

Abstract
Aims
Previous pharmacokinetic characterization of a transporter probe cocktail containing digoxin (P‐gp), furosemide (OAT1, OAT3), metformin (OCT2, MATE1, MATE2‐K) and rosuvastatin (OATP1B1, OATP1B3, BCRP) in healthy subjects showed increases in rosuvastatin systemic exposure compared to rosuvastatin alone. In this trial, the doses of metformin and furosemide as putative perpetrators were reduced to eliminate their drug–drug interaction (DDI) with rosuvastatin.
Methods
In a randomized, open‐label, single‐centre, five‐treatment, five‐period crossover trial, 30 healthy male subjects received as reference treatments separately 0.25 mg digoxin, 1 mg furosemide, 10 mg metformin and 10 mg rosuvastatin, and as test treatment all four drugs administered together as a cocktail. Primary pharmacokinetic endpoints were AUC0‐tz (area under the plasma concentration–time curve from time zero to the last quantifiable concentration) and C max (maximum plasma concentration) of each probe drug.
Results
Geometric mean ratios and 90% confidence intervals of test (cocktail) to reference (single drug) for AUC0‐tz were 96.4% (88.2–105.3%) for digoxin, 102.6% (93.8–112.3%) for furosemide, 97.5% (93.5–101.6%) for metformin and 105.0% (96.4–114.4%) for rosuvastatin, indicating lack of interaction. The same analysis for C max and for pharmacokinetic parameters of urinary excretion of all cocktail components also indicated no DDI.
Conclusions
Digoxin (0.25 mg), furosemide (1 mg), metformin (10 mg) and rosuvastatin (10 mg) exhibit no mutual pharmacokinetic interactions and are well tolerated administered as a cocktail. The cocktail is thus optimized and has the potential to be used as a screening tool for clinical investigation of transporter‐mediated DDI.
Keywords: drug interactions, drug transporters, pharmacokinetics, Phase I
What is Already Known about this Subject
Probe drug cocktails for investigating transporter‐based drug–drug interactions (DDI) are a promising approach to reduce the number of clinical studies performed in drug development.
In two previous clinical investigations of a new four‐component transporter cocktail based on in vitro studies, minor mutual interactions between the cocktail components remained.
What this Study Adds
In this modified cocktail, absence of DDI was shown between digoxin (0.25 mg), furosemide (1 mg), metformin (10 mg) and rosuvastatin (10 mg) when administered together compared to dosing as single substances.
The cocktail is thus optimized and has the potential to be used as a screening tool for further clinical studies investigating transporter‐mediated DDI.
Introduction
During development of new molecular entities (NMEs), characterization of potential drug–drug interaction (DDI) liabilities with involvement of drug transporters is required 1, 2, 3. In particular, regulatory authorities request a thorough assessment of the propensity of an NME to cause DDIs by inhibition of those drug transporters with compelling clinical evidence for relevance 4, including http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=768 (P‐gp; ATP‐binding cassette transporter family, subfamily B, member 1 [ABCB1]), organic anion transporting polypeptide 1B1 (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=238), http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=238, organic anion transporter 1 (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1025), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1027, organic cation transporter 2 (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1020), multidrug and toxin extrusion protein 1 (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=236), http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=236, and breast cancer resistance protein (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=792; ABCG2) 5, 6, 7. Recently, interest in the use of the probe drug cocktail approach in transporter DDI clinical trials has increased considerably 8, 9, 10, 11. Such cocktails allow the effect of an NME on the pharmacokinetics of several probe drugs with different transporter specificities to be investigated simultaneously in a single trial, thus reducing the number of clinical DDI studies in the development programme.
In a previous Phase I clinical DDI trial, we investigated mutual interactions within a transporter probe drug cocktail containing http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4726 0.25 mg (as a probe for P‐gp), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4839 5 mg (OAT1, OAT3), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4779 500 mg (OCT2, MATE1, MATE2‐K) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2954 10 mg (OATP1B1, OATP1B3, BCRP) 8. Based on in vitro investigations 12, it was expected that interactions between the individual probe substrates would be absent in vivo, which is an important prerequisite 5, 11. It was found that such mutual interactions were indeed essentially absent, with the notable exception of approximately 40% increases in C max (maximum plasma concentration) and AUC0‐tz (area under the plasma concentration–time curve from zero to the last quantifiable concentration) of rosuvastatin in the four‐probe cocktail compared to rosuvastatin administered alone. Based on the hypothesis that metformin and possibly furosemide were the most likely perpetrators of this effect on rosuvastatin, we performed a second DDI trial investigating pairwise interactions in which it was demonstrated that reduction of the metformin dose to 50 mg or 10 mg and the furosemide dose to 1 mg eliminated the interactions with rosuvastatin 9. The present trial was conducted to confirm the absence of mutual interactions between all probe substrates in the full four‐component cocktail with reduced doses of furosemide and metformin, thus optimizing this cocktail for further use in the investigation of transporter‐mediated DDI.
Methods
Subjects
Thirty healthy male subjects aged 18–55 years and with a body mass index of 18.5–29.9 kg m−2 were eligible to participate in this trial (EudraCT no. 2016‐001893‐14, http://clinicaltrials.gov identifier NCT02854527). Women were not included, to avoid any potential interference of hormonal cycle or hormone‐based contraceptives with the trial results. The clinical trial protocol was approved by the Independent Ethics Committee of the State Chamber of Physicians of Baden‐Württemberg, Stuttgart, Germany, and the Federal Institute for Drugs and Medicinal Products (BfArM), Bonn, Germany. The trial was conducted in accordance with the principles of the Declaration of Helsinki and the International Council for Harmonization guidelines for Good Clinical Practice. All subjects provided written informed consent.
Trial objectives, design and treatments
The main trial objective was to investigate the mutual pharmacokinetic DDI of 0.25 mg digoxin, 1 mg furosemide, 10 mg metformin hydrochloride, and 10 mg rosuvastatin when given together as a probe cocktail (the test treatment). The four separate reference treatments were 0.25 mg digoxin, 1 mg furosemide, 10 mg metformin hydrochloride and 10 mg rosuvastatin orally. The design was a randomized, open‐label, single‐centre, five‐treatment, five‐period, 10‐sequence crossover trial.
