

Abstract
Resveratrol, a dietary product present in grapes, vegetables and berries, regulates several signaling pathways that control cell division, cell growth, apoptosis and metastasis. Malignant melanoma proliferates more readily in comparison with any other types of skin cancer. In the present study, the anti-cancer effect of resveratrol on melanoma cell proliferation was evaluated. Treating A375SM cells with resveratrol resulted in a decrease in cell growth. The alteration in the levels of cell cycle-associated proteins was also examined by western blot analysis. Treatment with resveratrol was observed to increase the gene expression levels of p21 and p27, as well as decrease the gene expression of cyclin B. In addition, the generation of reactive oxygen species (ROS) and endoplasmic reticulum (ER) stress were confirmed at the cellular and protein levels using a 2′,7′-dichlorofluorescein diacetate assay, TUNEL assay and western blot analysis. Resveratrol induced the ROS-p38-p53 pathway by increasing the gene expression of phosphorylated p38 mitogen-activated protein kinase, while it induced the p53 and ER stress pathway by increasing the gene expression levels of phosphorylated eukaryotic initiation factor 2α and C/EBP homologous protein. The enhanced ROS-p38-p53 and ER stress pathways promoted apoptosis by downregulating B-cell lymphoma-2 (Bcl-2) expression and upregulating Bcl-2-associated X protein expression. In conclusion, resveratrol appears to be an inducer of ROS generation and ER stress, and may be responsible for growth inhibition and cell cycle arrest of A375SM melanoma cells.
Keywords: resveratrol, melanoma cells, cell cycle arrest, reactive oxygen species, endoplasmic reticulum stress, apoptosis
Introduction
Malignant melanoma is known for its exceptionally high mortality rate among all types of skin cancer. Malignant melanoma occurs due to an intricate interaction between endogenous and exogenous factors. In total, >65% of malignant melanoma cases are influenced by sun exposure and ~12% of cases are caused by genetic factors, such as mutations of critical genes (including cyclin-dependent kinase inhibitor 2A, melanocortin 1 receptor and DNA repair genes) (1). A great number of melanoma patients notably acquired driver oncogenic mutations in genes that encode proteins associated with growth factor receptor signaling pathways, such as the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase and phosphoinositide 3-kinase/protein kinase B pathways (2,3). Treatments for advanced melanoma have been investigated for the last decade. Surgical removal is the primary treatment for melanoma due to its noticeable appearance on the skin, and early removal of melanoma increases the chance of preventing metastasis. Chemotherapy, immunotherapy and molecular-targeted therapies are also well known treatment methods for melanoma, which have increased the survival rate of melanoma patients; however, similar to other types of cancer, resistance to these treatments has been rising (4).
Resveratrol, a trans-3,4′,5-trihydroxystilbene, is a dietary phenol present in numerous plants and dietary supplements, and is commonly found in grapes. Resveratrol has been reported to have an impact on every stage of carcinogenesis. In addition, it serves as a chemopreventive agent due to its potential to mediate signaling pathways that manage cell division, cell growth, apoptosis, angiogenesis, metastasis and inflammation (5,6). There have been numerous studies on resveratrol as an ideal anticancer molecule, as it provokes a cytotoxic effect on cancer cells, while it does not affect nonmalignant cells (7–9). The main resveratrol-mediated chemotherapeutic mechanism is apoptosis associated with the activation of p53, a tumor suppressor, and induced activation of death receptor Fas/CD85/APO-1 in diverse cancer cells (10). Resveratrol is also know to exhibit a preventive role in heart disease, since it prevents coagulation and platelet aggregation, modifies eicosanoid synthesis and mediates lipoprotein metabolism (11).
Endoplasmic reticulum (ER) is the primary site for protein folding and transporting, and maintenance of cellular functions. ER stress occurs when cell homeostasis collapses. The unfolded protein response (UPR) is a response to ER stress that involves several different stress signaling and inflammatory pathways (12). Failure to resolve ER stress leads to apoptosis. One ER stress-induced apoptosis pathway involves the activation of C/EBP homologous protein (CHOP), a transcription factor, by ER stress and the promotion of apoptosis by downregulation of B-cell lymphoma 2 (Bcl-2) (13,14). ER stress also stimulates intracellular reactive oxygen species (ROS) production, and these species then reinforce ER stress-mediated apoptosis (15). Oxidative stress is caused by excess production of ROS and leads to cell death. ROS consist of cytotoxic molecules, including superoxide (O2−), hydrogen peroxide (H2O2), singlet oxygen (1/2 O2) and the hydroxyl radical (•OH). These ROS can damage all cellular components, including proteins, lipids and DNA, and further cause disruptions in normal cellular signaling (16). Oxidative and antioxidative stress also occurs when the balance between pro-oxidants and antioxidants collapses (17,18). While excessive ROS generation induces oxidative stress, excessive reduction in the oxidative stress level by antioxidants induces antioxidative stress, which also has harmful effects on human health.
In the present study, it was attempted to determine whether resveratrol induces ER stress-mediated apoptosis to suppress cell growth. In addition, its effect on the intracellular ROS level in the A375SM cell line was examined.
Materials and methods
Reagents and chemicals
Resveratrol was purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany) and was dissolved in dimethyl sulfoxide (DMSO; Junsei Chemical Co., Ltd., Tokyo, Japan).
