

Abstract
Aims
Adrenomedullin (ADM) is an important regulator of endothelial barrier function and vascular tone, and may represent a novel treatment target in sepsis. The non‐neutralizing ADM antibody adrecizumab has shown promising results in preclinical sepsis models. In the present study, we investigated the safety, tolerability and pharmacokinetics (PK)/pharmacodynamics of adrecizumab in a first‐in‐man study and in a second study during experimental human endotoxaemia.
Methods
Forty‐eight healthy male volunteers were enrolled in two randomized, double‐blind, placebo‐controlled phase I studies. In both studies, subjects received placebo or one of three doses of adrecizumab (n = 6 per group). In the second study, a bolus of 1 ng kg–1 endotoxin was followed by infusion of 1 ng kg–1 h–1 endotoxin for 3 h to induce systemic inflammation, and the study medication infusion started 1 h after endotoxin bolus administration.
Results
Adrecizumab showed an excellent safety profile in both studies. PK analyses showed proportional increases in the maximum plasma concentration of adrecizumab with increasing doses, a small volume of distribution, a low clearance rate and a terminal half‐life of ~14 days. adrecizumab elicited a pronounced increase in plasma ADM levels, whereas levels of mid‐regional pro‐adrenomedullin remained unchanged, indicating that de novo synthesis of ADM was not influenced. In the second study, no effects of adrecizumab on cytokine clearance were observed, whereas endotoxin‐induced flu‐like symptoms resolved more rapidly.
Conclusions
Administration of adrecizumab is safe and well tolerated in humans, both in the absence and presence of systemic inflammation. These findings pave the way for further investigation of adrecizumab in sepsis patients.
Keywords: adrecizumab, adrenomedullin, antibody, endotoxaemia, sepsis, shock
What is Already Known about this Subject
Previous work in animal models of septic shock and systemic inflammation has shown treatment with the adrenomedullin antibody adrecizumab to have promising effects.
This included reduced organ dysfunction and vasopressor demand, improved vascular barrier function and enhanced survival.
What this Study Adds
The data presented in the present manuscript demonstrate the excellent first‐in‐human safety and tolerability of adrecizumab, both during non-inflammatory conditions in a first‐in‐human study as well as during systemic inflammation induced by intravenous endotoxin administration in healthy volunteers.
This study provides valuable information on the pharmacokinetic properties and pharmacodynamic effects of adrecizumab, which may also contribute to elucidating the mechanism of action.
Overall, the present work paves the way for future research with adrecizumab in sepsis patients.
Introduction
Sepsis is a major health problem for patients with infectious diseases worldwide, with increasing incidence and a high mortality rate 1, 2, 3. It is defined as life‐threatening organ dysfunction caused by a dysregulated host response to infection 4. Sepsis‐induced vascular effects include vasodilation and loss of vascular barrier function 5. This results in hypotension, tissue oedema and, ultimately, lethal organ dysfunction. Besides (supportive) therapies such as antibiotics, mechanical ventilation and vasopressors, there are currently no adjuvant therapies available.
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=683 (ADM) is a free circulating peptide hormone, which is involved in the regulation of vascular tone and stabilization of the endothelial barrier 6, 7, 8. During sepsis and most pronounced during septic shock, elevated concentrations of circulating ADM are observed, which correlate with disease severity and mortality 9, 10. However, correlation does not imply causation, and increased levels of ADM could also represent a (failing) compensatory response. Mechanistic studies actually indicate that ADM can exert both beneficial and detrimental effects in sepsis. Therefore, ADM is referred to as a ʻdouble‐edgedʼ sword in sepsis. On one hand, preclinical studies in animal models of systemic inflammation and sepsis have shown that ADM administration restores vascular barrier function through effects on endothelial cells, thereby reducing detrimental tissue oedema 11, 12, 13, 14. On the other hand, ADM has also been reported to induce vasodilation and hypotension 15, 16, 17, which could in theory further aggravate hypotension in patients with septic shock. It was thus hypothesized that modulation of ADM with antibodies could be beneficial, if it would retain or even potentiate the beneficial effects of ADM while negating its potentially detrimental vasodilatory effects. Interestingly, a highly specific non‐neutralizing mouse monoclonal antibody (HAM1101) was previously shown to improve survival in cecal ligation and puncture (CLP)‐induced sepsis in mice 18. In addition, in a fully resuscitated murine CLP‐induced septic shock model, treatment with this antibody resulted in reduced vasopressor demand and improved organ function 19. These promising results led to the development of a humanized antibody for further clinical investigation (HAM8101, later named adrecizumab). In lipopolysaccharide (LPS)‐induced systemic inflammation in rats and CLP‐induced sepsis in mice, adrecizumab attenuated vascular leakage and vascular dysfunction, as well as improved survival 20. Extensive preclinical safety and toxicological studies did not reveal any safety concerns (unpublished data).
The present work describes two phase I studies in which the first‐in‐human safety, tolerability and pharmacokinetics (PK) and pharmacodynamics (PD) of single, escalating intravenous doses of adrecizumab were investigated. The first study was conducted in healthy male volunteers during normal non‐inflammatory conditions. The second study was conducted during systemic inflammation evoked by experimental human endotoxaemia. The experimental endotoxaemia model is a safe and reproducible method for inducing a controlled transient systemic inflammatory response in humans by intravenous administration of E. coli endotoxin (LPS) 21.