The investigational medicinal products were digoxin (Lanicor® 0.25 mg tablet, Teofarma S.r.l., Italy), furosemide (Lasix® liquidum 10 mg ml−1 oral solution, Sanofi‐Aventis Deutschland GmbH, Germany), metformin hydrochloride (MetfoLiquid GeriaSan® 500 mg/5 ml oral solution, Infectopharm Arzneimittel und Consilium GmbH, Germany), and rosuvastatin (Crestor® 10 mg film‐coated tablet, AstraZeneca GmbH, Germany). The treatments were administered to each of the 30 subjects according to one of the 10 different randomly assigned treatment sequences, with three subjects per sequence.
After an overnight fast of at least 10 h, the trial medications were administered to the subjects in a standing position as single oral doses together with a total of 320 ml water. The doses of furosemide and metformin were prepared by adding 0.1 ml of the respective oral solution via a microsyringe to a mixing bottle containing 20 ml water immediately before administration. The subjects drank this 20 ml solution, then the bottle was rinsed with further 20 ml of water which the subjects also drank. In the cocktail, furosemide and metformin were administered from separate mixing bottles. Administration of diluted oral solution was followed by further 280 ml water (treatment with furosemide or metformin alone) or by further 240 ml water together with rosuvastatin and digoxin tablets (cocktail treatment). During the first 4 h after medication administration, no food was allowed and subjects were not allowed to lie down.
Pharmacokinetics
Blood samples for the measurement of plasma concentrations of the administered medications were taken using K3‐EDTA as anticoagulant from a forearm vein of each subject before dosing and at 20 min, 40 min, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 10, 12, 24, 36, 48, 72 and 96 h after dosing. In the treatment periods with furosemide and metformin alone, the last samples were taken at 24 h and 48 h, respectively. Urine samples were obtained before dosing and in the time intervals 0–4 h, 4–8 h, 8–12 h, 12–24 h and 24–36 h. In the treatment period with furosemide alone, the last urine fraction collected was 12–24 h. Pharmacokinetic parameters were calculated using standard noncompartmental methods with the software Phoenix WinNonlin® Professional, version 6.3 (Certara, Princeton, NJ, USA).
For all analytes, the primary pharmacokinetic endpoints were AUC0–tz and C max and the secondary endpoint AUC0–∞ (area under the plasma concentration–time curve extrapolated to infinite time). Further endpoints were t max (time to attainment of maximum plasma concentration), t 1/2 (terminal elimination half‐life), CLR (renal clearance) and fe (fraction excreted unchanged in urine).
Bioanalytical methods
Plasma and urine concentrations of the drug analytes were determined by fully validated 13, 14 LC–MS/MS (liquid chromatography–tandem mass spectrometry) methods using the isotope‐labelled internal standards [2H3]digoxin, [2H5]furosemide, [2H6]metformin and [13C1,2H4]rosuvastatin. Analyses were performed at Covance Laboratories Ltd., Harrogate, UK, for digoxin and at SGS Cephac Europe, St. Benoît, France, for the remaining analytes. The methods were described in previous publications 8, 9 except those for digoxin which are commercially available; further details specific to the present trial, including the assay performance data, are provided in the Supporting Information bioanalysis.
Safety and tolerability assessment
Safety and tolerability were assessed based on adverse events (AE), 12‐lead ECGs and vital signs. Clinically relevant findings in 12‐lead ECGs and vital signs were to be reported as AEs. Safety laboratory was done at screening and at end of trial. The treated set (consisting of all subjects treated with at least one study drug) was used for safety analyses. AEs were analysed according to the concept of treatment‐emergent AEs and the number of subjects with AEs. AEs occurring within 8 days after study drug intake were defined as treatment‐emergent and assigned to the corresponding treatment. Concomitant diagnoses and AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 19.1.
Statistical methods
The pharmacokinetic parameters of digoxin, furosemide, metformin and rosuvastatin were compared when given together as a cocktail (test treatment) to when given alone (reference treatment) by computing the test/reference ratios of the adjusted geometric means (GMR, geometric mean ratio) and their two‐sided 90% confidence intervals (CI) for the primary (AUC0–tz, C max) and secondary (AUC0–∞) pharmacokinetic endpoints and for the urinary parameters CLR and fe. This method corresponds to the two one‐sided t‐tests procedure, each at the 5% significance level.
The statistical model used was analysis of variance (ANOVA) on the logarithmic scale and included the effects sequence, subjects within sequence, period and treatment. The effect ‘subjects within sequence’ was defined as random, whereas the other effects were fixed. No significance level adjustment for multiple comparisons was applied. The pharmacokinetic parameter analysis set consisted of all treated subjects who provided at least one evaluable primary or secondary endpoint. The statistical analyses were performed using SAS® (version 9.4) by SAS Institute Inc., Cary, NC, USA).
The sample size calculation for this exploratory trial was based on the expected precision of the GMR estimate (defined as the ratio of the upper 90% CI limit over the point estimate) and assuming a within‐subject geometric coefficient of variation (gCV) in the range of 10–30% for the primary pharmacokinetic endpoints as observed in previous trials 8, 9. The dropout rate was expected to be considerably less than 10 and evenly distributed across the 10 treatment sequences. Assuming an intra‐individual gCV of 20% and 30 (20) evaluable subjects, the precision was expected to be 1.10 (1.13). Thus, for an assumed GMR of 100%, the 90% CI was expected to range from 91% to 110% (89–113%). The calculation was performed as described by Kupper and Hafner 15 using R version 3.2.2.
For exploratory purposes, no‐effect boundaries of 80–125% were assumed, and lack of DDI was concluded if the 90% CI of the primary pharmacokinetic parameters were included within this interval 5.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 16, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 17.