Cell culture and media
A malignant melanoma cell line, A375SM, was obtained from the Korean Cell Line Bank (Seoul, Korea) and cultured in Dulbecco's modified Eagle's medium (DMEM; HyClone; GE Healthcare Life Sciences, Logan, UT, USA) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS; Rocky Mountain Biologicals, Inc., Missoula, MT, USA), 10 U/ml penicillin and 100 µg/ml streptomycin (Cellgro Mediatech; Corning Incorporated, Corning, NY, USA), and 10 mM HEPES (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37°C in 5% CO2 and 95% air in a humidified cell incubator. Melanoma cells were trypsinized using 0.05% trypsin/0.02% EDTA (GE Healthcare Life Sciences).
Cell viability assay
The cell viability test was conducted for 6 days. On the first day, A375SM cells (3×103 cells/well) were seeded in 96-well plates (SPL Life Science, Seoul, Korea) and cultured in DMEM supplemented with FBS, 1% penicillin and streptomycin, and 1% HEPES at 37°C in a humidified atmosphere with 5% CO2. The following day, A375SM cells were treated with concentrations of resveratrol ranging between 10−2 and 10 µM in DMEM supplemented with 0.1% DMSO for 4 days. On the third day of this treatment, the media were changed to a fresh version of the same media. The day after the four days of treatments were completed (day 6), an EZ-cytox Cell Viability Assay kit (DoGen Bio Co., Ltd., Seoul, Korea) was used to verify the cell viability in each well. An ELISA plate reader (VERSAmax; Molecular Devices, LLC, Sunnyvale, CA, USA) was used to measure the absorbance at 480 nm. Resveratrol (1 µM) was expected to produce a sufficient effect on ER-stress and ROS production and was applied for subsequent analysis; a dose-dependent effect was also investigated by using 1 and 10 µM resveratrol as 10 µM resveratrol did not induce cell damage >45%.
Western blot assay
The A375SM cell line was incubated in medium containing 0.1% DMSO and resveratrol (1 and 10 µM) for 48 h. Next, radioimmunoprecipitation assay buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% Triton X-100 (Sigma-Aldrich; Merck KGaA), 0.1% sodium dodecyl sulfate and 0.5% deoxycholic acid (Sigma-Aldrich; Merck KGaA) mixed with protease and phosphatase inhibitors was used for harvesting proteins for western blot analysis. The extracted proteins were incubated overnight at 4°C and then centrifuged at 18,430 × g for 1 h at 4°C. The selected protein concentration was obtained using a mixture of bicinchoninic acid and copper (II) sulfate (both from Sigma-Aldrich; Merck KGaA). Subsequently, 50 µg total protein was used for 10% SDS-polyacrylamide gel electrophoresis, and then the separated proteins were transferred to a polyvinylidene fluoride membrane (Bio-Rad Laboratories, Inc., Hercules, CA USA). The membrane was subsequently incubated overnight at 4°C with the following monoclonal primary antibodies: mouse monoclonal anti-p21 (1:1,000; Abcam, Cambridge, MA, USA; ab188224), anti-cyclin B (1:1,000; Abcam; ab2949), anti-cyclin E (1:1,000; Abcam; ab98952), anti-p53 (1:1,000; Santa Cruz Biotechnology, Inc., Dallas, TX, USA; sc-126), anti-phosphorylated (p)-p38 MAPK (1:1,000; Abcam; ab4822), anti-CHOP (1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA; 2895), anti-Bcl-2-associated X protein (Bax; 1:1,000; Santa Cruz Biotechnology, Inc.; sc-7480), anti-Bcl-2 (1:200; Cell Signaling Technology, Inc.; 15071), anti-GAPDH (1:12,000; Abcam; ab130099), rabbit monoclonal anti-nuclear factor erythroid 2-related factor 2 (Nrf2; 1:2,000; Abcam; ab62352), anti-p27 (1:1,000; Abcam; ab32034) and anti-p-eukaryotic initiation factor 2α (eIF2α; 1:1,000; Cell Signaling Technology, Inc.; 9721). The primary antibody binding was observed by incubating for 1 h at 4°C with a secondary antibody, including anti-mouse IgG (1:3,000; Thermo Fisher Scientific, Inc.; 170-6516) or anti-rabbit IgG (1:3,000; Thermo Fisher Scientific, Inc.; 170-6515). A West-Q Chemiluminescent Substrate Plus kit (GenDEPOT, Inc., Barker, TX, USA) was utilized for protein detection. The band densities on the membrane were utilized to quantify each protein expression through CS Analyzer 4 software ver. 2.1.2 (Atto Corporation, Tokyo, Japan). Each experiment was conducted at least three times, and the expression levels of the aforementioned proteins were normalized to that of GADPH protein.
Measurement of ROS generation
An assay using 2′,7′-dichlorofluorescein diacetate (DCF-DA) was conducted to measure the cellular levels of ROS in the A375SM cell line. Briefly, A375SM cells were seeded at a density of 3×105 cells per well in a 6-well plate with the culture medium. After 48 h of incubation, the culture medium was replaced with medium containing either 0.1% DMSO or resveratrol (1 and 10 µM). As a positive control for ROS production, 2 ml of 3% H2O2 solution was added to the A375SM cell line for 15 min. A new medium containing DCF-DA solution in DMEM was substituted for 30 min. Subsequently, each well was washed with PBS, and the A375SM cell line was visualized using a fluorescence microscope (IX-73 inverted microscope; Olympus Corporation, Tokyo, Japan). The amount of ROS formed by the resveratrol treatment was quantified using CellSens Dimension software ver. 1.13 (Olympus Corporation).
