

Abstract
Objective
To assess the contribution of noncoding repeat expansions in Japanese patients with amyotrophic lateral sclerosis (ALS).
Methods
Sporadic ALS in Western countries is frequently associated with noncoding repeat expansions in the C9ORF72 gene. Spinocerebellar ataxia type 8 (SCA8) is another noncoding repeat disease caused by expanded CTA/CTG repeats in the ATXN8OS gene. Although the involvement of upper and lower motor neurons in SCA8 has been reported, a positive association between SCA8 and ALS remains unestablished. Spinocerebellar ataxia type 36 is a recently identified disease caused by noncoding repeat expansions in the NOP56 gene and is characterized by motor neuron involvement. We collected blood samples from 102 Japanese patients with sporadic ALS and analyzed the ATXN8OS gene by the PCR–Sanger sequencing method and the C9ORF72 and NOP56 genes by repeat-primed PCR assay.
Results
Three patients with ALS (3%) had mutations in the ATXN8OS gene, whereas no patient had a mutation in the C9ORF72 or NOP56 gene. The mutation-positive patients were clinically characterized by neck weakness or bulbar-predominant symptoms. None of our patients had apparent cerebellar atrophy on MRI, but 2 had nonsymptomatic abnormalities in the white matter or putamen.
Conclusions
Our finding reveals the importance of noncoding repeat expansions in Japanese patients with ALS and extends the clinical phenotype of SCA8. Three percent seems small but is still relatively large for Japan, considering that the most commonly mutated genes, including the SOD1 and SQSTM1 genes, only account for 2%–3% of sporadic patients each.
Amyotrophic lateral sclerosis (ALS) mostly occurs sporadically. However, more than 10% of sporadic patients examined in Japan and in Western countries had mutations in various genes causative for familial ALS.1–3 Noncoding six-base (GGGGCC) repeat expansions in the C9ORF72 gene are causative for sporadic ALS and are frequent in Western countries (4%–21% of all sporadic ALS).2,3 By contrast, only small numbers of sporadic patients in Japan had mutations in the C9ORF72 gene (<0.5%).4 Thus, the significance of noncoding repeat expansions in ALS has not been established in Japan.
Spinocerebellar ataxia type 8 (SCA8) is an autosomal dominant neurodegenerative disease, caused by noncoding CTA/CTG repeat expansions in the ATXN8OS (ATAXIN 8 OPPOSITE STRAND) gene.5,6 The pathogenicity of the expanded allele has been proven using a transgenic mouse model.7 Many patients had pure cerebellar ataxia, whereas some had parkinsonism.5,6,8 Several studies have suggested the involvement of upper and lower motor neurons in SCA8, but a positive association between SCA8 and ALS remains unestablished.9,10
Spinocerebellar ataxia type 36 (SCA36) is a recently identified disease, caused by noncoding six-base (GGCCTG) repeat expansions in the NOP56 gene.11 Motor neuron involvement becomes apparent during the disease course.
Identification of repeat abnormalities by current exome or whole-genome analyses in ALS remains challenging, although efforts are underway.12 In this report, we analyzed the ATXN8OS, C9ORF72, and NOP56 genes in 102 Japanese patients with sporadic ALS and in 10 patients with mutations in various genes causative for ALS.