Results
Subjects
Thirty healthy white male subjects were randomized in the trial and treated. Their median age (min–max) was 36.5 (20–55) years and the mean (standard deviation) body mass index was 25.93 (1.90) kg m−2. Two subjects (6.7%) did not complete the planned observation time. One subject was lost to follow‐up after completion of period 1 (furosemide alone). A further subject discontinued trial participation due to an AE (see ‘Safety and tolerability’ section below) that occurred during the washout after period 3, the subject having taken metformin, furosemide and rosuvastatin in periods 1, 2 and 3, respectively.
Pharmacokinetics
Digoxin
Geometric mean plasma concentration–time profiles of digoxin when given alone (reference treatment) or as part of the test cocktail were essentially superimposable (Figure 1A). Maximum plasma concentrations occurred at a median t max of 1 h for both treatments, and the urinary excretion parameters fe0–36 and CLR,0–36 were comparable between the treatments (Table S1). The adjusted geometric means, GMRs and 90% CIs for AUC0–tz and C max (primary endpoints) and AUC0–∞ (secondary endpoint) based on the ANOVA model are given in Table 1. The pharmacokinetic parameters of digoxin as part of the test cocktail and as a single drug were similar. The GMRs ranged from 93% to 97% for the three endpoints. The 90% CIs included 100% and were within the standard bioequivalence acceptance range of 80–125%, indicating a lack of interaction. GMRs and 90% CIs of the urinary pharmacokinetic parameters CLR,0–36 and fe0–36 also showed lack of interaction (Table S2).
Figure 1.
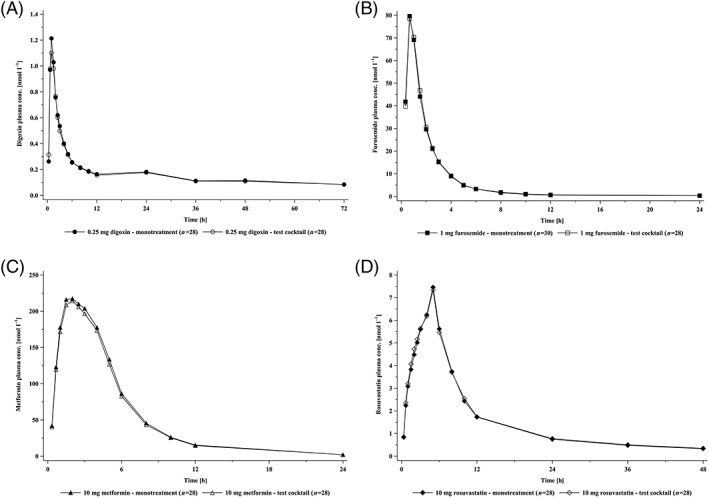
Geometric mean plasma concentration–time profiles of digoxin 0.25 mg (A), furosemide 1 mg (B), metformin 10 mg (C) and rosuvastatin 10 mg (D) after oral dosing alone (closed symbols) and in combination as a cocktail (open symbols)
Table 1.
Adjusted geometric means, geometric mean ratios and 90% confidence intervals (CI) for primary and secondary pharmacokinetic parameters of digoxin administered alone or in the four‐component cocktail
| Endpoint | Digoxin in test cocktail (T) | Digoxin alone (R) | Ratio T/R | 90% CI | gCVa | ||
|---|---|---|---|---|---|---|---|
| N b | Adj. geom. mean | N | Adj. geom. mean | [%] | [%] | [%] | |
| AUC 0–tz [nmol·h l −1 ] | 28 | 11.55 | 28 | 11.98 | 96.39 | (88.22; 105.33) | 19.4 |
| C max [nmol l −1 ] | 28 | 1.26 | 28 | 1.36 | 93.17 | (83.49; 103.97) | 24.1 |
| AUC 0–∞ [nmol·h l −1 ] | 27 | 17.67 | 28 | 18.30 | 96.53 | (92.08; 101.20) | 10.1 |
Within‐subject geometric coefficient of variation.
N for AUC0–∞ less than N for AUC0–tz due to insufficient bioanalytically quantifiable plasma concentrations in some subjects at late sampling times to allow determination of the terminal half‐life.
Furosemide
Geometric mean plasma concentration–time profiles of furosemide when given alone or as part of the test cocktail were essentially superimposable (Figure 1B). Maximum plasma concentrations occurred at a median t max of 40 min for both treatments, and the urinary excretion parameters fe0–24 and CLR,0–24 were comparable between the treatments (Table S3). The adjusted geometric means, GMRs and 90% CIs for AUC0–tz and C max (primary endpoints) and AUC0–∞ (secondary endpoint) based on the ANOVA model are given in Table 2. The pharmacokinetic parameters of furosemide as part of the test cocktail and as a single drug were similar. The GMRs ranged from 97% to 104% for the three endpoints. The 90% CIs included 100% and were within the standard bioequivalence acceptance range of 80–125%, indicating a lack of interaction. The GMR and 90% CIs of the urinary pharmacokinetic parameter fe0–24 also showed lack of interaction. For CLR,0–24, the lower 90% confidence limit was slightly below 80% due to an abnormally high urinary excretion value from one subject after treatment with furosemide alone (Table S4).
Table 2.
Adjusted geometric means, geometric mean ratios and 90% confidence intervals (CI) for primary and secondary pharmacokinetic parameters of furosemide administered alone or in the four‐component cocktail
| Endpoint | Furosemide in test cocktail (T) | Furosemide alone (R) | Ratio T/R | 90% CI | gCVa | ||
|---|---|---|---|---|---|---|---|
| N b | Adj. geom. mean | N b | Adj. geom. mean | [%] | [%] | [%] | |
| AUC 0–tz [nmol·h l −1 ] | 28 | 163.61 | 30 | 159.43 | 102.62 | (93.82; 112.25) | 20.4 |
| C max [nmol l −1 ] | 28 | 86.28 | 30 | 82.99 | 103.96 | (93.60; 115.46) | 24.0 |
| AUC 0–∞ [nmol·h l −1 ] | 20 | 156.38 | 20 | 160.55 | 97.40 | (90.87; 104.41) | 9.6 |
Within‐subject geometric coefficient of variation.