TUNEL assay
Apoptotic melanoma cells were detected using a DeadEnd™ fluorometric TUNEL assay kit (Promega Corporation, Madison, WI, USA) as described in the manufacturer's protocol. Briefly, melanoma cells were seeded at a density of 3×105 cells per well in a 6-well plate. After 48 h of incubation with medium containing 0.1% DMSO and resveratrol (1 and 10 µM), cells were fixed with 3.7% formaldehyde for 25 min and incubated with recombinant terminal deoxynucleotidyl transferase incubation buffer for 1 h at 37°C. The cells were then stained with DAPI (Invitrogen; Thermo Fisher Scientific, Inc.), and both apoptotic and DAPI-stained cells were visualized using a fluorescence microscope (IX-73 inverted microscope, Olympus Corporation). ImageJ software ver. 1.49 (National Institutes of Health, Bethesda, MD, USA) was used for merging the images of DAPI and TUNEL staining. The number of apoptotic A375SM cells produced by resveratrol treatment was quantified using the CellSens Dimension software ver. 1.13 (Olympus Corporation).
Statistical analysis
All experiments were performed a minimum of three times, and the resulting data were analyzed with the GraphPad Prism software ver. 5 (GraphPad Software, Inc., San Diego, CA, USA). All data are presented as the mean ± standard deviation, and were analyzed using one-way analysis of variance, followed by Dunnett's test. Differences with P-values of <0.05 were recognized as statistically significant.
Results
Cell proliferation of A375SM was repressed by resveratrol
To evaluate the effects of resveratrol on A375SM cell proliferation, the cells were treated with 0.1% DMSO (control) or resveratrol (10−2, 10−1, 1, 5 and 10 µM) for 4 days. On day 6 of the treatment, EZ-cytox was added to measure the cell viability. Resveratrol significantly suppressed the cell viability of the melanoma cell line in a dose-dependent manner (Fig. 1). Based on the results of the cell viability assay, the resveratrol concentrations of 1 and 10 µM were selected for further experiments.
Figure 1.
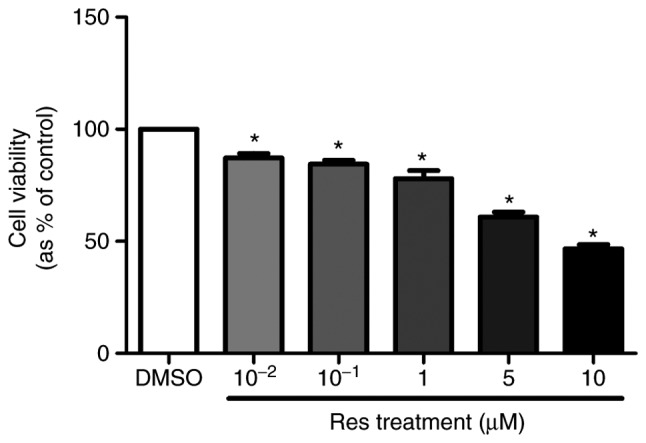
Effect of resveratrol on the viability of the A375SM cell line. A375SM cells (3×103 cells/well) were seeded into 96-well plates. The following day, the cells were treated with 0.1% DMSO (control) or resveratrol (between 10−2 and 10 µM) for 5 days. EZ-cytox was then added on the last day of the treatment to measure the cell viability, with 0.1% DMSO used as a control. Data are represented as the mean ± standard deviation. *P<0.05 vs. control (0.1% DMSO). Res, resveratrol; DMSO, dimethyl sulfoxide.
Resveratrol induced cell cycle arrest of melanoma cell line
A western blot assay was performed on the A375SM melanoma cell line to examine whether resveratrol influenced the protein expression of genes controlling the cell cycle progression. Melanoma cells were treated with the control or resveratrol (1 and 10 µM) for 48 h, and then the protein levels of cell cycle-associated genes, including p21, p27, cyclin E and cyclin B, were quantified. It was observed that the expression levels of cyclin-dependent kinase inhibitors p21 and p27 were significantly increased in a dose-dependent manner (Fig. 2). By contrast, the expression of cyclin B was markedly decreased, whereas the expression of cyclin E did not exhibit any significant difference among melanoma cells that were treated with the control and resveratrol (1 and 10 µM), as shown Fig. 2B. According to previous studies, upregulation of cyclin-dependent kinase inhibitors, p21 and p27 arrests the cell cycle at the G2/M phase by deregulating cyclin B1 levels, inducing G2 arrest to hinder the replication of damaged DNA (19,20). These results indicate that resveratrol may activate p21 and p27 to suppress the G2/M phase of the cell cycle in A375SM melanoma cells.
Figure 2.