Methods
General
Firstly, a first‐in‐human phase I, randomized, double‐blind, placebo‐controlled study was conducted to evaluate single escalating intravenous (i.v.) doses of adrecizumab in healthy male subjects. Next, a second phase I, randomized, double‐blind, placebo‐controlled study was conducted to evaluate single escalating i.v. doses of adrecizumab in healthy male subjects during experimental human endotoxaemia (details provided below). Both studies were conducted at a single site (the Department of Intensive Care Medicine at the Radboud University Medical Center in Nijmegen, the Netherlands), and were carried out in accordance with the Declaration of Helsinki and Good Clinical Practice standards. The study protocols were approved by the local ethics committee of the Radboud University Medical Center (approval numbers 2016–2283 and 2016–2740) prior to recruitment and inclusion of subjects, and registered at http://clinicaltrials.gov (NTC02991508 and NTC03083171).
Study medication
Adrecizumab is a non‐neutralizing humanized high‐affinity immunoglobulin (Ig) G1κ full‐length antibody directed against the N‐terminus of ADM. Adrecizumab was produced using Chinese hamster ovary cells under good manufacturing practice conditions. Adrecizumab and placebo were supplied by the study sponsor (Adrenomed AG, Hennigsdorf, Germany) as a solution for injection in identical sterile single‐use vials, containing 10.4 ml solution, allowing for an extractable volume of 10 ml. Adrecizumab (20 mg ml–1) was dissolved in a vehicle consisting of histidine‐hydrochloride monochloride, glycine and water. The placebo solution consisted of the identical vehicle. Vials were manufactured by Glycotope Biotechnology GmbH (Heidelberg, Germany). Manufacturing, packaging, quality control and preparation were described in an Investigational Medicinal Product Dossier. Randomization of subjects (using a predetermined randomization list) and preparation of study medication was performed by an independent and unblinded research team, who were not involved in any other aspect of the studies. Dose selection for the present human studies (0.5, 2.0 and 8.0 mg kg–1 adrecizumab) was based on preclinical data, showing a no adverse events level (NOAEL) of 400 mg kg–1 and 100 mg kg–1 for rats and cynomolgus monkeys, respectively. Using a conversion factor of 6.2 for the rat and 3.1 for the monkey, the human equivalent doses are 65 mg kg–1 and 32 mg kg–1, providing a safety margin factor of 130 or 64 to the proposed starting dose of 0.5 mg kg–1. In addition, a therapeutic dose of 2.0 mg kg–1 was regarded as sufficient, based on various preclinical efficacy studies (18, 19, 20 and other, as yet unpublished, data). Including an additional higher dose of 8.0 mg kg–1 in humans provides the opportunity to demonstrate potential dose dependency of this higher dosage.
Subjects
After giving written consent, male subjects, aged 18–35 years, with a body mass index (BMI) between 18 kg m–2 and 30 kg m–2 were included. Before participation, health status was determined by past medical history, physical examination, 12‐lead electrocardiography (ECG) and safety laboratory tests at a screening visit. Exclusion criteria included atopic constitution, use of any medication, flu‐like symptoms 14 days prior to the studies, significant blood loss and/or participation in any other clinical trial within 90 days prior to the studies. For the experimental endotoxaemia study, previous participation in endotoxaemia trials was an additional exclusion criterion. The use of recreational drugs was prohibited 7 days prior to and during the study and in the subsequent 90‐day follow‐up period. Alcohol and tobacco use was prohibited 24 h before and after the experimental day.
Study procedures
The general study procedures are depicted in Figure 1 and were virtually identical for both studies, except for the administration of LPS in the endotoxaemia study. Fasted subjects were admitted to the research unit in the morning (07:30 h). An intravenous cannula was placed for infusion of fluids, as well as administration of endotoxin and study drug. An intra‐arterial cannula was placed for continuous blood pressure monitoring and frequent blood withdrawal. Vital signs were continuously monitored until discharge. In each study, 24 eligible subjects were assigned to one of three dose groups: 0.5, 2 and 8 mg kg–1 body weight. Each dose group consisted of eight subjects randomly assigned to receive either adrecizumab or placebo (n = 6 active study drug, n = 2 placebo). Frequent blood samples were collected during the first 8 h after study drug administration, for analyses of safety laboratory parameters as well as PK and PD parameters. Subjects were discharged 8 h after study drug administration, after assessment and confirmation of their fitness by the investigator. Owing to the expected long half‐life (T½) of adrecizumab, subjects returned for further follow‐up visits after 1, 7, 14, 28, 60 and 90 days.
Figure 1.