N for AUC0–∞ less than N for AUC0–tz due to insufficient bioanalytically quantifiable plasma concentrations in some subjects at late sampling times to allow determination of the terminal half‐life.
Metformin
Geometric mean plasma concentration–time profiles of metformin when given alone or as part of the test cocktail were essentially superimposable (Figure 1C). Maximum plasma concentrations occurred at a median t max of 2 h for both treatments, and the urinary excretion parameters fe0–36 and CLR,0–24 were comparable between the treatments (Table S5). The adjusted geometric means, GMRs and 90% CIs for AUC0–tz and C max (primary endpoints) and AUC0–∞ (secondary endpoint) based on the ANOVA model are given in Table 3. The pharmacokinetic parameters of metformin as part of the test cocktail and as a single drug were similar. The GMRs ranged from 97% to 98% for the three endpoints. The 90% CIs included 100% and were within the standard bioequivalence acceptance range of 80–125%, indicating a lack of interaction. GMRs and 90% CIs of the urinary pharmacokinetic parameters CLR,0–24 and fe0–24 also showed lack of interaction (Table S6).
Table 3.
Adjusted geometric means, geometric mean ratios and 90% confidence intervals (CI) for primary and secondary pharmacokinetic parameters of metformin administered alone or in the four‐component cocktail
| Endpoint | Metformin in test cocktail (T) | Metformin alone (R) | Ratio T/R | 90% CI | gCVa | ||
|---|---|---|---|---|---|---|---|
| N | Adj. geom. mean | N | Adj. geom. mean | [%] | [%] | [%] | |
| AUC 0–tz [nmol·h l −1 ] | 28 | 1283.80 | 29 | 1316.79 | 97.49 | (93.54; 101.61) | 9.0 |
| C max [nmol l −1 ] | 28 | 225.16 | 29 | 229.17 | 98.25 | (91.85; 105.09) | 14.7 |
| AUC 0–∞ [nmol·h l −1 ] | 28 | 1290.93 | 29 | 1324.08 | 97.50 | (93.58; 101.58) | 8.9 |
Within‐subject geometric coefficient of variation.
Rosuvastatin
