

Abstract
Liver transplantation were reported in patients with classic maple syrup urine disease in the literature. Branched chain alpha keto acid dehydrogenase activity can be improved in patients after transplant, and a protein-restricted diet is usually not needed. The first patient was a boy aged 2,5 years who presented with frequent ketosis attacks and epileptic seizures, and the second patient was an 11-month-old boy who also presented with frequent ketosis episodes, both despite adherence to diet therapy. Both patients received liver transplantations from live donors. A low protein diet was no longer required and no decline in cognitive functions was observed in either patient in the follow-up. We wanted to present these cases to show that despite a normal diet, plasma levels of branched- chain amino acids remained normal without any decline in cognitive function after liver transplantation in patients with classic maple syrup urine disease patients.
Keywords: Child, liver transplantation, maple syrup urine disease patients
Introduction
Currently, classic maple syrup urine disease (MSUD) can still cause severe neurologic disorders and sudden death despite advances in developing medical nutritional therapies (1). The picture of acute metabolic intoxication can lead to brain herniation due to brain edema and cardiopulmonary arrest. In the chronic period, increased levels of branched-chain amino acids and disrupted keto acid homeostasis can lead to disorders in neurotransmitter metabolism, chronic cognitive disorders, and mental retardation by altering amino acid uptake in the brain (2).
These cases are presented with the objective of demonstrating that worsening of cognitive functions did not occur and plasma branched-chain amino acid levels remained within normal limits in our patients with classic-type MSUD despite a free diet after liver transplantation.
Case 1
A 2.5-year-old male patient was diagnosed as having MSUD in the neonatal period and was followed up by Istanbul University, Cerrahpaşa Medical Faculty, Division of Pediatric Nutrition and Metabolism. The patient presented to our clinic with the objective of liver transplantation because of frequently repeating ketosis and epileptic seizures, though he successfully complied with medical nutritional therapies. Physical examination revealed no pathology except for neuromotor developmental retardation. Liver synthesis functions were found to be normal. His neuromotor development was compatible with nine months in the psychomotor development test performed at the chronologic age of 29 months before transplantation. Mutation analysis was performed with the objective of obtaining a definite diagnosis, which revealed homozygous deletion in the 4th exon of the BCKDHA gene in the location of 19q13.2. As a result of organ transplantation counsel, live-donor transplantation (the patient’s father) was planned. Protein loading, which is not a classic method, was performed (3 g/kg/day) in order to evaluate the ability of the donor to remove branched-chain amino acids (BCAA) from the blood. The father, who did not have an increase in plasma BCAA levels, was considered to be appropriate as a live donor. The left lateral segment of the live donor was used in liver transplantation. The plasma amino acid levels before transplantation were as follows: valine: 745 mmol/L (Normal [N]: 90–300), leucine: 870 mmol/L (N: 65–220), and isoleucine: 620 mmol/L (N: 26–100). On the 1st day after transplantation, the BCAA levels were found to still be increased (valine 450 mmol/L, leucine 750 mmol/L, and isoleucine 410 mmol/L). Therefore, nutritional therapy was initiated with a special formula poor in natural protein and BCAAs. The amount of natural protein was gradually increased (0.5 g/kg/day) approximately at the end of the first week and 2 g/kg/day was reached on the 14th day after operation. Plasma BCAA levels measured at this time were found to be within the normal limits (valine: 90, leucine: 87 and isoleucine: 72 mmol/L) (Figure 1). The patient, whose general status was well, was discharged. At the end of the 24th month after transplantation, no metabolic attack was observed in the patient whose general status was well and he was consuming protein freely. Neuromotor development was found to be compatible with 32 months on the psychomotor development test performed at the chronologic age of 54 months. Our patient is using tacrolimus (0.15 mg/kg/day, oral) and his outpatient follow-up is continuing.
Figure 1.