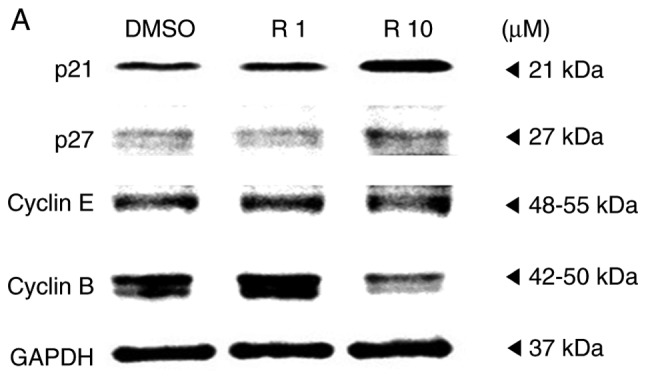

Resveratrol altered the expression levels of cell cycle-associated proteins in A375SM cells treated with DMSO (control) or resveratrol (1.0 and 10 µM) for 48 h. (A) The expression levels of p21, p27, cyclin E and cyclin B were measured using a western blot assay following protein extraction. (B) Each band was quantified and normalized by GAPDH expression, with 0.1% DMSO used as a control. Data of three single experiments are represented as the mean ± standard deviation. *P<0.05 vs. control (0.1% DMSO). Res, resveratrol; DMSO, dimethyl sulfoxide.
Resveratrol elevated ROS generation and ER stress of melanoma cell line
The study further evaluated the ROS generation and ER stress on the A375SM melanoma cell line when exposed to resveratrol. Cellular ROS production in the A375SM cell line exposed to 1 or 10 µM resveratrol for 48 h was measured using a DCF-DA assay, using hydrogen peroxide as a positive control. Fig. 3 displays the induced ROS generation on the A375SM cell line, and the DCF positive cells were significantly increased in a dose-dependent manner. Western blot analysis was also conducted to verify the altered expression rates of the ROS and ER stress-associated proteins Nrf2, p-eIF2α and CHOP as a result of resveratrol treatment, compared with the control. The expression of the anti-oxidant factor Nrf2 was significantly decreased in a dose-dependent manner in resveratrol-treated cells compared with that in the control (Fig. 4), which likely resulted from the increase in ROS production and ER stress. However, treatment with 10 µM resveratrol significantly increased the expression levels of p-eIF2α and CHOP, which are ER stress-associated apoptosis markers, as demonstrated in Fig. 4. These results indicated that resveratrol may induce ROS generation and ER stress to hinder the anti-oxidative effects of resveratrol and enhance the apoptosis of melanoma cells.
Figure 3.

ROS generation in melanoma cells was increased due to resveratrol exposure. (A) Intracellular ROS production in the A375SM cell line was measured using a DCF-DA assay. A375SM cells were treated with DMSO (control) or resveratrol (1.0 and 10 µM) for 48 h. Cellular ROS production was observed using a fluorescence microscope. (B) The amount of ROS formed by resveratrol treatment was quantified using CellSens Dimension software. Data of three single experiments are represented as the mean ± standard deviation. *P<0.05 vs. control (0.1% DMSO). Res, resveratrol; DMSO, dimethyl sulfoxide; ROS, reactive oxygen species; DCF-DA, 2′,7′-dichlorofluorescein diacetate.
Figure 4.
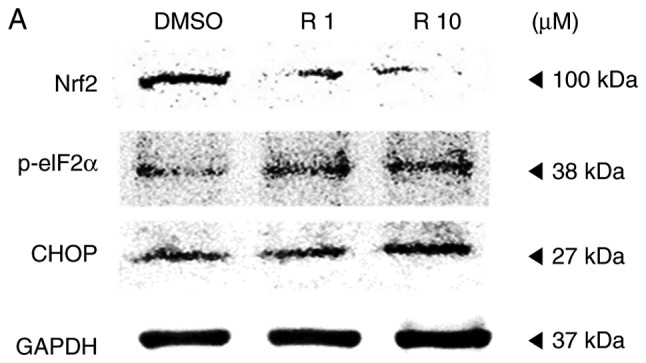

Resveratrol altered the expression levels of reactive oxygen species and endoplasmic reticulum stress-associated proteins in A375SM cells. (A) Cells were treated with DMSO (control) or resveratrol (1.0 and 10 µM) for 48 h. The expression levels of Nrf2, p-eIF2α and CHOP were measured using a western blot assay following protein extraction. (B) Each band was quantified and normalized by GAPDH expression, with 0.1% DMSO used as a control. Data of three single experiments are represented as the mean ± standard deviation. *P<0.05 vs. control (0.1% DMSO). Res, resveratrol; DMSO, dimethyl sulfoxide; Nrf2, nuclear factor erythroid 2-related factor 2; p-eIF2α, phosphorylated eukaryotic initiation factor 2α; CHOP, C/EBP homologous protein.