Geometric mean plasma concentration–time profiles of rosuvastatin when given alone or as part of the test cocktail were essentially superimposable (Figure 1D). Maximum plasma concentrations occurred at a median t max of 5 h for both treatments, and the urinary excretion parameters fe0–36 and CLR,0–36 were comparable between the treatments (Table S7). The adjusted geometric means, GMRs and 90% CIs for AUC0–tz and C max (primary endpoints) and AUC0–∞ (secondary endpoint) based on the ANOVA model are given in Table 4. The pharmacokinetic parameters of rosuvastatin as part of the test cocktail and as a single drug were similar. The GMRs ranged from 104% to 108% for the three endpoints. The 90% CIs included 100% and were within the standard bioequivalence acceptance range of 80–125%, indicating a lack of interaction. GMRs and 90% CIs of the urinary pharmacokinetic parameters CLR,0–36 and fe0–36 also showed lack of interaction (Table S8).
Table 4.
Adjusted geometric means, geometric mean ratios and 90% confidence intervals (CI) for primary and secondary pharmacokinetic parameters of rosuvastatin administered alone or in the four‐component cocktail
| Endpoint | Rosuvastatin in test cocktail (T) | Rosuvastatin alone (R) | Ratio T/R | 90% CI | gCVa | ||
|---|---|---|---|---|---|---|---|
| N b | Adj. geom. mean | N b | Adj. geom. mean | [%] | [%] | [%] | |
| AUC 0–tz [nmol·h l −1 ] | 28 | 81.93 | 29 | 78.02 | 105.01 | (96.39; 114.40) | 18.8 |
| C max [nmol l −1 ] | 28 | 8.14 | 29 | 7.80 | 104.28 | (94.95; 114.53) | 20.6 |
| AUC 0–∞ [nmol·h l −1 ] | 22 | 97.39 | 25 | 90.48 | 107.63 | (97.04; 119.39) | 19.4 |
Within‐subject geometric coefficient of variation.
N for AUC0–∞ less than for AUC0–tz due to insufficient bioanalytically quantifiable plasma concentrations in some subjects at late sampling times to allow determination of the terminal half‐life.
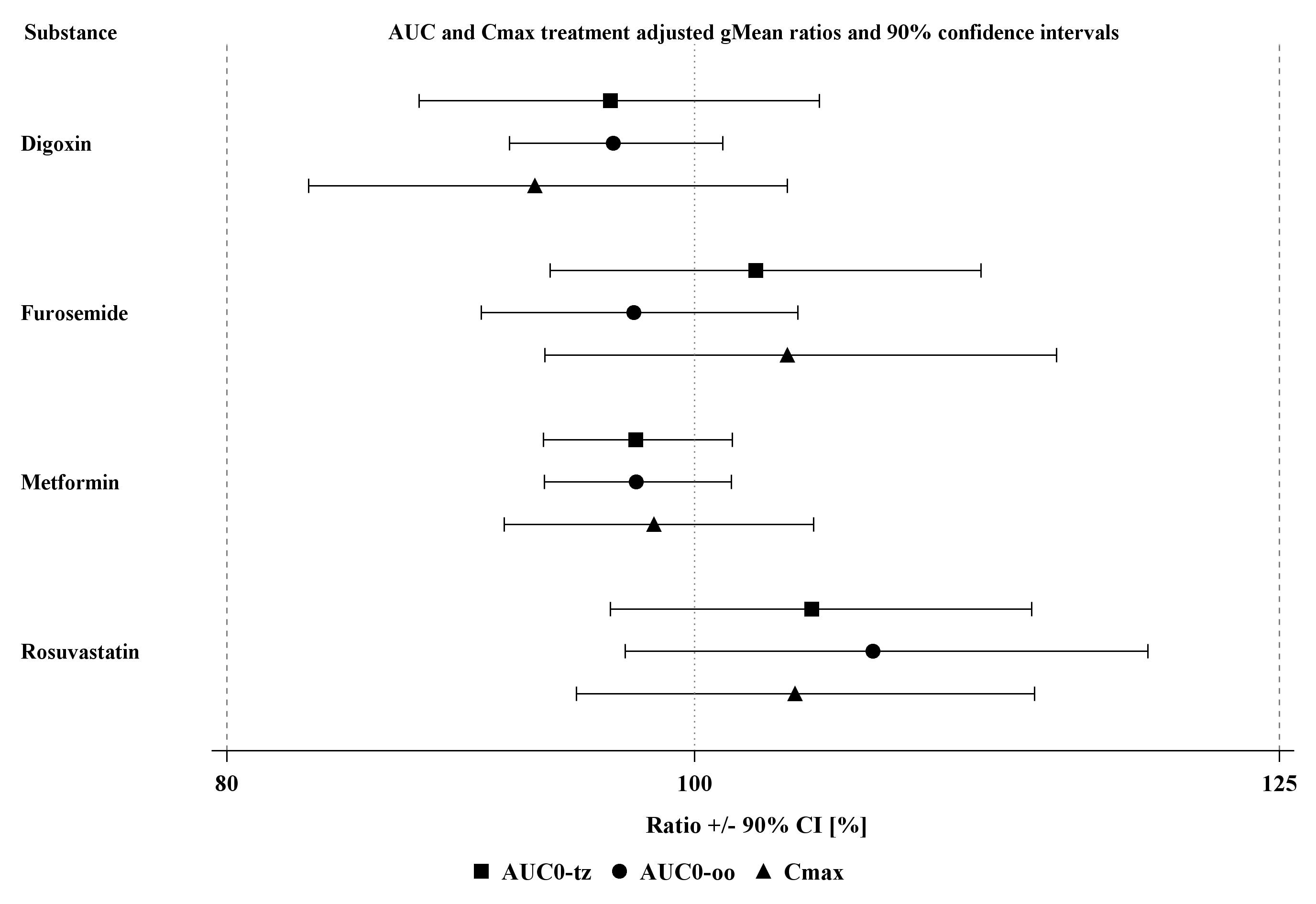
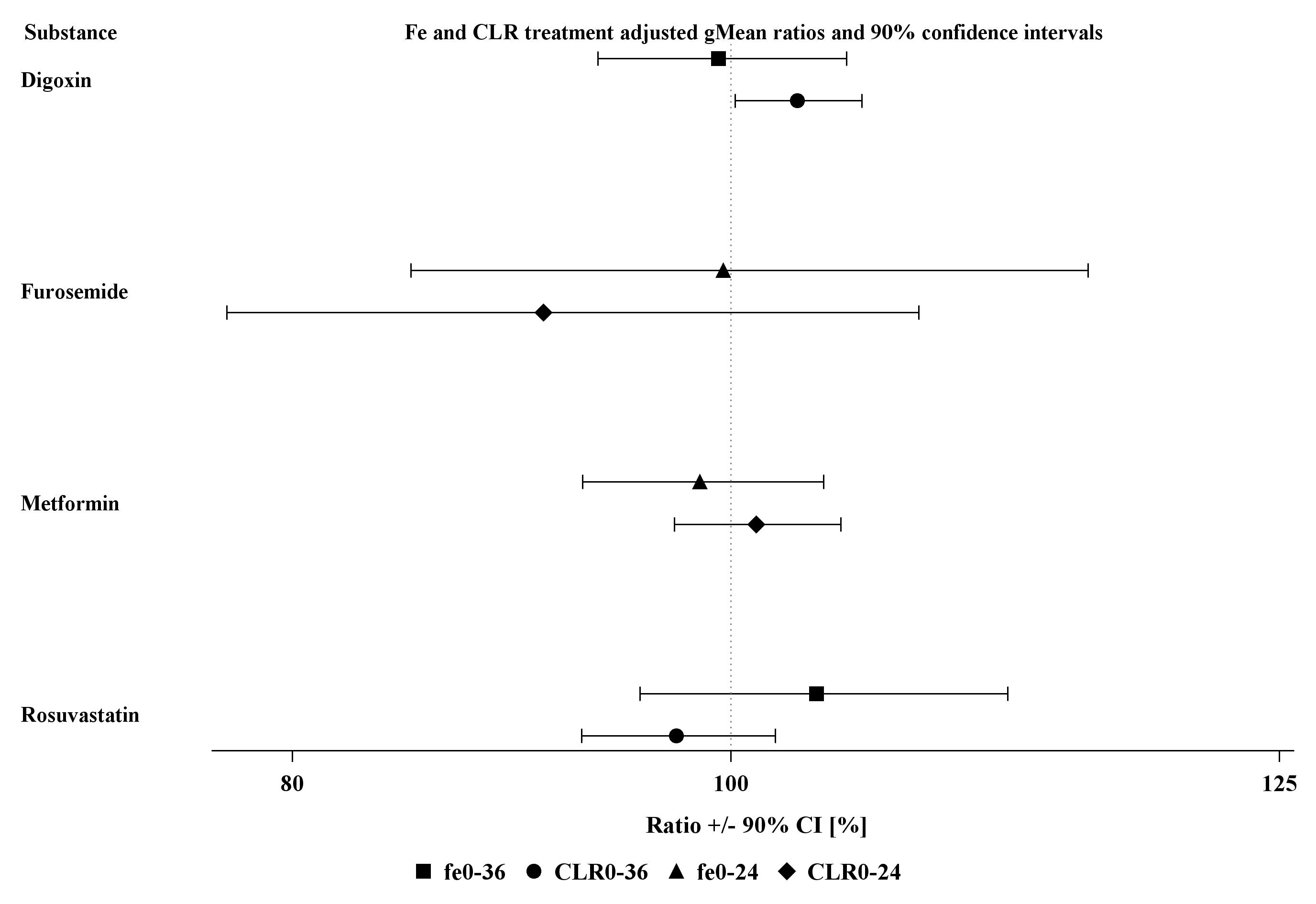
Forest plots summarizing the GMRs and 90% CIs for plasma and urinary pharmacokinetic parameters of all four probe drugs are given in Figures S1 and S2.