Methods
Genetic testing
All patients and controls were Japanese and were enrolled in this study from the Kinki region in Japan between 2005 and 2017. Blood samples were collected from 102 Japanese patients with sporadic ALS who had no mutations in the SOD1, FUS, TARDBP, SQSTM1, VCP, OPTN, UBQLN2, ATXN1, ATXN2, SMN1, or AR genes.2,11,13,14 DNA was extracted with a DNA extraction kit (Qiagen Inc, Germantown, MD), and the region containing the CTA/CTG repeat of the ATXN8OS gene was amplified using PCR, as described previously.5,15 The amplified products were purified with gel electrophoresis and subjected to Sanger sequencing. We analyzed the C9ORF72 and NOP56 genes by repeat-primed PCR assay, as described previously.3,11 The normal number of CTA/CTG repeats in the ATXN8OS gene ranges from 15 to 50, whereas repeats of length 80 or more are pathogenic. In several reports, expansions of more than 50 CTA/CTG repeats, including intermediate expansions, were stated to cause ataxia at some point in the life; however, there were no clinical details.16,17 We also examined 10 patients who had either sporadic or familial ALS with mutations in the SOD1 (n = 3, 1 man and 2 women), TARDBP (n = 1, 1 man), FUS (n = 1, 1 woman), VCP (n = 2, 1 man and 1 woman), or SQSTM1 (n = 3, 1 man and 2 women) genes.1,13,14 When mutations in the ATXN8OS, C9ORF72, or NOP56 gene were detected, the ERBB4 and COQ2 genes and genes for SCA3, SCA6, SCA7, SCA12, SCA17, SCA31, and DRPLA were additionally examined.15,18 The patients had a diagnosis of clinically definite, probable, or possible ALS, as defined in the revised El Escorial diagnostic criteria. Patients with sporadic ALS included 68 men and 34 women with an age of 65 ± 12 years (mean ± SD). The initial symptoms occurred in the limb muscles in 76 patients, in the bulbar muscles in 20, in the neck muscles in 3, in the respiratory muscles in 2, and in the facial muscles in 1. A control group consisting of 200 apparently healthy controls (116 men and 84 women; mean age ± SD, 71 ± 7 years) was also studied. We performed haplotype analyses in patients with ATXN8OS mutations using several microsatellite markers, including YI18, D13S1296, JJ9, JJ10, and JJ12 surrounding the CTA/CTG repeats with distances of 284, 57, 17, 20, and 52 kb, respectively.16 DNA from relatives of patients with ALS was not available. We additionally examined 2 ataxic sisters with SCA8 (aged 36 and 39 years) to compare the haplotype data with those for ALS.
Standard protocol approvals, registrations, and patient consents
This study was approved by our institutional review board. All patients provided written informed consent.
Results
Results of genetic testing of our patients
Genetic testing revealed that 3 patients with ALS had mutations in the ATXN8OS gene (figure 1), but none in the C9ORF72 gene or in the NOP56 gene. The clinical information of our 3 patients with ALS and 1 reported Korean patient with an ALS-like phenotype is summarized in table 1, and the detailed clinical history of our patients is described as follows. None of the controls had mutations in the ATXN8OS, C9ORF72, or NOP56 genes. Controls had 26 ± 4 repeats (mean ± SD), ranging from 18 to 32, in the ATXN8OS gene. Double mutations in different genes were not found in our cohort. Patient 1 had 223 repeats (CTA11CTG212), and patient 2 had 170 repeats (CTA9CTG161). Patient 3 had 91 CTA/CTG repeats with an interruption (CTA19CTG3CTA1CTG68). Interruption by CTA in a sequence of CTG repeats has previously been reported, but its significance is unknown.17 The haplotype analyses identified apparently different haplotypes in the patients with ALS, although their haplotypes were similar in part. By contrast, ataxic sisters with SCA8 had the same haplotype except for the numbers of CTG repeats in the ATXN8OS gene (table 2). These results suggested that the 3 patients with ALS seemed unrelated.
Figure 1. Agarose gel electrophoretic analysis for the ATXN8OS gene in patients (Pt) 1–3.