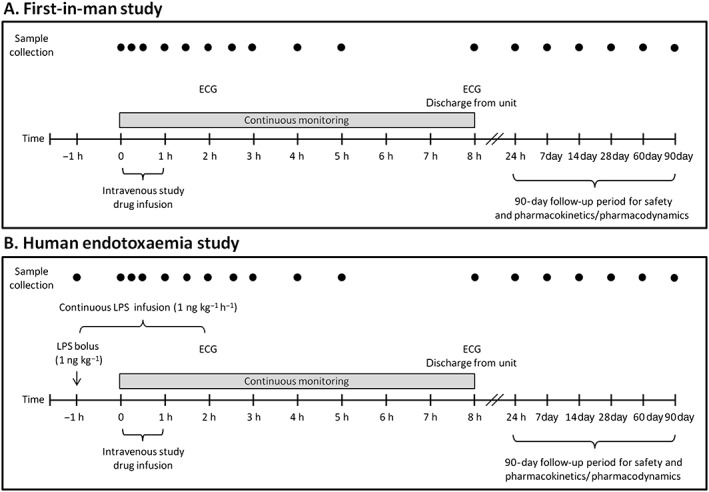
Schematic overview of study procedures for the first‐in‐human study (A) and the human endotoxaemia study (B). ECG, electrocardiography; LPS, lipopolysaccharide
Induction of systemic inflammation in the human endotoxaemia study
In the present study, an endotoxaemia model, applying continuous infusion of LPS for 3 h, was used because this is thought to be a better representation of the inflammatory response as observed in patients with sepsis than a bolus administration of LPS 22. Systemic inflammation was induced by a bolus administration of 1 ng kg–1 Escherichia coli type O113 LPS (List Biological Laboratories Inc., Campbell, CA, USA) 1 h prior to study drug administration. This bolus was followed by a continuous intravenous infusion of LPS 1 ng kg–1 h–1 for 3 h to induce systemic inflammation. To prevent vasovagal responses, subjects received prehydration with 1.5 l glucose 2.5%/NaCl 0.45% over the course of 45 min 23.
Safety parameters
Safety and tolerability were the primary endpoints of both studies. Frequent safety and tolerability assessments were performed on the study drug administration day for both studies until discharge and during the 90‐day follow‐up period. Safety parameters included blood pressure, heart rate and peripheral oxygen saturation [recorded from a Philips MP50 patient monitor (Philips, Eindhoven, the Netherlands); on the study drug administration day, data were sampled every 30 s by a custom inhouse‐developed data recording system], temperature (FirstTemp Genius 2; Sherwood Medical, St Louis, MO, USA), 12‐lead ECG (Philips PageWriter Trim III, Philips, Amsterdam, the Netherlands) and routine haematology and biochemistry laboratory tests. An adverse event (AE) was defined as any untoward medical occurrence in a clinical trial subject administered a study product, which did not necessarily have a causal relationship with this treatment. AEs were recorded throughout the study and follow‐up period. All AEs were judged by the investigator with regard to severity (mild, moderate or severe) and their relation to the study drug (unrelated, possible, probably or definite). In the human endotoxaemia study, common symptoms of endotoxaemia (headache, abdominal pain, back pain, fever and muscle aches) were not regarded as AEs unless they were of abnormal severity or duration. In order to minimize the risks for subjects in both studies, dosage groups were tested sequentially if the previous dose was tolerated without relevant side effects. In each dose group, the first four subjects were tested consecutively, with 48 h between experimental days. In both studies, an independent data safety monitoring board reviewed safety data, including vital signs, laboratory parameters and AEs, and approved to continue the study with the next dose groups.
Sample collection
Blood was drawn in ethylenediaminetetraacetic acid‐anticoagulated vacutainers and centrifuged (10 min, 2000 g, 4°C), after which plasma was stored at −80°C until analysis.
PK analysis
For both studies, PK analysis was performed on samples collected at the following time points relative to study drug administration: immediately prior to administration and then 15, 30, 60 and 90 min; 2, 3, 4 and 8 h; and 1, 7, 14, 28, 60 and 90 days after administration. PK analysis was performed by Aurigon Toxicological Research Center (Dunakeszi, Hungary) under good laboratory practice conditions. Free (unbound) adrecizumab was quantified in the human plasma samples using a validated luminescence immunoassay, with a quantification limit of 0.85 μg ml–1. The highest observed plasma concentration was defined as Cmax. The area under the plasma concentration–time curve from t0 to the last measurement (AUC0–t) was calculated using the linear trapezoidal rule. The elimination rate constant (λz) was calculated by log linear regression of concentrations observed during the terminal phase of elimination. The AUC from t0 to infinity AUC(0–∞) was calculated as the sum of AUC0–t and the extrapolated area using the last measured concentration (C(last)) and the elimination rate constant by taking the formula (C(last))/λz. The terminal T½ (T½λ) was calculated by dividing the natural logarithm of 2 by λz. Clearance (CL) was calculated as dose/AUC(0–∞). The apparent volume of distribution during the terminal phase (Vz) was calculated as Cl/λz. The individual and mean plasmaconcentration–time curves were evaluated using the noncompartmental method for infusion administration. PK analysis was performed using validated Phoenix WinNonlin Version 6.3 software (Pharsight Corporation, St Louis, MO, USA).