Resveratrol-induced apoptosis on melanoma cell line
Melanoma cells that underwent apoptosis due to resveratrol treatment were detected using a DeadEnd™ fluorometric TUNEL assay kit, and the protein levels were verified using a western blot assay. A375SM cells were treated with the control or resveratrol (1 and 10 µM) for 48 h. Treatment with resveratrol displayed increased cell death compared with that observed in the control, as shown in Fig. 5. In addition, the higher concentration of resveratrol was correlated with a marked increase in melanoma cell death (Fig. 5) compared with 1 µM resveratrol. The apoptosis-associated proteins p38, p53, Bax and Bcl-2 were also observed using a western blot assay to support the TUNEL assay findings. The expression levels of p53, a tumor suppressor and mediator of programmed cell death, and of p-p38, which is upstream of and targets p53, were increased in resveratrol-treated melanoma cells, as shown in Fig. 6. When cells are under stress, p53 interacts with the anti-apoptotic members Bcl-2 and Bcl-xL and counterbalances their expression. This counterbalance activates apoptosis through the induction of mitochondrial outer membrane permeabilization factors, such as Bax, Bak and BH3-only (21). In the present study, the expression of the anti-apoptotic protein Bcl-2 was suppressed, likely due to the activation of p53. By contrast, the expression of the pro-apoptotic protein Bax was increased in a dose-dependent manner (Fig. 6). These results imply that the apoptosis that occurred in melanoma cells treated with resveratrol was influenced by the phosphorylation of p38 and activation of p53, which inhibited the expression of anti-apoptotic factors and activated pro-apoptotic factors.
Figure 5.

Resveratrol induced apoptosis at the cellular level in A375SM cells. (A) Resveratrol induced apoptosis in A375SM cells as determined by a fluorescence microscope. Cells were treated with 1.0 and 10 µM resveratrol, or with the control for 48 h, and then a TUNEL assay was performed. ImageJ software ver. 1.49 was used to merge the images of DAPI and TUNEL. (B) The number of apoptotic A375SM cells produced by resveratrol treatment was quantified using CellSens Dimension software. Data of three single experiments are represented as the mean ± standard deviation. *P<0.05 vs. control (0.1% DMSO). #P<0.05 vs. 1 µM resveratrol. Res, resveratrol; DMSO, dimethyl sulfoxide.
Figure 6.
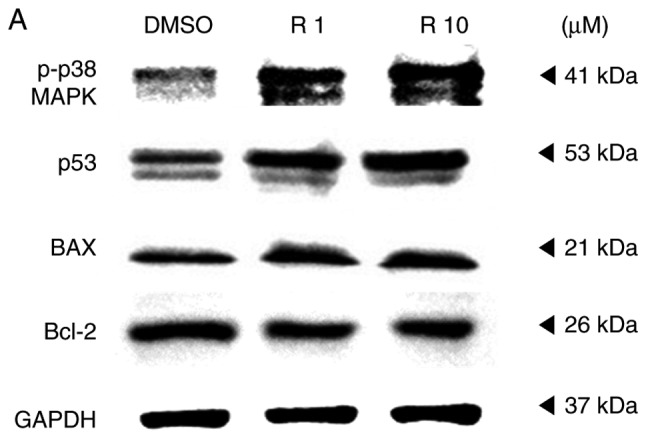

Resveratrol altered the expression of apoptosis-associated proteins in A375SM cells. (A) Cells were treated with DMSO (control) or resveratrol (1.0 and 10 µM) for 48 h, and the expression levels of p-p38 MAPK, p53, Bax and Bcl-2 were measured using a western blot assay following protein extraction. (B) Each band was quantified and normalized by GAPDH expression, with 0.1% DMSO used as a control. Data of three single experiments are represented as the mean ± standard deviation. *P<0.05 vs. control (0.1% DMSO). Res, resveratrol; DMSO, dimethyl sulfoxide; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein; MAPK, mitogen-activated protein kinase.
Discussion
Melanoma diagnosed at an early stage can be effectively treated with surgical removal and radiation therapy (4,22). Although a vast range of treatment strategies are available for melanoma, ranging from chemotherapy to molecular-targeted therapies, treatment resistance is unavoidable in melanoma patients. Therefore, identifying novel methods for the treatment or prevention of melanoma has become a focus point of cancer research.
According to a review published in 2011, resveratrol exhibited a chemopreventive role in various diseases, including cancer (23). Therefore, the current study examined how resveratrol, a compound found in various types of food, influences melanoma at the cellular and protein levels. It was observed that resveratrol inhibited melanoma cell proliferation at a concentration of >10−2 µM. The concentrations 1 and 10 µM were selected to further examine the effect of resveratrol in a dose-dependent manner. Resveratrol was demonstrated to activate the expression of p21 and p27, which promoted cell cycle arrest in melanoma cells. Furthermore, the effects of resveratrol on cyclin B and cyclin E were assessed, both of which are highly expressed in melanoma that exhibits metastatic tendencies (24). Resveratrol suppressed the expression of cyclin B, but did not have a significant effect on the expression of cyclin E.
In the current study, the generation of cellular ROS in melanoma cells was observed using a DCF-DA assay. The density of DCF-positive melanoma cells was increased in a dose-dependent manner, which implies that A375SM cells cultured with resveratrol generated a higher amount of ROS. Resveratrol increased the cytosolic ROS generation by >5-fold (1 µM resveratrol) and 15-fold (10 µM resveratrol) as compared with that in the control. Although the glutathione/glutathione disulfide (GSH/GSSG) ratio was not measured, it can easily be assumed that the increased ROS generation by resveratrol reduced the GSH/GSSG ratio compared with the control, and placed the melanoma cells under oxidative stress.