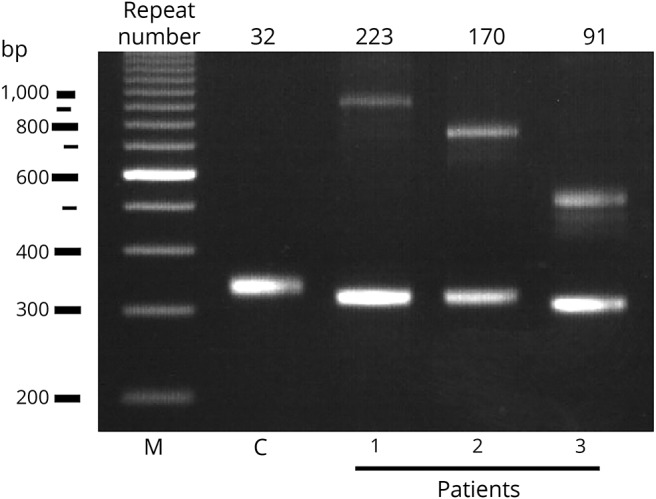
Three patients had expansions of the CTA/CTG repeat as indicated, whereas controls (C) had a normal repeat size. M, 100 base pair (bp) marker.
Table 1.
Patients with ALS or an ALS-like phenotype associated with ATXN8OS mutations

Table 2.
Result of haplotype analyses in patients with mutations in the ATXN8OS gene

Clinical and imaging information for the 3 patients with ATXN8OS-related ALS and 1 patient with an ALS-like phenotype
The results of MRI are summarized in figure 2, A-C. The detailed clinical histories of our patients are described below.
Figure 2. Imaging results for patients who had ALS with mutations in the ATXN8OS gene.

(A.a–A.d) Results of MRI (Philips 1.5T, TE/TR = 100/4200 for axial T2-weighted image) of patient 1 (Pt 1) at the age of 47 years (2 years after onset). Atrophy of the cerebellum (A.a), pons (A.b), hippocampus (A.c, arrows), or frontal lobes (A.d) is not obvious; however, ischemic changes (A.d, arrows) in the white matter can be observed. (B.a–B.d) Results of MRI (SIEMENS Symphony 1.5T, TE/TR = 83/3400 for axial T2-weighted image and TE/TR = 12/450 for coronal T1-weighted image) of patient 2 (Pt 2) at the age of 68 years (2 months after onset). Because this patient was not able to complete the MRI examination, the sequences, angles, and resolutions were limited. Atrophy of the cerebellum (B.a, B.b), pons (B.c), hippocampus (B.c, arrows), or frontal lobes (B.d) is not obvious. (C.a–C.d) Results of MRI (SIEMENS Symphony 1.5T, TE/TR = 89/4200 for axial T2-weighted image) of patient 3 (Pt 3) at the age of 75 years (9 years after onset). Atrophy of the cerebellum (C.a), pons (C.b), hippocampus (C.c, arrows), or frontal lobes (C.d) is not obvious; however, high intensities were observed in the putamen (arrowhead).
Patients 1
A 56-year-old woman, with 223 repeats, who had dysarthria since the age of 45 years visited a local hospital. She had no family history or previous history of neurologic disease. The symptoms progressed over 1 year. She visited the local hospital again at the age of 46 years, with atrophy of the tongue, an increased jaw jerk reflex, increased deep tendon reflexes (DTRs) in all 4 limbs, and positive plantar responses. She was then referred to our hospital. A needle electromyogram revealed neurogenic changes, with polyphasic or high-amplitude neuromuscular units with long durations in the tongue, left deltoid, and left tibialis anterior muscles. During the following year, at the age of 47 years, dysarthria, dysphagia, and neck flexor weakness became severe, but mild weakness of the limbs was seen only in the left upper extremity. Fasciculation was observed in the tongue and all 4 limbs. She was unable to perform a spirometry because of weak lip seal. She underwent percutaneous endoscopic gastrostomy (PEG) at the age of 47 years. She then showed progressive respiratory failure and weakness and atrophy in all 4 limbs. She underwent tracheostomy with mechanical ventilation at the age of 48 years. Her eyeball movement was not limited and without nystagmus. At the age of 52 years, she was completely bedridden and moved only her fingers with very limited ranges, communicating with a computer-based tool. Cerebellar ataxia, parkinsonism, sensory disturbance, or autonomic failure was not apparent during the disease course. The clinical diagnosis, according to the El Escorial diagnostic criteria, fulfilled the criteria for definite ALS.