PD analysis
Concentrations of total ADM (adrecizumab bound and unbound) were measured using the Spingotest® bio‐ADM assay 24. Mid‐regional pro‐Adrenomedullin (MR‐proADM) was measured using the B·R·A·H·M·S MR‐proADM KRYPTOR assay (BRAHMS GmbH, Hennigsdorf, Germany). Endothelin‐1 was analysed using an enzyme‐linked immunosorbent assay (QuantiGlo®, R&D systems, Minneapolis, MN, USA). Noradrenaline, adrenaline and dopamine levels were measured using routine analysis methods (high‐pressure liquid chromatography with fluorometric detection, as described previously 25). Plasma renin was analysed using a radioimmunoassay (RENIN III, Cisbio, Codolet, France). In the endotoxaemia study, circulating concentrations of the inflammatory cytokines tumour necrosis factor alpha (TNF‐α), interleukin (IL) 6, IL‐8, IL‐10, granulocyte‐colony stimulating factor, monocyte chemoattractant protein 1 and interferon gamma‐induced protein 10 were determined batchwise using a simultaneous Luminex assay (Milliplex, Millipore, Billerica, MA, USA) according to the manufacturers' instructions.
Endotoxaemia‐induced symptoms
Subjects scored endotoxaemia‐induced flu‐like symptoms (the ‘sickness score’) every 30 min throughout the study drug administration day on a numerical response scale ranging from 0 to 10 (0 meaning no complaint at all, 10 extremely severe complaints). LPS‐induced flu‐like symptoms were not considered to be AEs unless they were of abnormal severity or duration, as judged by the blinded investigators.
Statistical analysis
Data were tested for normality using the Kolmogorov–Smirnov test, and all data were normally distributed. Demographic data were expressed as mean ± standard deviation, and other data were expressed as mean ± standard error of the mean. Differences between placebo and adrecizumab groups were tested pair‐wise using the interaction term from two‐way analysis of variance (ANOVA) with repeated measures in the factor time. The dose proportionality of Cmax and AUC0–∞ of adrecizumab was assessed using one‐way ANOVA followed by a Bonferroni post‐hoc test in case a P‐value <0.05 indicated nonproportionality. Calculations and statistical analyses were performed using GraphPad Prism version 5 for Windows (Graphpad Software Inc., La Jolla, CA, USA). In case of missing data, values were imputed by the mean of the two adjacent data points. A two‐tailed P‐value of <0.05 was considered statistically significant.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to Pharmacology 26, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 27.
Results
Study population and subject disposition
A total of 48 subjects were included and randomized in the two studies. The baseline characteristics are listed in Table 1. All participating subjects received study medication as intended, completed the study and 90‐day follow‐up period, and were deemed compliant with the study protocol. No subjects were replaced.
Table 1.
Subject characteristics of the first‐in‐human study (A) and the human endotoxaemia study (B)
| A. First‐in‐human | Placebo | 0.5 mg kg–1 | 2 mg kg–1 | 8 mg kg–1 |
|---|---|---|---|---|
| (n = 6) | (n = 6) | (n = 6) | (n = 6) | |
| Age (years) | 23 ± 2 | 23 ± 2 | 21 ± 1 | 23 ± 3 |
| BMI (kg m–2) | 25 ± 4 | 23 ± 2 | 22 ± 2 | 23 ± 2 |
| Weight (kg) | 80 ± 13 | 75 ± 7 | 76 ± 9 | 76 ± 9 |
| Height (cm) | 181 ± 10 | 182 ± 3 | 185 ± 3 | 184 ± 5 |
| B. Endotoxaemia | Placebo | 0.5 mg kg–1 | 2 mg kg–1 | 8 mg kg–1 |
|---|---|---|---|---|
| (n = 6) | (n = 6) | (n = 6) | (n = 6) | |
| Age (years) | 22 ± 1 | 22 ± 3 | 23 ± 3 | 23 ± 3 |
| BMI (kg m–2) | 24 ± 1 | 23 ± 2 | 25 ± 2 | 26 ± 3 |
| Weight (kg) | 80 ± 6 | 76 ± 4 | 86 ± 8 | 87 ± 11 |
| Height (cm) | 184 ± 5 | 181 ± 3 | 187 ± 7 | 185 ± 9 |
All presented parameters were determined during the screening visit and are presented as mean ± standard deviation. BMI, body mass index
Safety and tolerability
Safety and tolerability were the primary endpoints of both studies. Administration of a single dose of adrecizumab was well tolerated by all subjects in all dose groups, for both studies, and did not result in any safety concerns. All reported AEs were transient, except for one (new‐onset type 1 diabetes mellitus, requiring ongoing insulin treatment, detailed below). One serious AE (SAE) was reported in the human endotoxaemia study (the afore‐mentioned type 1 diabetes). All variations in laboratory parameters, vital signs and 12‐lead ECG were deemed not clinically significant.
First‐in‐human study
A total of 37 AEs were reported over the 90‐day study period (Table 2A). Thirty‐four AEs were judged to be mild, whereas three AEs were moderate (these three were deemed ‘unrelated’ to the study drug) and required treatment: one case of a sexually transmitted disease (Chlamydia infection), one dislocated shoulder and one subject with repeated nose bleeds (details of all AEs are listed in Table S1A). Fifteen AEs (41%) were judged by the blinded investigators to be ‘unrelated’ to the study drug, whereas the rest was deemed ‘possibly’ related (59%). Twelve AEs were observed in the placebo group, whereas six, 11 and eight AEs were found in the 0.5, 2 and 8 mg kg–1 adrecizumab groups, respectively. Commonly reported AEs were nasopharyngitis and headaches, the latter occurring predominantly in the placebo group (see Table S1A).