Although resveratrol is known to have an antioxidant effect, recent studies have demonstrated that resveratrol exhibits both antioxidant and prooxidant properties, depending on its concentration and the cell type (25,26). It has also been proposed that the pro-oxidant action may be an important action mechanism of the anticancer and apoptosis-inducing properties of resveratrol (27). Correspondingly, the present results observed that resveratrol displayed apoptosis-inducing properties in the melanoma cell line, A375SM, by acting as a pro-oxidant that promotes ROS formation.
Nrf2 is a mediator of cellular resistance to oxidants and hyperactivation of the Nrf2 pathway establishes a favorable environment for normal and malignant cells (28). In addition, Nrf2 has been considered to protect the human body against cancer (29–31). However, certain tumor types persistently express Nrf2, which allows the cancer to proliferate and gain resistance to oxidants and anticancer drugs (32). Nrf2 is notable for its role as a regulator of cellular defense mechanisms against oxidative stress; however, recent studies revealed the dual nature of Nrf2 (29,33,34). Though it has a protective role against cancer, constant expression of Nrf2 gave rise to strong resistance of cancer to chemotherapeutic drugs (28). Since resveratrol-treated melanoma cells demonstrated a decrease in Nrf2 expression in the present study, it is hypothesized that chemotherapy-resistant melanoma cells may regain sensitivity to chemotherapy with exposure to resveratrol. Considering that Nrf2 is an anti-oxidant factor, its reduced protein expression by resveratrol also supports the occurrence of resveratrol-induced oxidative stress. In the context of an existing melanoma, constant expression of Nrf2 gives rise to resistance to chemotherapeutic drugs. Therefore, the decrease in Nrf2 expression caused by resveratrol may prevent the development of such resistance and thereby increase the sensitivity of melanoma cells to chemotherapy. Similar to Nrf2, which has the dual effects of protection against cancer and enhancement of chemoresistance, resveratrol also has a dual nature, namely anti-oxidant and pro-oxidant activities. Thereby, it is concluded that resveratrol displayed an anti-melanoma effect through its pro-oxidant activity and reduction of the chemoresistance assigned by Nrf2.
In addition to increasing intracellular ROS levels, resveratrol also enhances ER stress (12). The UPR is modulated by three ER membrane-associated proteins: PKR-like ER kinase (PERK), inositol-requiring enzyme 1, and activating transcription factor-6. Phosphorylation of eIF2α by the PERK kinase modulates its translational response and promotes apoptotic cell death (35–37). In the present study, the expression levels of p-eIF2α and CHOP were significantly increased in A375SM cells treated with a high concentration of resveratrol, thereby promoting programmed cell death.
Subsequent to confirming the impact of resveratrol on the generation of ROS and ER stress, the present study further determined that resveratrol induced the ROS-mediated p38-p53 pathway in melanoma cells and promoted apoptosis mediated by the ROS-p38-p53 and ER stress pathway (38). The density of TUNEL-positive cells was increased in a dose-dependent manner. It was further demonstrated that resveratrol induced the mitochondrial apoptotic pathway in melanoma cells through the ROS-p38-p53 pathway by increasing the protein expression of p-p38 MAPK, and through the p53 and ER stress pathway by increasing the protein expression of p-eIF2α and CHOP. The enhanced ROS-p38-p53 and ER stress pathways promoted apoptosis by downregulating Bcl-2 expression and upregulating Bax expression (13).
In the present study, it was revealed that resveratrol induced oxidative stress in melanoma cancer cells by promoting ROS formation. The ROS-mediated oxidative stress induced by resveratrol led to ER stress and mitochondrial dysfunction, both of which induced the apoptosis of A375SM melanoma cells via different pathways. Although it was not established which is the main cause of the resveratrol-induced melanoma cell toxicity and the resultant cell death, ER stress and mitochondrial dysfunction can be considered as mechanisms of resveratrol in terms of the induction of apoptosis in melanoma cells.
Taken together, these results revealed that resveratrol generated intracellular ROS and ER stress in melanoma cells. As canonical steps, eIF2α was phosphorylated, activating CHOP, which induces an ER stress-mediated apoptosis pathway. Furthermore, elevated ROS production led to the phosphorylation of p38 MAPK and activation of p53. Activated p53 promoted cell cycle arrest by activating p21 and p27, enhancing cell cycle arrest in the G2/M phase by suppressing the expression of cyclin B. The activated p53 and CHOP then accelerated apoptosis by impeding Bcl-2 expression, upregulating the expression of Bax, as shown in Fig. 7. In addition, the decreased expression of Nrf2 caused by resveratrol should be studied in order to determine whether it decreases melanoma resistance to chemotherapeutic agents.
Figure 7.

Summary of the role of resveratrol in activating apoptosis and cell cycle arrest through enhancing ER stress and ROS generation. Resveratrol impeded the growth of A375SM cells via stimulating cell cycle arrest and apoptosis by elevating the levels of p38, p53, and Bax, and decreasing the level of Bcl-2. Resveratrol increased the intracellular ROS production and ER stress-mediated apoptosis (p-eIF2α and CHOP) through deactivation of the anti-oxidant factor Nrf2. Therefore, resveratrol accelerated cell cycle arrest and apoptosis by boosting the ROS production and ER stress. ROS, reactive oxygen species; ER, endoplasmic reticulum; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein; p-eIF2α, phosphorylated eukaryotic initiation factor 2α; Nrf2, nuclear factor erythroid 2-related factor 2; CHOP, C/EBP homologous protein; MAPK, mitogen-activated protein kinase.