Patient 2
A 68-year-old man with 170 repeats noticed weakness of the neck. Two months later, he visited our hospital at the age of 68 years. He had no previous or family history of neurologic diseases. Weight loss (10 kg in 2 months) was reported. The first neurologic examination revealed very mild weakness of neck flexion and flexion of the bilateral proximal upper limbs. No nystagmus was noted. A spirogram revealed that % vital capacity was 81.9%. A needle electromyogram revealed neurogenic changes in the cranial nerve territory and upper and lower limbs, with fibrillation in the right tibialis anterior muscle and fasciculation in the left trapezius and right deltoid muscle. One month later, dysphagia, dysarthria, and respiratory failure became apparent and progressed very rapidly. Two months later, atrophy of the face and limb muscles became apparent. DTRs were normal in the atrophied upper limbs, but increased in the lower limbs, with bilateral extensor planter response. The clinical diagnosis, according to the El Escorial diagnostic criteria, fulfilled the criteria for probable ALS. Videofluorography showed moderate involvement of oral and pharyngeal phases, with penetration into the larynx. The patient then received a nasal ventilator and underwent PEG about 4 months after onset. Although weakness in the 4 limbs remained mild (manual muscle testing 4/5), dysarthria and respiratory failure progressed with reduced vital capacity (49.1%). He refused tracheostomy and died of respiratory failure at age 68 years, about 5 months after onset. An autopsy was not performed.
Patient 3
A 76-year-old woman with 91 repeats noticed head dropping when walking at the age of 66 years. Ten months later (at the age of 67 years), she experienced dysphagia and weight loss, dropping from 78 to 50 kg in half a year. She had a history of complete resection of breast cancer at age 67 years. She had no previous or family history of neurologic diseases. Neurologic examinations revealed severe dysphagia with aspiration, dysarthria, facial weakness, and severe tongue atrophy. No nystagmus was noted. Mild weakness was observed in the neck and the right upper limb. DTRs were normal in the bilateral upper limbs and in the right patella, with decreases in the left patella tendon and bilateral Achilles tendon reflexes. Bilateral extensor planter responses were positive. A needle electromyogram revealed neurogenic changes in all the examined muscles, including the right trapezius, biceps brachii, the first dorsal interosseous, rectus femoris, tibialis anterior, and tongue muscles. Fibrillation potentials were present in the tibialis anterior and the first dorsal interosseous. At the age of 68 years, she began to receive noninvasive positive pressure ventilation because of reduced vital capacity (66.2%) and underwent PEG for tube feeding. Cerebellar ataxia, parkinsonism, sensory disturbance, and autonomic failure were not apparent. MRI revealed no apparent cerebral atrophy, but with faint T2-hyperintense signals in the putaminal rim (not shown). Weakness of the upper limbs progressed slowly. She showed right vocal cord paralysis at the age of 69 years and then left paralysis at age 70 years, while the airway remained open. She had aspiration pneumonia and underwent tracheostomy at the age of 71 years, 5 years after onset, but she became independent of mechanical ventilation during the day after recovery from pneumonia. She used a ventilator only during the night for safety. The diagnostic criteria fulfilled the criteria for possible ALS, but with atypically slow progression. MRI at the age of 75 years is shown in figure 2C. She lived alone, dealt with tube feeding and nighttime ventilation by herself, and walked independently at the age of 76 years, more than 10 years after onset.