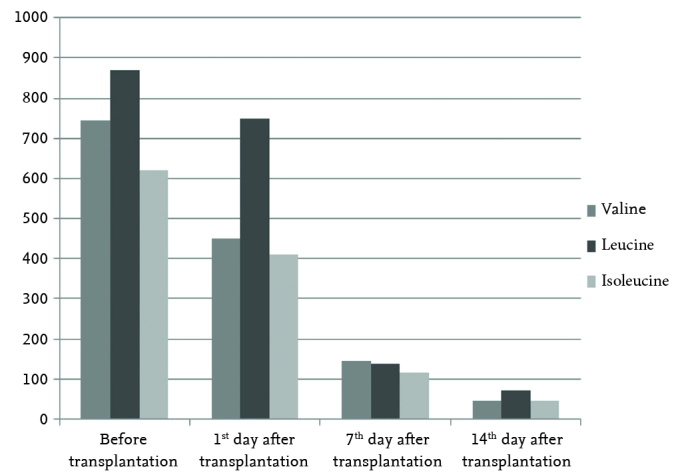
Valine, leucine, and isoleucine levels before and after transplantation in our first case
Case 2
An 11-month-old male patient was diagnosed as having MSUD in the neonatal period and was being followed up in Gazi University, Faculty of Medicine, Division of Pediatric Nutrition and Metabolism. He presented to our clinic with the objective of liver transplantation because of very frequent ketosis episodes despite successful compliance with medical nutritional therapies. The patient’s neuromotor development and physical examination findings were found to be normal. Liver synthesis functions were normal. His neuromotor development was compatible with seven months in the psychomotor development test performed at the chronologic age of nine months before transplantation. Mutation analysis revealed a homozygous deletion in the 7th exon in the BCKDHB gene in the location of 6q14.1. As a result of organ transplantation counsel, live-donor transplantation (the patient’s mother) was planned. The mother, whose plasma BCAA levels were found to be normal after protein loading (3 g/kg/day), was considered to be an appropriate live-donor. The left lateral segment of the live donor was used in liver transplantation. The plasma amino acid levels before transplantation were as follows: valine: 780 mmol/L, leucine: 950 mmol/L and isoleucine: 640 mmol/L. On the first day after transplantation, BCAA levels were observed to still be increased (valine: 410, leucine: 450 and isoleucine: 380 mmol/L). Low natural protein and low-BCAA nutritional therapy was initiated. The amount of natural protein consumed was increased from 0.5g/kg/day on the fifth day to 2 g/kg/day on the 14th day by increasing the amount 0.5g/kg/day daily. At this point, the plasma BCAA levels were found to be normal (valine: 45, leucine: 70 and isoleucine: 45 mmol/L) (Figure 2). The patient, whose general status was well, was discharged. His neuromotor development was compatible with 18 months in the psychomotor development test performed at the chronologic age of 24 months after transplantation. Our patient is receiving tacrolimus (0.15 mg/kg/day, oral) and his outpatient follow-up is still continuing. Written informed consent was obtained from the patients’ parents.
Figure 2.
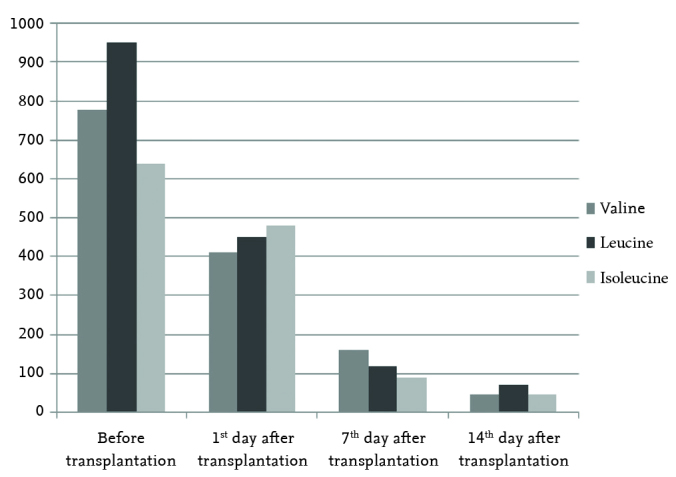
Valine, leucine, and isoleucine levels before and after transplantation in our second case
Discussion
In congenital metabolic diseases of the liver, the number of patients undergoing liver transplantation is gradually increasing (3). These metabolic diseases are caused by deficiency of enzymes which are completely (e.g., ornithine transcarbamylase deficiency and citrullinemia) or partially (e.g., propionic acidemia and MSUD) synthesized in the liver (4). Liver transplantation is performed in patients with MSUD; it has been reported that a free diet can be initiated and the picture of metabolic episode that might develop with acute stress can be prevented after transplantation (5).
The liver constitutes 9–13% of the total branched-chain keto acid dehydrogenase enzyme complex (BCAADEC) in the body (6). Mazariegos et al. (1) reported that liver transplantation could prevent potential new brain injury, though it could not heal previous brain injury, based on the neuropsychiatric tests performed before and after transplantation in 37 patients with MSUD who were followed up for 45.±2.2 years after liver transplantation. Low protein nutritional therapy was not required and acute metabolic episode was not observed in these patients. No metabolic episodes were observed and regression in cognitive functions did not occur at the 24-month follow-up visit in our first patient and at the 11-month follow-up visit in our second patient.
Neuropsychiatric disorders and slowly-progressing neuromotor retardation may be observed in patients with MSUD. Sudden acute metabolic episodes may develop despite application of medical nutritional therapies with limitation of natural protein and BCAA-free formulas. These episodes may lead to severe neurologic findings and even death. Elective liver transplantation has become an alternative therapeutic option for diet therapy to prevent acute metabolic episodes in patients with MSUD (5).
