on the editorial boards of Neurologia i Neurochirurgia Polska, European Journal of Clinical and Experimental Medicine, Clinical and Experimental Medical Letters, and Wiadomosci Lekarskie; holds and has contractual rights for receipt of future royalty payments from patents for “A Novel Polynucleotide Involved in Heritable Parkinson's Disease”; and receives royalties from publishing Parkinsonism & Related Disorders (Elsevier, 2016, 2017) and the European Journal of Neurology (Wiley Blackwell, 2016, 2017). P.A. Low receives research support from the NIH (P01 NS44233, U54 NS065736, R01 NS092625, and UL1 TR000135), FDA (R01 FD004789), Cure MSA Foundation, and Mayo Funds and serves as a clinical editor of Autonomic Neuroscience. W. Singer reports no disclosures. G. Bu received support from P50AG016574, RF1AG051504, R01AG027924, R01AG035355, R01AG046205, and P01NS074969 and a grant from the Cure Alzheimer's fund. D.W. Dickson receives support from P50-AG016574, P50-NS072187, P01-AG003949, and CurePSP: Foundation for PSP|CBD and Related Disorders. D.W. Dickson is an editorial board member of Acta Neuropathologica, Annals of Neurology, Brain, Brain Pathology, and Neuropathology and is editor-in-chief of the American Journal of Neurodegenerative Disease and International Journal of Clinical and Experimental Pathology. O.A. Ross received support from R01 NS078086, P50-NS072187, and U54 NS100693 and the Michael J. Fox Foundation. O.A. Ross is an editorial board member of the American Journal of Neurodegenerative Disease, Neurologia i Neurochirurgia Polska, and Molecular Neurodegeneration. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Ahmed Z, Asi YT, Sailer A, et al. The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol Appl Neurobiol 2012;38:4–24. [DOI] [PubMed] [Google Scholar]
- 2.Scholz SW, Bras J. Genetics underlying atypical parkinsonism and related neurodegenerative disorders. Int J Mol Sci 2015;16:24629–24655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sailer A, Scholz SW, Nalls MA, et al. A genome-wide association study in multiple system atrophy. Neurology 2016;87:1591–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jay TR, von Saucken VE, Landreth GE. TREM2 in neurodegenerative diseases. Mol Neurodegener 2017;12:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koga S, Parks A, Uitti RJ, et al. Profile of cognitive impairment and underlying pathology in multiple system atrophy. Mov Disord 2017;32:405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Chen X, Guo X, et al. Assessment of TREM2 rs75932628 association with Parkinson's disease and multiple system atrophy in a Chinese population. Neurol Sci 2015;36:1903–1906. [DOI] [PubMed] [Google Scholar]
- 9.Simons M, Lyons DA. Axonal selection and myelin sheath generation in the central nervous system. Curr Opin Cel Biol 2013;25:512–519. [DOI] [PubMed] [Google Scholar]
- 10.Painter MM, Atagi Y, Liu CC, et al. TREM2 in CNS homeostasis and neurodegenerative disease. Mol Neurodegener 2015;10:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poliani PL, Wang Y, Fontana E, et al. TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest 2015;125:2161–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koga S, Aoki N, Uitti RJ, et al. When DLB, PD, and PSP masquerade as MSA: an autopsy study of 134 patients. Neurology 2015;85:404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]