Discussion
This study found that 3 sporadic patients who fulfilled the diagnosis of ALS had mutations in the ATXN8OS gene. Haplotype analyses suggested that the patients seemed unrelated. By contrast, no patient had a mutation in the C9ORF72 or NOP56 gene, which was consistent with the results of other studies conducted in Japan.11,19 The ATXN8OS gene has been viewed as causative for pure cerebellar ataxia and parkinsonian disorders, both of which do not always affect motor neurons as in ALS. However, a systematic review reported that about half of patients with SCA8 had hyperreflexia,20 an upper motor neuron sign. The phenotype of the reported patient from Korea mimicked ALS with involvement of both upper and lower motor neurons, although several atypical findings, such as mild atrophy of the cerebellum on MRI, vertical nystagmus, unsteady gait, and a family history of SCA8, precluded a diagnosis of ALS.9 A more evident involvement of lower motor neurons in SCA8 was described in an autopsied patient with atypical SCA8 and subsequent motor neuron disease. Neuronal loss and gliosis were found in the cranial motor nucleus with basophilic inclusions immunoreactive for TDP43, a protein causative of familial ALS.10 These findings suggest that SCA8 occasionally affects upper and lower motor neurons, a prerequisite for a diagnosis of ALS.
We found that the predominant clinical symptoms of patients with ATXN8OS-related ALS were neck weakness and bulbar signs including dysarthria and dysphagia. Neck weakness was a rare initial symptom in our cohort (only 3/102) and in a reported study (2%).21 Of interest, neck weakness during the disease process was a sign closely associated with bulbar symptoms.21 Bulbar-onset ALS, or ALS with bulbar sign within 1 year of onset, has been associated with poor prognosis.4 As shown in table 1, 3 of the 4 patients indeed showed rapid progression. However, we found that 1 patient with a relatively small repeat size had severe and progressive bulbar symptoms at the beginning but remained ambulant and independent of mechanical ventilation during the day, more than 10 years after onset. Thus, clinical severity may vary and might be associated with repeat sizes, which should be confirmed by future studies.
Two of our patients had apparently nonsymptomatic MRI abnormalities: patient 1 had T2-hyperintense lesions in the cerebral white matter, and patient 3 had T2-hyperintense lesions in the putaminal rim. The white matter lesions observed in patient 1 at the age of 47 years were a finding relatively rare in individuals before the age of 50 years. However, a reported 22-year-old patient who had SCA8 with 102 CTA/CTG repeats had T2-hyperintense lesions in the periventricular white matter.22 Of interest, white matter lesions have been reported in some patients with other noncoding repeat diseases such as C9ORF72-related ALS23 and myotonic dystrophy type 1.24 These might suggest the importance of noncoding repeat expansions in glial degeneration. The T2-hyperintense lesions in the putaminal rim in patient 3 (without parkinsonism) was a finding often seen in multiple system atrophy of parkinsonian type, but also found in some control individuals.25 Such an MRI finding has not been described in SCA8. Although additional accumulation of patients will be needed to draw firm conclusions, the MRI abnormalities might be a clue to the associations of the ATXN8OS gene, as with nonsymptomatic atrophy in the hippocampus in VCP-related ALS.14,26
The observed finding that mutations in a single gene are associated with multiple phenotypes, including ALS and spinocerebellar ataxia, has been described in several other genes. In the ATXN1 gene for spinocerebellar ataxia type 1 or the ATXN2 gene for spinocerebellar ataxia type 2 (SCA2), intermediate triplet repeat expansions or sometimes full expansions encoding polyglutamine proteins have been found in patients with ALS.27–29 In addition, 2 independent reports described that ataxia and ALS phenotypes were present within the same family with ATXN2 mutations.29,30 The molecular mechanisms underlying ATXN1-related or ATXN2-related disorders may include altered clearance of cytosolic misfolded proteins, a mechanism shared with that for general ALS.27,31 Very recent studies demonstrated that suppression of ATXN2 expression by antisense oligonucleotides improved motor functions in SCA2 animal models32 and prolonged the lifespan of TDP43-related ALS models.33 These findings support the idea that 2 different phenotypes may be at least partly related to a similar pathomechanism, which can be a therapeutic target.