Table 2.
Overview of adverse events in the first‐in‐human study (A) and the human endotoxaemia study
| A. First‐in‐human (n = 24) | Placebo (n = 6) | 0.5 mg kg–1 (n = 6) | 2 mg kg–1 (n = 6) | 8 mg kg–1 (n = 6) | Overall |
|---|---|---|---|---|---|
| Adverse events (AEs) | 12 (32%) | 6 (16%) | 11 (30%) | 8 (22%) | 37 (100%) |
| Severe AEs (SAEs) | 0 | 0 | 0 | 0 | 0 |
| Discontinued study drug due to (S)AEs | 0 | 0 | 0 | 0 | 0 |
| AEs of mild intensity | 12 (35%) | 6 (18%) | 8 (23.5%) | 8 (24%) | 34 (100%) |
| AEs of moderate intensity | 0 | 0 | 3 (100%) | 0 | 3 (100%) |
| AEs of severe intensity | 0 | 0 | 0 | 0 | 0 |
| Unrelated AEs | 6 (40%) | 3 (20%) | 4 (27%) | 2 (13%) | 15 (100%) |
| Possibly related AEs | 6 (27%) | 3 (14%) | 7 (32%) | 6 (27%) | 22 (100%) |
| Probably related AEs | 0 | 0 | 0 | 0 | 0 |
| Definitely related AEs | 0 | 0 | 0 | 0 | 0 |
| B. Endotoxaemia | Placebo (n = 6) | 0.5 mg kg–1 (n = 6) | 2 mg kg–1 (n = 6) | 8 mg kg–1 (n = 6) | Overall (n = 24) |
|---|---|---|---|---|---|
| AEs | 4 (14.8%) | 8 (29.6%) | 8 (29.6%) | 7 (25.9%) | 27 (100%) |
| Severe AEs (SAEs) | 0 | 0 | 1 (100%) | 0 | 1 (100%) |
| Discontinued due to (S)AEs | 0 | 0 | 0 | 0 | 0 |
| AEs of mild intensity | 4 (16%) | 8 (32%) | 7 (28%) | 6 (24%) | 25 (100%) |
| AEs of moderate intensity | 0 | 0 | 0 | 1 (100%) | 1 (100%) |
| AEs of severe intensity | 0 | 0 | 1 (100%) | 0 | 1 (100%) |
| Unrelated AEs | 1 (5.6%) | 5 (27.8%) | 7 (38.9%) | 5 (27.8%) | 18 (100%) |
| Possibly related AEs | 3 (33%) | 3 (33%) | 1 (11%) | 2 (22%) | 9 (100%) |
| Probably related AEs | 0 | 0 | 0 | 0 | 0 |
| Definitely related AEs | 0 | 0 | 0 | 0 | 0 |
Human endotoxaemia study
A total of 27 AEs were observed over the 90‐day study period (Table 2B). Twenty‐five AEs were mild, one was moderate and required treatment (a traumatic carpal bone fracture during the follow‐up period; details of all AEs are listed in Table S1B) and one was severe and also required treatment (new‐onset type 1 diabetes). Eighteen (67%) AEs were judged to be ‘unrelated’ to the study drug by blinded investigators, and the nine others were deemed to be ‘possibly related’ (33%). Four AEs were observed in the placebo group, compared with eight, eight and six in the 0.5, 2 and 8 mg kg–1 adrecizumab groups, respectively. Commonly reported AEs were nasopharyngitis and headaches.
One SAE (new‐onset type 1 diabetes mellitus) was reported in a subject in the 2.0 mg kg–1 group. Briefly, this subject experienced weight loss, starting approximately 2 months after study drug administration, and increased thirst and diuresis starting approximately 5 months after study drug administration. Eventually, approximately 8 months after study drug administration, the subject was admitted to the hospital for acute dehydration and a total of 18 kg weight loss. Laboratory results showed hyperglycaemia, ketonuria and elevated glycated haemoglobin, and the subject was diagnosed with type 1 diabetes mellitus. After treatment with insulin and intravenous fluids, he recovered rapidly (<24 h). Currently, no long‐term complications have been reported, although the subject remains dependent on exogenous insulin administration. The severity was graded as ʻsevereʼ. To investigate the possible relationship with the administration of the study drug, specific type 1 diabetes antibodies were determined on 4 April 2018 in spare baseline samples (taken on 3 January and 6 February 2017, prior to study participation). Causality with adrecizumab was deemed ʻunrelatedʼ because a strong presence of specific type 1 diabetes antibodies was demonstrated in the baseline blood samples obtained prior to adrecizumab administration (anti‐islet cell antigen, anti‐zinc transporter 8, anti‐glutamic acid decarboxylase and anti‐islet antigen 2 antibodies were all positive at baseline, indicating that the disease was already developing prior to participation in the study, several months before the first clinical signs became apparent).
PK
Plasma adrecizumab concentration–time profiles for both studies are presented in Figure 2, and PK parameters are summarized in Table 3.
Figure 2.