Safety and tolerability
Treatment‐emergent AEs were reported by 15 out of the 30 subjects (50.0%; Table S9). All AEs were of mild or moderate intensity. One AE assessed as drug‐related by the investigator (mild headache, reversible) was reported during the test cocktail treatment by one of 18 subjects (3.6%). No other AEs were assessed as drug‐related. One subject was reported with a serious AE (SAE; i.e. exposure to toxic agent, mild intensity, requiring hospitalization overnight for surveillance) during the digoxin period. This SAE was not related to study drug or study procedures and the subject recovered. One subject displayed asymptomatic extrasystoles of bigeminy type prior to scheduled dosing in period 4, that recovered within less than 1 h. This subject was not dosed in periods 4 and 5 with digoxin and cocktail, respectively. Extrasystoles of bigeminy type may occur in healthy volunteers 18, and there is no reasonable possibility for a relationship to the treatments the subject had received in periods 1, 2 and 3 (metformin, furosemide and rosuvastatin). A listing of treatment‐emergent AEs is given in Table S9.
Discussion
The results of this clinical DDI trial show the lack of mutual pharmacokinetic interaction between the drug transporter probe cocktail components digoxin 0.25 mg, furosemide 1 mg, metformin 10 mg and rosuvastatin 10 mg in normal male subjects (Tables 1, 2, 3, 4, Supplementary Tables S1–S8). In the cocktail approach, the substrates should exhibit no mutual interactions, regardless of whether drug metabolizing enzymes or transporters are being investigated 5, 6, 7. This lack of interaction was demonstrated in the present trial by assessment of both plasma and urinary pharmacokinetic data and is summarized in the forest plots in Figures S1 and S2. This trial thus concludes the in vivo process of designing a novel four‐component cocktail that is free of mutual pharmacokinetic interaction, which was initiated in a first in human dose‐finding trial 8.
In that first trial, the cocktail was essentially free of relevant mutual interactions, with the notable exception of an increase of rosuvastatin C max and AUC0–tz by approximately 40% when given together with digoxin (0.25 mg), metformin (500 mg) and furosemide (5 mg) 8. On the assumption that probably metformin, or possibly also furosemide, were the perpetrators of that limited interaction, we performed a second trial to explore the effect of different single oral doses of metformin and furosemide on a single oral dose of 10 mg rosuvastatin in pairwise combinations 9. The results of that second trial indicated that the increase of rosuvastatin plasma concentrations in the first cocktail trial was primarily caused by metformin at a single oral dose of 500 mg. Moreover, the results suggested that furosemide at a single oral dose of 5 mg may have contributed slightly to this interaction. Decreased doses of metformin (10 or 50 mg) or furosemide (1 mg) had no effect on rosuvastatin C max and AUC0–tz when tested as pairwise interactions (metformin–rosuvastatin or furosemide–rosuvastatin).
Based on these results, in the current (third) trial, we reduced the doses of metformin and furosemide in the cocktail to 10 mg and 1 mg respectively and investigated this optimized cocktail for mutual interactions at the level of plasma concentrations and urinary excretion data. Importantly, the increases of approximately 40% in C max and AUC0–tz of rosuvastatin 10 mg that were observed in our first trial when dosed in the original cocktail compared to dosing alone 8, could be completely eliminated in the present trial. This effect can be attributed to reduction of the doses of metformin and possibly also furosemide. In addition, in the original cocktail in our first trial, a 19% decrease in furosemide C max was observed compared to dosing alone 8. Based on pharmacokinetic data (isolated decrease of C max with unchanged furosemide AUC and renal clearance), it was suggested that this interaction was caused by an extrarenal effect, possibly a decrease of the rate rather than the extent of intestinal furosemide absorption 8. Literature data suggested that metformin was the most likely perpetrator of this slight interaction 19. Consistent with this, furosemide C max and AUC0–tz were unchanged in the optimized cocktail investigated in the current trial (Table 2), evidently as a result of the 50‐fold reduction in the metformin dose.
Increases in metformin plasma concentrations could be caused by coadministration with the cocktail of test NMEs that are potent OCT/MATE inhibitors. This effect was simulated in our second trial by increasing the metformin dose fivefold from 10 mg to 50 mg 9, which produced increases of 4.1‐fold in metformin AUC0–tz and 3.6‐fold in C max. These had no effect on rosuvastatin systemic exposure 9, and cover the range of metformin systemic exposure increases that have so far been attributed to OCT/MATE inhibition 20, 21, 22. Therefore, the use of the 10 mg metformin dose in the cocktail should allow for an approximately fourfold no‐effect margin with respect to a secondary effect on rosuvastatin AUC and C max, if an NME that is coadministered as a potential perpetrator increases the plasma exposure of metformin. Similarly, test NMEs that are potent inhibitors of OAT1 or OAT3 could increase the systemic exposure of furosemide (and decrease its renal clearance). In our second trial, increasing the furosemide dose from 1 mg to 5 mg produced proportionate fivefold increases in furosemide AUC0–tz and C max 9. This was greater than the maximum increase in furosemide plasma exposure reported in the literature when it was administered together with the potent OAT inhibitor probenecid (a factor of 3.6) 23, and caused only minimal increases (16–18%) in rosuvastatin AUC and C max 9.
A limitation of the optimized cocktail tested in this trial is that the metformin and furosemide doses used are subtherapeutic. Thus, it is so far unclear whether they would react similarly if administered in therapeutic doses in the presence of an NME that inhibits transporters. This limitation is currently being addressed in a follow‐up DDI trial that investigates the effect of well‐described inhibitors of drug transporters on the cocktail. This trial will include, amongst others, a comparison of the effects of inhibitors of OCT/MATE or OAT on metformin and furosemide pharmacokinetics at cocktail doses and at therapeutic doses. Another cocktail focused on transporter‐based DDI has recently been reported. Prueksaritanont et al. 10 described a microdose substrate probe drug cocktail developed for the detection of interactions mediated by inhibition of OATP1B, BCRP, P‐gp and CYP3A, but not aimed at addressing OATs, OCT2 and MATEs.