Because noncoding repeat expansions may have some common pathomechanisms, including formation of RNA foci and repeat-associated non-ATG translation,34 a therapeutic approach to C9ORF72-related ALS may also be applicable to ATXN8OS-related ALS. In C9ORF72-related ALS, suppression of abnormal transcription by antisense oligonucleotides is an ongoing clinical project.35 A similar method was recently reported in SCA36.36 In another study, suppression of toxicity in an abnormal ATXN8OS transcript by a certain protein in vivo exerted a therapeutic effect.37 Although the ATXN8OS gene, reported to have bidirectional transcripts, may have a more complex pathomechanism,7 suppression of at least 1 pathologic pathway might help slow the disease process.
In this study, we found that 3 patients with ALS had mutations in the ATXN8OS gene. The coincidental occurrence of SCA8 and ALS is possible because mutations in the aforementioned gene have been infrequently found in controls.5,6,38 However, the repeat sizes found in patients with ALS, 91-223 repeats, were not found in the reported control alleles in Japan (n = 654).38 The relatively low prevalence of SCA8 (0.7/100,000) and ALS (5/100,000) in Japan suggests that their coincidental coexistence is unlikely to happen in the 3 presumably unrelated patients. Three percent seems small, but is still relatively large for Japan, because the most commonly mutated genes, including the SOD1 and SQSTM1 genes, each account for 2%–3% of sporadic patients.13,19 Because ATXN8OS mutations were not found in general ALS in US,39 analysis of certain subtypes, such as cervical or bulbar-onset types, might be needed. Our results may improve the understanding of the pathomechanism of ALS and extend the clinical phenotype of SCA8.
Acknowledgment
The authors thank Dr. Abe of Department of Neurology, Okayama University for technical assistance for the analysis of the NOP56 gene.
Glossary
- ALS
amyotrophic lateral sclerosis
- DTR
deep tendon reflex
- PEG
percutaneous endoscopic gastrostomy
- SCA
spinocerebellar ataxia
Author contributions
M. Hirano and K. Saigoh contributed to acquisition of the data, analysis or interpretation of the data, and drafting or revising the manuscript for intellectual content. M. Samukawa and C. Isono contributed to design or conceptualization of the study, acquisition of the data, and analysis or interpretation of the data. Y. Nakamura and S. Kusunoki contributed to design or conceptualization of the study, analysis or interpretation of the data, and drafting or revising the manuscript for intellectual content.
Study funding
This study was partly supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to MH16K09705).
Disclosure
M. Hirano has received funding for speaker honoraria from Daiichi Sankyo, Eisai, Otsuka Pharmaceutical, Sanofi, and Shire; has received Grants-in-Aid from Japan's Ministry of Education, Culture, Sports, Science and Technology; and has received research support from Kindai University and the Japan ALS Association. M. Samukawa, C. Isono, K. Saigoh, and Y. Nakamura report no disclosures. S. Kusunoki has received funding for speaker honoraria from Teijin Pharma, Nihon Pharmaceuticals, Japan Blood Products Organization, Biogen, Novartis, Dainippon Sumitomo Pharma, Kyowa Kirin, Ono Pharmaceutical, Pfizer, Alexion, and Chugai; serves on the editorial boards of the Journal of Neuroimmunology, Clinical and Experimental Neurology, and the Neurology and Clinical Neuroscience; and has received research support from Novartis, Dainippon Sumitomo Pharma, Sanofi, Japan Blood Products Organization, Otsuka, Kyowa Kirin, Daiichi Sankyo, Eisai, Takeda, and Nihon Pharmaceuticals, as well as from Japan's Ministry of Education, Culture, Sports, Science and Technology and their Agency for Medical Research and Development. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Okazaki M, Suzuki H, Takahashi Y, et al. Novel mutation in the SOD1 gene in a patient with early-onset, rapidly progressive amyotrophic lateral sclerosis. Neurol Clin Neurosci 2017;5:189–191. [Google Scholar]