Although these outcomes revealed novel insight that may be helpful for melanoma treatment, there was a degree of uncertainty in the present study. For instance, ROS involves a diverse range of cell outcomes, including pyroptosis and apoptosis, which are associated with DNA fragmentation that exhibits positive results in a TUNEL assay. However, p53-mediated cell death usually occurs by apoptosis rather than pyroptosis, as it was observed in the current study. Although pyroptosis was not investigated herein, the lack of a mechanism by which p53-mediated cell death leads to pyroptosis results in the conclusion that resveratrol induced A375SM melanoma cell death via ROS formation and p53-mediated apoptosis. Increased cytosolic ROS generation by resveratrol was also identified through the DCF-DA assay; however, this assay did not demonstrate the exact site of ROS formation in the cell and the types of ROS. For more specific representation of the role of resveratrol in the melanoma microenvironment and its association with ROS (the main cause of resveratrol-induced melanoma cell toxicity and death), follow-up experiments examining pyroptosis, associations with calcium ions and the ratio of GSH/GSSG will be required, along with suitable in viv experiments, prior to the application of resveratrol in clinical studies.
In conclusion, the present study demonstrated that resveratrol impeded the viability of melanoma cells by activating the expression of both p21 and p27, which suppressed the expression of cyclin B and promoted cell cycle arrest. Furthermore, resveratrol increased the generation of cellular ROS and simultaneously induced the ER stress pathway in melanoma cells. These results reveal a potential use for resveratrol in the treatment for melanoma.
Acknowledgments
Not applicable.
Funding
This study was supported by the Global Research and Development Center (GRDC) Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (MEST) of the Republic of Korea (2017K1A4A3014959). In addition, this study was also supported by a NRF grant funded by the MEST of the Republic of Korea (2017R1D1A1A09000663).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
JH, SK and KH made substantial contributions to conception and design, and/or acquisition of data, and/or analysis and interpretation of data. JH, JK and KC participated in drafting the article or revising it critically for important intellectual content. All authors gave final approval of the version to be submitted and any revised version.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Bandarchi B, Ma L, Navab R, Seth A, Rasty G. From melanocyte to metastatic malignant melanoma. Dermatol Res Pract. 2010;2010 doi: 10.1155/2010/583748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogelstein B, Kinzler KW. The genetic evolution of melanoma. N Engl J Med. 2016;374:996. doi: 10.1056/NEJMc1515834. [DOI] [PubMed] [Google Scholar]
- 3.Wellbrock C, Arozarena I. The complexity of the ERK/MAP-kinase pathway and the treatment of melanoma skin cancer. Front Cell Dev Biol. 2016;4:33. doi: 10.3389/fcell.2016.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heo JR, Kim NH, Cho J, Choi KC. Current treatments for advanced melanoma and introduction of a promising novel gene therapy for melanoma (Review) Oncol Rep. 2016;36:1779–1786. doi: 10.3892/or.2016.5032. [DOI] [PubMed] [Google Scholar]
- 5.Kraft TE, Parisotto D, Schempp C, Efferth T. Fighting cancer with red wine? Molecular mechanisms of resveratrol. Crit Rev Food Sci Nutr. 2009;49:782–799. doi: 10.1080/10408390802248627. [DOI] [PubMed] [Google Scholar]
- 6.Lee GA, Hwang KA, Choi KC. Roles of dietary phytoestrogens on the regulation of epithelial-mesenchymal transition in diverse cancer metastasis. Toxins (Basel) 2016;8:E162. doi: 10.3390/toxins8060162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marcsek ZL, Kocsis Z, Szende B, Tompa A. Effect of formaldehyde and resveratrol on the viability of Vero, HepG2 and MCF-7 cells. Cell Biol Int. 2007;31:1214–1219. doi: 10.1016/j.cellbi.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 8.Reagan-Shaw S, Mukhtar H, Ahmad N. Resveratrol imparts photoprotection of normal cells and enhances the efficacy of radiation therapy in cancer cells. Photochem Photobiol. 2008;84:415–421. doi: 10.1111/j.1751-1097.2007.00279.x. [DOI] [PubMed] [Google Scholar]
- 9.Baarine M, Thandapilly SJ, Louis XL, Mazué F, Yu L, Delmas D, Netticadan T, Lizard G, Latruffe N. Pro-apoptotic versus anti-apoptotic properties of dietary resveratrol on tumoral and normal cardiac cells. Genes Nutr. 2011;6:161–169. doi: 10.1007/s12263-011-0232-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Athar M, Back JH, Kopelovich L, Bickers DR, Kim AL. Multiple molecular targets of resveratrol: Anti-carcinogenic mechanisms. Arch Biochem Biophys. 2009;486:95–102. doi: 10.1016/j.abb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 12.Rojas C, Pan-Castillo B, Valls C, Pujadas G, Garcia-Vallve S, Arola L, Mulero M. Resveratrol enhances palmitate-induced ER stress and apoptosis in cancer cells. PLoS One. 2014;9:e113929. doi: 10.1371/journal.pone.0113929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. The role of the PERK/eIF2α/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr Mol Med. 2016;16:533–544. doi: 10.2174/1566524016666160523143937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yen YP, Tsai KS, Chen YW, Huang CF, Yang RS, Liu SH. Arsenic induces apoptosis in myoblasts through a reactive oxygen species-induced endoplasmic reticulum stress and mitochondrial dysfunction pathway. Arch Toxicol. 2012;86:923–933. doi: 10.1007/s00204-012-0864-9. [DOI] [PubMed] [Google Scholar]