Although the liver constitutes only 9–13% of the total BCAADEC function in the body, liver transplantation provides the necessary improvement for metabolism of BCAAs in patients with MSUD (6, 7). Metabolic disease is controlled in most patients with MSUD who undergo liver transplantation (1). Enzyme activity may increase after transplantation and low-protein medical nutritional therapy is mostly no longer required. Diaz et al. (8) reported that liver transplantation prevented potential new brain injury, though it could not improve previous brain injury in patients with MSUD who they followed up for a mean time period of 12.2 years (5–12 years) after liver transplantation and was an efficient treatment method for patients with classic MSUD (9). On the other hand, Al-Shamsi et al. (10) reported that acute metabolic episodes occurred in the early period after heterozygous living-donor liver transplantation in a patient with MSUD in a case report.
Liver transplantation should be considered as a therapeutic option in patients with severe enzyme deficiency, as in our patients, but it should be kept in mind that each patient should be evaluated individually when making the final transplantation decision (8, 9).
In conclusion, it was shown that the plasma levels of branched-chain amino acids remained within normal limits and cognitive functions did not deteriorate following liver transplantation despite a free diet in our patients with classic-type MSUD in our case presentation.
Footnotes
Informed Consent: Written informed consent were obtained from patient who participated in these case.
Peer-review: Externally peer-reviewed.
Author Contributions: Concept - A.B., R.A., E.S. Design - E.S., İ.A., H.E., A.Y.; Supervision - R.A., A.Y., A.D.; Funding - M.K., H.E.; Materials - H.E., İ.A., M.K.; Data Collection and/or Processing - İ.A., E.S., M.K., H.E.; Analysis and/or Interpretation - A.B., R.A., A.D.; Literature Review - A.B., E.S.; Writing - A.B.; Critical Review - R.A., A.D., A.Y.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The authors declared that this study has received no financial support.
References
- 1.Mazariegos GV, Holmes Morton D, Sindhi R, et al. Liver transplantation for classical maple syrup urine disease: long-term follow-up in 37 patients and comparative United Network for Organ Sharing experience. J Pediatr. 2012;160:116–21. doi: 10.1016/j.jpeds.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffmann B, Helbling C, Schadewaldt P, et al. Impact of longitudinal plasma leucine levels on the intellectual outcome in patients with classic MSUD. Pediatr Res. 2006;59:17–20. doi: 10.1203/01.pdr.0000190571.60385.34. [DOI] [PubMed] [Google Scholar]
- 3.Moini M, Mistry P, Schilsky ML. Liver transplantation for inherited metabolic disorders of the liver. Curr Opin Organ Transplant. 2010;15:269–76. doi: 10.1097/MOT.0b013e3283399dbd. [DOI] [PubMed] [Google Scholar]
- 4.Shneider BL, Vockley J, Mazariegos GV. Trading places: liver transplantation as a treatment, not a cure, for metabolic liver disease. Liver Transpl. 2011;17:628–30. doi: 10.1002/lt.22293. [DOI] [PubMed] [Google Scholar]
- 5.Strauss KA, Mazariegos GV, Sindhi R, et al. Elective liver transplantation for the treatment of classical maple syrup urine disease. Am J Transplant. 2006;6:557–64. doi: 10.1111/j.1600-6143.2006.01467.x. [DOI] [PubMed] [Google Scholar]
- 6.Suryawan A, Hawes JW, Harris RA, et al. A molecular model of human branched-chain amino acid metabolism. Am J Clin Nutr. 1998;68:72–81. doi: 10.1093/ajcn/68.1.72. [DOI] [PubMed] [Google Scholar]
- 7.Wendel U, Saudubray JM, Bodner A, et al. Liver transplantation in maple syrup urine disease. Eur J Pediatr. 1999;158:S60–4. doi: 10.1007/PL00014324. [DOI] [PubMed] [Google Scholar]
- 8.Díaz VM, Camarena C, de la Vega Á, et al. Liver transplantation for classical maple syrup urine disease: long-term follow-up. J Pediatr Gastroenterol Nutr. 2014;59:636–9. doi: 10.1097/MPG.0000000000000469. [DOI] [PubMed] [Google Scholar]
- 9.Hahn LG, Morgan JE. A case of liver transplantation in maple syrup urine disease. In: Morgan JE, Baron IS, Ricker JH, editors. Casebook of clinical neuropsychology. New York: Oxford University Press; 2011. pp. 164–71. [Google Scholar]
- 10.Al-Shamsi A, Baker A, Dhawan A, et al. Acute metabolic crises in maple syrup urine disease after liver transplantation from a related heterozygous living donor. JIMD Rep. 2016 Apr 28;30:59–62. doi: 10.1007/8904_2016_532. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]