- 2.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishiura H, Takahashi Y, Mitsui J, et al. C9ORF72 repeat expansion in amyotrophic lateral sclerosis in the Kii peninsula of Japan. Arch Neurol 2012;69:1154–1158. [DOI] [PubMed] [Google Scholar]
- 5.Koob MD, Moseley ML, Schut LJ, et al. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat Genet 1999;21:379–384. [DOI] [PubMed] [Google Scholar]
- 6.Day JW, Schut LJ, Moseley ML, Durand AC, Ranum LP. Spinocerebellar ataxia type 8: clinical features in a large family. Neurology 2000;55:649–657. [DOI] [PubMed] [Google Scholar]
- 7.Moseley ML, Zu T, Ikeda Y, et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet 2006;38:758–769. [DOI] [PubMed] [Google Scholar]
- 8.Baba Y, Uitti RJ, Farrer MJ, Wszolek ZK. Sporadic SCA8 mutation resembling corticobasal degeneration. Parkinsonism Relat Disord 2005;11:147–150. [DOI] [PubMed] [Google Scholar]
- 9.Kim JS, Son TO, Youn J, Ki CS, Cho JW. Non-ataxic phenotypes of SCA8 mimicking amyotrophic lateral sclerosis and Parkinson disease. J Clin Neurol 2013;9:274–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yokoyama T, Ishiyama M, Hasegawa K, Uchihara T, Yagishita S. Novel neuronal cytoplasmic inclusions in a patient carrying SCA8 expansion mutation. Neuropathology 2014;34:27–31. [DOI] [PubMed] [Google Scholar]
- 11.Kobayashi H, Abe K, Matsuura T, et al. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet 2011;89:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dolzhenko E, van Vugt J, Shaw RJ, et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res 2017;27:1895–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirano M, Nakamura Y, Saigoh K, et al. Mutations in the gene encoding p62 in Japanese patients with amyotrophic lateral sclerosis. Neurology 2013;80:458–463. [DOI] [PubMed] [Google Scholar]
- 14.Hirano M, Nakamura Y, Saigoh K, et al. VCP gene analyses in Japanese patients with sporadic amyotrophic lateral sclerosis identify a new mutation. Neurobiol Aging 2015;36:e1601–e1606. [DOI] [PubMed] [Google Scholar]
- 15.Isono C, Hirano M, Sakamoto H, Ueno S, Kusunoki S, Nakamura Y. Differential progression of dysphagia in heredity and sporadic ataxias involving multiple systems. Eur Neurol 2015;74:237–242. [DOI] [PubMed] [Google Scholar]
- 16.Ikeda Y, Dalton JC, Moseley ML, et al. Spinocerebellar ataxia type 8: molecular genetic comparisons and haplotype analysis of 37 families with ataxia. Am J Hum Genet 2004;75:3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ayhan F, Ikeda Y, Dalton JC, Day JW, Ranum LPW. Spinocerebellar ataxia type 8. In: Pagon RA, Adam MP, Ardinger HH, editors. GeneReviews®. Seattle, WA: University of Washington; 1993. [Google Scholar]
- 18.Hirano M, Yamagishi Y, Yanagimoto S, Saigoh K, Nakamura Y, Kusunoki S. Time course of radiological imaging and variable interindividual symptoms in amyotrophic lateral sclerosis and frontotemporal dementia associated with p.Arg487His mutation in the VCP gene. Eur Neurol 2017;78:78–83. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura R, Sone J, Atsuta N, et al. Next-generation sequencing of 28 ALS-related genes in a Japanese ALS cohort. Neurobiol Aging 2016;39:219.e1–219.e8. [DOI] [PubMed] [Google Scholar]
- 20.Zeman A, Stone J, Porteous M, Burns E, Barron L, Warner J. Spinocerebellar ataxia type 8 in Scotland: genetic and clinical features in seven unrelated cases and a review of published reports. J Neurol Neurosurg Psychiatry 2004;75:459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Konno T, Shiga A, Tsujino A, et al. Japanese amyotrophic lateral sclerosis patients with GGGGCC hexanucleotide repeat expansion in C9ORF72. J Neurol Neurosurg Psychiatry 2013;84:398–401. [DOI] [PubMed] [Google Scholar]