When establishing a new cocktail, one focus must be on safety and tolerability. This was recently illustrated in a trial using a CYP phenotyping cocktail containing single doses of tramadol, omeprazole, losartan and caffeine 24. These are, as single substances, usually safe and well‐tolerated probes. However, with the cocktail, unacceptable adverse effects were observed in several subjects that were not expected based on the safety profiles of the single substances 24. In the transporter probe drug cocktail proposed in the present work, based on mechanisms of action, the safety profile and the doses used, we do not expect relevant pharmacodynamic interaction to occur between the probe drugs. In line with this expectation, in the current study, treatment‐emergent AEs after the four cocktail drugs given alone or in combination as a cocktail reflected commonly occurring events in healthy volunteer trials. An exception was the SAE ‘exposure to toxic agent’, which, however, occurred during the ambulatory period of the treatment with digoxin alone, and was not related to trial procedures or the drugs administered in this trial. A single AE of mild headache occurring 5 h after cocktail treatment, with a duration of ~3 h and not requiring therapy, was assessed as possibly drug‐related by the investigator due to the close proximity in time to the cocktail administration and because no definitive other cause could be determined. In the current trial, digoxin, furosemide, metformin and rosuvastatin at the doses tested alone or in combination as a cocktail were safe and well tolerated.
With the trial reported here, the transporter cocktail was optimized by adjusting the dose of individual components, thereby eliminating the previously reported pharmacokinetic DDI. The optimized cocktail, consisting of 0.25 mg digoxin, 1 mg furosemide, 10 mg metformin and 10 mg rosuvastatin thus has the potential to be used as a screening tool for further clinical studies investigating transporter‐mediated DDI.
Conclusions
Digoxin (0.25 mg), furosemide (1 mg), metformin (10 mg) and rosuvastatin (10 mg) exhibit no mutual pharmacokinetic interactions and are well tolerated and safe when administered as a cocktail. The cocktail is thus optimized and has the potential to be used as a screening tool for further clinical studies investigating transporter‐mediated DDI. Before use in clinical development programmes, the sensitivity of the cocktail to inhibition of drug transporters needs to be investigated, which is currently ongoing.
Competing Interests
K.H. was contracted by Boehringer Ingelheim as an external statistician. All other authors are employees of Boehringer Ingelheim. The trial was funded by Boehringer Ingelheim Pharma GmbH & Co. KG, who acted as sponsor.
The authors thank Paul Tanswell for organizational and medical writing support during the preparation of this article. We also thank the team of the Human Pharmacology Centre, Boehringer Ingelheim's Phase I unit for highly professional clinical conduct of the study. The authors thank Sebastian Kandert for clinical monitoring, Christa Walk for data management, SGS Cephac Europe, St. Benoît, France and Covance Laboratories Ltd, Harrogate, UK, for bioanalytical work, and Kiran Maass for medical writing of the clinical trial report on which this publication is based.
Supporting information
Table S1 Additional pharmacokinetic parameters of digoxin administered alone and in the four‐component cocktail
Table S2 Adjusted by‐treatment geometric means and geometric mean ratios for urinary pharmacokinetic endpoints of digoxin given alone or as test cocktaila
Table S3 Additional pharmacokinetic parameters of furosemide administered alone and in the four‐component cocktaila
Table S4 Adjusted by‐treatment geometric means and geometric mean ratios for urinary pharmacokinetic endpoints of furosemide given alone or as test cocktaila
Table S5 Additional pharmacokinetic parameters of metformin administered alone and in the four‐component cocktail
Table S6 Adjusted by‐treatment geometric means and geometric mean ratios for urinary pharmacokinetic endpoints of metformin given alone or as test cocktaila
Table S7 Additional pharmacokinetic parameters of rosuvastatin administered alone or in the four‐component cocktail
Table S8 Adjusted by‐treatment geometric means and geometric mean ratios for urinary pharmacokinetic endpoints of rosuvastatin given alone or as test cocktaila
Table S9 Treatment‐emergent adverse events (AEs) during on‐treatment periods by preferred terms – treated set
Figure S1 Forest plot of geometric mean ratios (test/reference) of AUC0‐tz, AUC0‐oo and C max, with 90% confidence intervals (CI) for the four cocktail probe drugs. Test = cocktail, reference = probe drug alone
Figure S2 Forest plot of geometric mean ratios (test/reference) of fe0–36, fe0–24, CLR,0–36 and CLR,0–24, with 90% confidence intervals (CI) for the four cocktail probe drugs. Test = cocktail, reference = probe drug alone
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Stopfer, P. , Giessmann, T. , Hohl, K. , Hutzel, S. , Schmidt, S. , Gansser, D. , Ishiguro, N. , Taub, M. E. , Sharma, A. , Ebner, T. , and Müller, F. (2018) Optimization of a drug transporter probe cocktail: potential screening tool for transporter‐mediated drug–drug interactions. Br J Clin Pharmacol, 84: 1941–1949. 10.1111/bcp.13609.
PI Statement Thomas Giessmann was the principal investigator for this trial.
References
- 1. König J, Müller F, Fromm MF. Transporters and drug–drug interactions: important determinants of drug disposition and effects. Pharmacol Rev 2013; 65: 944–966. [DOI] [PubMed] [Google Scholar]
- 2. Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, et al Membrane transporters in drug development. Nat Rev Drug Discov 2010; 9: 215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zolk O, Fromm MF. Transporter‐mediated drug uptake and efflux: important determinants of adverse drug reactions. Clin Pharmacol Ther 2011; 89: 798–805. [DOI] [PubMed] [Google Scholar]
- 4. Hillgren KM, Keppler D, Zur AA, Giacomini KM, Stieger B, Cass CE, et al Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin Pharmacol Ther 2013; 94: 52–63. [DOI] [PubMed] [Google Scholar]
- 5. US‐FDA . Guidance for industry: clinical drug interaction studies – study design, data analysis, and clinical implications (draft guidance). 2017. Available at https://www.fda.gov/downloads/drugs/guidances/ucm292362.pdf (accessed 17 November 2017).
- 6. Ministry of Health, Labor and Welfare, Tokyo, Japan . MHLW guideline on drug interactions for drug development and appropriate provision of drug information (final draft) (2014).