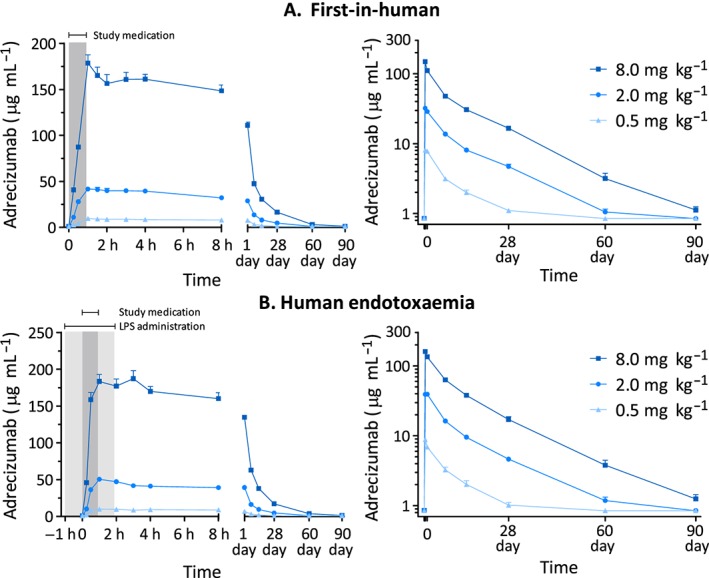
Plasma concentration–time profiles of adrecizumab in the first‐in‐human study (A) and the human endotoxaemia study (B). Data are expressed as mean ± standard error of the mean. The dark grey area indicates the study drug administration period, and the light grey period the lipopolysaccharide (LPS) infusion period
Table 3.
Pharmacokinetic parameters of adrecizumab in the first‐in‐human study (A) and in the human endotoxaemia study (B)
| A. First‐in‐human | 0.5 mg kg–1 (n = 6) | 2 mg kg–1 (n = 6) | 8 mg kg–1 (n = 6) |
|---|---|---|---|
| Cmax (μg ml–1) | 9.71 ± 0.86 | 44.1 ± 4.50 | 179 ± 21.10 |
| AUC0‐∞ (μg*h ml–1) a | 2610 ± 222 | 10 700 ± 1870 | 39 000 ± 3340 |
| Vz (ml kg–1) a | 94.4 ± 8.98 | 95.9 ± 9.70 | 107 ± 11.50 |
| T½λ (h) a | 340 ± 6.3 | 352 ± 51.3 | 361 ± 49.9 |
| CL (ml h–1kg–1) a | 0.193 ± 0.02 | 0.193 ± 0.04 | 0.206 ± 0.02 |
| B. Endotoxaemia | 0.5 mg kg–1 (n = 6)b | 2 mg kg–1 (n = 6)b | 8 mg kg–1 (n = 6) |
|---|---|---|---|
| Cmax (μg ml–1) | 10.6 ± 2.07 | 51.0 ± 6.28 | 203 ± 17.00 |
| AUC0–∞ (μg*h ml–1) | 2120 ± 660 | 12 500 ± 1010 | 46 100 ± 5390 |
| Vz (ml kg–1) | 78.4 ± 11.5 | 86.9 ± 10.3 | 93.6 ± 9.9 |
| T½λ (h) | 233 ± 85.3 | 377 ± 67.3 | 372 ± 46.4 |
| CL (ml h–1 kg–1) | 0.260 ± 0.10 | 0.162 ± 0.01 | 0.176 ± 0.02 |
Data are expressed as mean ± standard deviation. AUC0−∞, area under the plasma concentration–time curve from time zero to infinity; Cmax, highest observed plasma concentration; CL, total clearance calculated; T½λ, elimination half‐life; Vz, volume of distribution characterized by the terminal phase
One subject from the 0.5 mg kg–1 group was excluded from these variables because the elimination phase was not well characterized
The T½ and the T½‐dependent parameters for two subjects (one from the 0.5 mg kg–1 group and one from the 2.0 mg kg–1 group) were excluded from the mean calculation because their elimination phase was not well characterized
First‐in‐human study
In all three dose groups, the Cmax was attained immediately after termination of infusion. Dose‐proportional increases in Cmax and AUC0–∞ were observed (Figure S1A). The T½ and the T½‐dependent parameters of two subjects were excluded from analysis because their elimination phase was not characterized well. The small CL value (~0.2 ml h–1 kg–1) indicates a slow overall elimination of adrecizumab from the circulation. A small Vz (~100 ml kg–1) indicates that adrecizumab predominantly remains within the circulation. The terminal elimination T½λ of adrecizumab was ~15 days.
Human endotoxaemia study
Similarly to the first‐in‐human study, values of Cmax were attained shortly after cessation of study drug infusion. Again, dose‐proportional increases in Cmax were observed (Figure S1B). However, dose proportionality was not observed for AUC0–∞ when comparing 0.5 mg kg–1 with 2.0 mg kg–1, and 0.5 mg kg–1 with 8.0 mg kg–1 adrecizumab (Figure S1B). PK parameters were virtually identical to those found in the first‐in‐human study, with the exception of a reduced T½λ (a mean of 10 days compared with 14 days; P = 0.02) and Vz (94 ml kg–1 compared with 78 ml kg–1; P = 0.03) in the 0.5 mg kg–1 dose group.