- 22.Kumar N, Miller GM. White matter hyperintense lesions in genetically proven spinocerebellar ataxia 8. Clin Neurol Neurosurg 2008;110:65–68. [DOI] [PubMed] [Google Scholar]
- 23.Oliveira Santos M, Caldeira I, Gromicho M, Pronto-Laborinho A, de Carvalho M. Brain white matter demyelinating lesions and amyotrophic lateral sclerosis in a patient with C9orf72 hexanucleotide repeat expansion. Mult Scler Relat Disord 2017;17:1–4. [DOI] [PubMed] [Google Scholar]
- 24.Hashimoto T, Tayama M, Miyazaki M, et al. Neuroimaging study of myotonic dystrophy: I. Magnetic resonance imaging of the brain. Brain Dev 1995;17:24–27. [DOI] [PubMed] [Google Scholar]
- 25.Tha KK, Terae S, Tsukahara A, et al. Hyperintense putaminal rim at 1.5 T: prevalence in normal subjects and distinguishing features from multiple system atrophy. BMC Neurol 2012;12:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirano M, Matsumura R, Nakamura Y, et al. Unexpectedly mild phenotype in an ataxic family with a two-base deletion in the APTX gene. J Neurol Sci 2017;378:75–79. [DOI] [PubMed] [Google Scholar]
- 27.Elden AC, Kim HJ, Hart MP, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010;466:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conforti FL, Spataro R, Sproviero W, et al. Ataxin-1 and ataxin-2 intermediate-length PolyQ expansions in amyotrophic lateral sclerosis. Neurology 2012;79:2315–2320. [DOI] [PubMed] [Google Scholar]
- 29.Tazen S, Figueroa K, Kwan JY, et al. Amyotrophic lateral sclerosis and spinocerebellar ataxia type 2 in a family with full CAG repeat expansions of ATXN2. JAMA Neurol 2013;70:1302–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baumer D, East SZ, Tseu B, et al. FTLD-ALS of TDP-43 type and SCA2 in a family with a full ataxin-2 polyglutamine expansion. Acta Neuropathol 2014;128:597–604. [DOI] [PubMed] [Google Scholar]
- 31.Park SH, Kukushkin Y, Gupta R, et al. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell 2013;154:134–145. [DOI] [PubMed] [Google Scholar]
- 32.Scoles DR, Meera P, Schneider MD, et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 2017;544:362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Becker LA, Huang B, Bieri G, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017;544:367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kearse MG, Todd PK. Repeat-associated non-AUG translation and its impact in neurodegenerative disease. Neurotherapeutics 2014;11:721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sareen D, O'Rourke JG, Meera P, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 2013;5:208ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsuzono K, Imamura K, Murakami N, et al. Antisense oligonucleotides reduce RNA foci in spinocerebellar ataxia 36 patient iPSCs. Mol Ther Nucleic Acids 2017;8:211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tripathi BK, Surabhi S, Bhaskar PK, Mukherjee A, Mutsuddi M. The RNA binding KH domain of Spoonbill depletes pathogenic non-coding spinocerebellar ataxia 8 transcripts and suppresses neurodegeneration in Drosophila. Biochim Biophys Acta 2016;1862:1732–1741. [DOI] [PubMed] [Google Scholar]
- 38.Izumi Y, Maruyama H, Oda M, et al. SCA8 repeat expansion: large CTA/CTG repeat alleles are more common in ataxic patients, including those with SCA6. Am J Hum Genet 2003;72:704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Figley MD, Thomas A, Gitler AD. Evaluating noncoding nucleotide repeat expansions in amyotrophic lateral sclerosis. Neurobiol Aging 2014;35:e931–e934. [DOI] [PMC free article] [PubMed] [Google Scholar]