- 16.Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012;5:9–19. doi: 10.1097/WOX.0b013e3182439613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poljsak B, Šuput D, Milisav I. Achieving the balance between ROS and antioxidants: When to use the synthetic antioxidants. Oxid Med Cell Longev. 2013;2013:956792. doi: 10.1155/2013/956792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sies H. Oxidative stress: From basic research to clinical application. Am J Med. 1991;91:31S–38S. doi: 10.1016/0002-9343(91)90281-2. [DOI] [PubMed] [Google Scholar]
- 19.Zhou Y, Wang K, Zhen S, Wang R, Luo W. Carfilzomib induces G2/M cell cycle arrest in human endometrial cancer cells via upregulation of p21(Waf1/Cip1) and p27Kip1. Taiwan J Obstet Gynecol. 2016;55:847–851. doi: 10.1016/j.tjog.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Tyner AL. A new year, a new role for p21. Cell Cycle. 2009;8:183. doi: 10.4161/cc.8.2.7824. [DOI] [PubMed] [Google Scholar]
- 21.Vaseva AV, Moll UM. The mitochondrial p53 pathway. Biochim Biophys Acta. 2009;1787:414–420. doi: 10.1016/j.bbabio.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maverakis E, Cornelius LA, Bowen GM, Phan T, Patel FB, Fitzmaurice S, He Y, Burrall B, Duong C, Kloxin AM, et al. Metastatic melanoma-a review of current and future treatment options. Acta Derm Venereol. 2015;95:516–524. doi: 10.2340/00015555-2035. [DOI] [PubMed] [Google Scholar]
- 23.Shukla Y, Singh R. Resveratrol and cellular mechanisms of cancer prevention. Ann NY Acad Sci. 2011;1215:1–8. doi: 10.1111/j.1749-6632.2010.05870.x. [DOI] [PubMed] [Google Scholar]
- 24.Georgieva J, Sinha P, Schadendorf D. Expression of cyclins and cyclin dependent kinases in human benign and malignant melanocytic lesions. J Clin Pathol. 2001;54:229–235. doi: 10.1136/jcp.54.3.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heiss EH, Schilder YD, Dirsch VM. Chronic treatment with resveratrol induces redox stress- and ataxia telangiectasia-mutated (ATM)-dependent senescence in p53-positive cancer cells. J Biol Chem. 2007;282:26759–26766. doi: 10.1074/jbc.M703229200. [DOI] [PubMed] [Google Scholar]
- 26.de la Lastra CA, Villegas I. Resveratrol as an antioxidant and pro-oxidant agent: Mechanisms and clinical implications. Biochem Soc Trans. 2007;35:1156–1160. doi: 10.1042/BST0351156. [DOI] [PubMed] [Google Scholar]
- 27.Singh A, Sati S, Mishra R. Resveratrol: Antioxidant-pro-oxidant. In J Tech Res Sci. 2016;1:106–112. [Google Scholar]
- 28.Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, Wondrak GT, et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29:1235–1243. doi: 10.1093/carcin/bgn095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Menegon S, Columbano A, Giordano S. The dual roles of NRF2 in cancer. Trends Mol Med. 2016;22:578–593. doi: 10.1016/j.molmed.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 30.Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid Redox Signal. 2010;13:1713–1748. doi: 10.1089/ars.2010.3221. [DOI] [PubMed] [Google Scholar]
- 31.Rachakonda G, Sekhar KR, Jowhar D, Samson PC, Wikswo JP, Beauchamp RD, Datta PK, Freeman ML. Increased cell migration and plasticity in Nrf2-deficient cancer cell lines. Oncogene. 2010;29:3703–3714. doi: 10.1038/onc.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moon EJ, Giaccia A. Dual roles of NRF2 in tumor prevention and progression: Possible implications in cancer treatment. Free Radic Biol Med. 2015;79:292–299. doi: 10.1016/j.freeradbiomed.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD. Dual roles of Nrf2 in cancer. Pharmacol Res. 2008;58:262–270. doi: 10.1016/j.phrs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sood R, Porter AC, Ma K, Quilliam LA, Wek RC. Pancreatic eukaryotic initiation factor-2alpha kinase (PEK) homologues in humans, Drosophila melanogaster and Caenorhabditis elegans that mediate translational control in response to endoplasmic reticulum stress. Biochem J. 2000;346:281–293. doi: 10.1042/bj3460281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: Cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol. 1998;18:7499–7509. doi: 10.1128/MCB.18.12.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu B, Cheng Y, Zhang B, Bian HJ, Bao JK. Polygonatum cyrtonem lectin induces apoptosis and autophagy in human melanoma A375 cells through a mitochondria-mediated ROS-p38-p53 pathway. Cancer Lett. 2009;275:54–60. doi: 10.1016/j.canlet.2008.09.042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.