PD
First‐in‐human study
Administration of adrecizumab did not influence heart rate, mean arterial pressure, peripheral oxygen saturation or temperature (Figure S2, all P > 0.05), and there were no significant differences between groups in routine haematological and biochemical safety laboratory measurements (data not shown). Of interest, adrecizumab administration resulted in a rapid and statistically significant dose‐dependent increase in total plasma ADM levels (note that the assay detects both bound and unbound ADM), whereas plasma levels of MR‐proADM (an inactive peptide originating from the same precursor as ADM) were not increased (Figure 3A), implying that the adrecizumab‐induced increase in total plasma ADM was not due to increased synthesis. We also measured plasma concentrations of renin, dopamine, noradrenaline, adrenaline and endothelin‐1 in the first‐in‐human study because these could theoretically counteract possible vasodilatory effects resulting from increased ADM levels. The plasma concentrations of none of these endogenous hormones were influenced relevantly by adrecizumab, as between‐group differences were only subtle and not dose dependent (see Figure S3).
Figure 3.
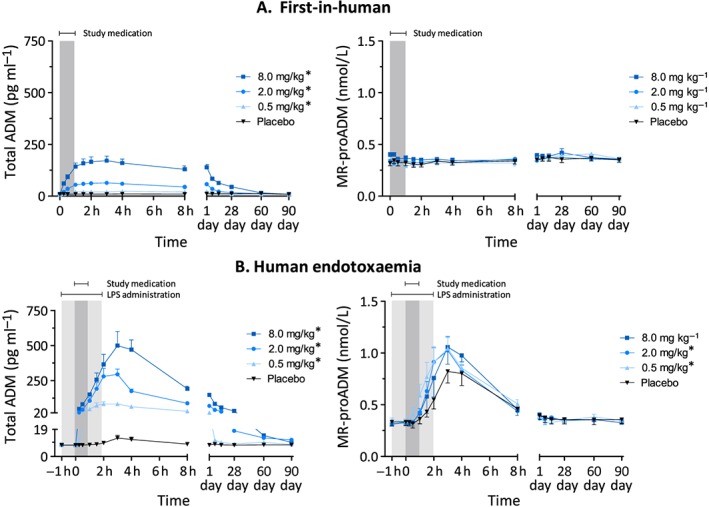
Plasma concentration–time profiles of adrenomedullin (AMD) and mid‐regional pro‐adrenomedullin (MR‐proADM) in the first‐in‐human study (A) and the human endotoxaemia study (B). Data are expressed as mean ± standard error of the mean. Differences between adrecizumab groups and placebo were evaluated using repeated measures two‐way analysis of variance, and interaction term P‐values are displayed
* P < 0.05
Human endotoxaemia study
LPS administration caused a transient increase in body temperature and heart rate, whereas mean arterial pressure decreased (Figure 4A‐C). Adrecizumab did not influence the LPS‐induced effects on blood pressure or heart rate, although a statistically significant effect was observed for the 0.5 mg kg–1 dose group for the change in body temperature over time (P = 0.02) (Figure 4). However, this difference was very slight, and because no effects for the higher doses were observed, this was likely to be a chance finding. LPS administration caused a transient increase in self‐reported flu‐like symptoms over time (Figure 4D). The highest sickness scores were observed in the placebo group, and resolution of symptoms was significantly more swift in the 8 mg kg–1 adrecizumab group compared with placebo (P = 0.01), whereas a trend was observed for the 0.5 mg kg–1 and 2 mg kg–1 dose groups (P = 0.08 and P = 0.07, respectively). Similarly to the first‐in‐human study, adrecizumab caused a dose‐dependent increase in total ADM levels (Figure 3B). Compared with the adrecizumab administration in the absence of LPS, much higher concentrations were reached during systemic inflammatory conditions, which could, at least in part, be explained by the LPS‐induced increased synthesis reflected by elevated MR‐proADM levels (Figure 3B). Expectedly, increased plasma concentrations of various cytokines and chemokines were observed after induction of endotoxaemia (Figure 5). Typically, cytokine responses showed very high inter‐individual variation. For instance, profound between‐subject and between‐group differences were observed in plasma levels of the archetypal proinflammatory cytokine, and main driver of the inflammatory response, TNF‐α, as early as during the first hour after endotoxin administration, when the study drug had not yet been administered. As these differences were already present prior to adrecizumab or placebo administration, it was not possible to interpret the adrecizumab‐induced effects on absolute cytokine levels. In order to evaluate the effects on cytokine kinetics (e.g. Cl) instead, we normalized cytokine levels to their peak concentrations on a per‐subject basis. As depicted in Figure 5, adrecizumab did not influence cytokine kinetics.
Figure 4.