- 7. EMA‐CHMP . Guideline on the investigation of drug interactions: final (CPMP/EWP/560/95/rev. 1 corr. 2). 2012. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf (accessed 13 March 2017).
- 8. Stopfer P, Giessmann T, Hohl K, Sharma A, Ishiguro N, Taub ME, et al Pharmacokinetic evaluation of a drug transporter cocktail consisting of digoxin, furosemide, metformin, and rosuvastatin. Clin Pharmacol Ther 2016; 100: 259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stopfer P, Giessmann T, Hohl K, Sharma A, Ishiguro N, Taub ME, et al Effects of metformin and furosemide on rosuvastatin pharmacokinetics in healthy volunteers: implications for their use as probe drugs in a transporter cocktail. Eur J Drug Metab Pharmacokinet 2018; 43: 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prueksaritanont T, Tatosian DA, Chu X, Railkar R, Evers R, Chavez‐Eng C, et al Validation of a microdose probe drug cocktail for clinical drug interaction assessments for drug transporters and CYP3A. Clin Pharmacol Ther 2017; 101: 519–530. [DOI] [PubMed] [Google Scholar]
- 11. Zhang L, Sparreboom A. Predicting transporter‐mediated drug interactions: commentary on: “Pharmacokinetic evaluation of a drug transporter cocktail consisting of digoxin, furosemide, metformin and rosuvastatin” and “Validation of a microdose probe drug cocktail for clinical drug interaction assessments for drug transporters and CYP3A”. Clin Pharmacol Ther 2017; 101: 447–449. [DOI] [PubMed] [Google Scholar]
- 12. Ebner T, Ishiguro N, Taub ME. The use of transporter probe drug cocktails for the assessment of transporter‐based drug–drug interactions in a clinical setting: proposal of a four component transporter cocktail. J Pharm Sci 2015; 104: 3220–3228. [DOI] [PubMed] [Google Scholar]
- 13. US‐FDA . Guidance for industry: bioanalytical method validation. 2001. Available at http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070107.pdf (accessed 13 March 2017).
- 14. EMA‐CHMP . Guideline on bioanalytical method validation EMEA/CHMP/EWP/192217/200921. 2011. Available at http://www.ema.europa.eu/ema/pages/includes/document/open_document.jsp?webContentId=WC500109686 (accessed 13 March 2017).
- 15. Kupper LL, Hafner KB. How appropriate are popular sample size formulas? Am Stat 1989; 43: 101–105. [Google Scholar]
- 16. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD, et al The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 2017; 174 (Suppl 1): S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hingorani P, Karnad DR, Rohekar P, Kerkar V, Lokhandwala YY, Kothari S. Arrhythmias seen in baseline 24‐hour Holter ECG recordings in healthy normal volunteers during phase 1 clinical trials. J Clin Pharmacol 2016; 56: 885–893. [DOI] [PubMed] [Google Scholar]
- 19. Glucophage (metformin hydrochloride tablets) 500 mg, 850 mg, and 1000 mg, Rx only (product information). Physician's Desk Reference 2001; 1005–1009. [Google Scholar]
- 20. Somogyi A, Stockley C, Keal J, Rolan P, Bochner F. Reduction of metformin renal tubular secretion by cimetidine in man. Br J Clin Pharmacol 1987; 23: 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Song IH, Zong J, Borland J, Jerva F, Wynne B, Zamek‐Gliszczynski MJ, et al The effect of dolutegravir on the pharmacokinetics of metformin in healthy subjects. J Acquir Immune Defic Syndr 2016; 72: 400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oh J, Chung H, Park SI, Yi SJ, Jang K, Kim AH, et al Inhibition of the multidrug and toxin extrusion (MATE) transporter by pyrimethamine increases the plasma concentration of metformin but does not increase antihyperglycaemic activity in humans. Diabetes Obes Metab 2016; 18: 104–108. [DOI] [PubMed] [Google Scholar]
- 23. Chennavasin P, Seiwell R, Brater DC, Liang WM. Pharmacodynamic analysis of the furosemide‐probenecid interaction in man. Kidney Int 1979; 16: 187–195. [DOI] [PubMed] [Google Scholar]
- 24. Pedersen RS, Damkier P, Christensen MM, Brosen K. A cytochrome P450 phenotyping cocktail causing unexpected adverse reactions in female volunteers. Eur J Clin Pharmacol 2013; 69: 1997–1999. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Additional pharmacokinetic parameters of digoxin administered alone and in the four‐component cocktail
Table S2 Adjusted by‐treatment geometric means and geometric mean ratios for urinary pharmacokinetic endpoints of digoxin given alone or as test cocktaila
Table S3 Additional pharmacokinetic parameters of furosemide administered alone and in the four‐component cocktaila
Table S4 Adjusted by‐treatment geometric means and geometric mean ratios for urinary pharmacokinetic endpoints of furosemide given alone or as test cocktaila
Table S5 Additional pharmacokinetic parameters of metformin administered alone and in the four‐component cocktail
Table S6 Adjusted by‐treatment geometric means and geometric mean ratios for urinary pharmacokinetic endpoints of metformin given alone or as test cocktaila
Table S7 Additional pharmacokinetic parameters of rosuvastatin administered alone or in the four‐component cocktail
Table S8 Adjusted by‐treatment geometric means and geometric mean ratios for urinary pharmacokinetic endpoints of rosuvastatin given alone or as test cocktaila
Table S9 Treatment‐emergent adverse events (AEs) during on‐treatment periods by preferred terms – treated set
Figure S1 Forest plot of geometric mean ratios (test/reference) of AUC0‐tz, AUC0‐oo and C max, with 90% confidence intervals (CI) for the four cocktail probe drugs. Test = cocktail, reference = probe drug alone
Figure S2 Forest plot of geometric mean ratios (test/reference) of fe0–36, fe0–24, CLR,0–36 and CLR,0–24, with 90% confidence intervals (CI) for the four cocktail probe drugs. Test = cocktail, reference = probe drug alone
Supporting info item
Supporting info item