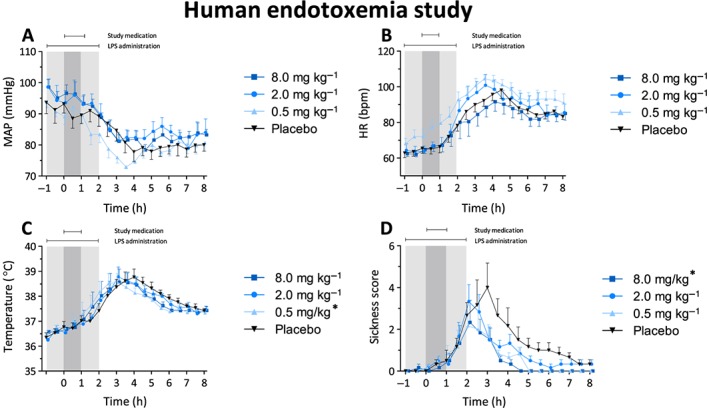
Clinical parameters from the human endotoxaemia study: mean arterial pressure (MAP) (A), heart rate (B), temperature (C) and sickness score (D). Data are expressed as mean ± standard error of the mean. Differences between adrecizumab groups and placebo were evaluated using repeated measures two‐way analysis of variance, and interaction term P‐values are displayed. LPS, lipopolysaccharide
* P < 0.05
Figure 5.
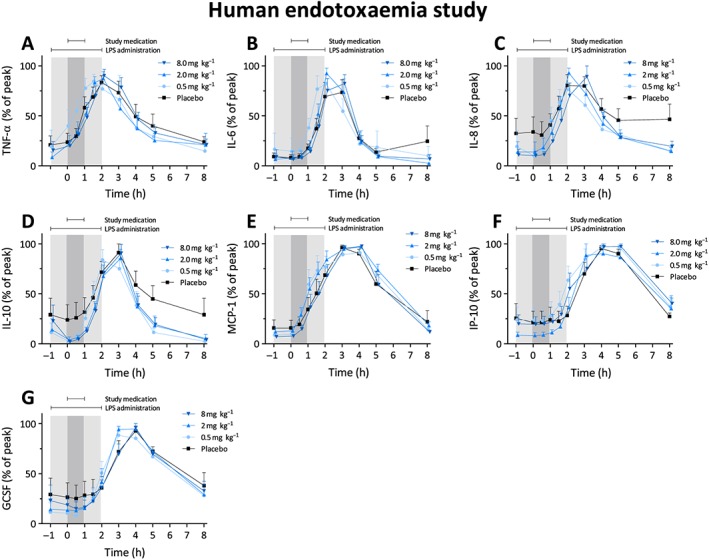
Cytokine clearance from the human endotoxaemia study. Data were normalized for the peak cytokine value and are expressed as mean ± standard error of the mean. Differences between adrecizumab groups and placebo were evaluated using repeated measures two‐way analysis of variance, and interaction term P‐values are displayed. GCSF, granulocyte‐colony stimulating factor; IL, interleukin; IP‐10, interferon gamma‐induced protein 10; LPS, lipopolysaccharide; MCP‐1, monocyte chemoattractant protein 1; TNF‐α, tumour necrosis factor alpha
* P < 0.05
Discussion
In the present study, we evaluated the safety, tolerability, PK and PD of the novel ADM‐binding antibody adrecizumab in a first‐in‐human study during non‐inflammatory conditions, and in a subsequent study during systemic inflammation using the experimental human endotoxaemia model. Adrecizumab was well tolerated and demonstrated an excellent safety profile throughout the investigated dose range. Our data revealed dose‐proportional maximum concentrations of adrecizumab which were reached almost immediately after cessation of study drug infusion, a low Vz, low CL and a long T½λ. Moreover, adrecizumab infusion induced a rapid and profound dose‐dependent increase in its target peptide ADM, which is thought to explain its mechanism of action 28. Adrecizumab did not affect vital signs or cytokine Cl, although it did result in a swifter resolution of flu‐like symptoms in the human endotoxaemia study.
Safety
One SAE occurred during the 90 day follow‐up period, and was demonstrated to be unrelated to the study drug. The majority of AEs were transient, of mild severity and evenly distributed among study groups. The few AEs of moderate severity that occurred (three in the first‐in‐human study, one in the human endotoxaemia study) were all deemed to be unrelated to the study drug by the blinded investigators. Moreover, we observed no ECG abnormalities, a normal local tolerability at the site of infusion and no clinically relevant changes in vital signs or safety laboratory values. Administration of adrecizumab during systemic inflammation also did not result in any safety concerns.
PK
Peak concentrations of adrecizumab were typically observed at the end of infusion and were dose proportional. A low CL and long T½λ of approximately 14 days were observed, which are typical for monoclonal antibodies 29, 30. A low volume of distribution, typical for IgG monoclonal antibodies with a large molecular weight 29, 30, indicates that adrecizumab predominantly remains confined to the blood compartment. Some PK parameters were influenced by systemic inflammation. A slightly lower volume of distribution and T½λ were observed for the lowest dose group of 0.5 mg kg–1 adrecizumab in the second study, performed under inflammatory conditions. In accordance with these observations, systemic exposure based on AUC0–∞ was slightly lower for the 0.5 mg kg–1 dose group in the present study. This observation can be explained neither by nonlinear protein binding of adrecizumab to its target peptide ADM, nor by increased binding of ADM–adrecizumab complexes due to inflammation‐induced upregulation of the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=49 13, 31 because adrecizumab is present in great excess over ADM in the blood. Therefore, these mechanisms cannot relevantly affect adrecizumab levels. A more plausible explanation is increased CL of adrecizumab through inflammation‐induced Fc receptor upregulation 29.
PD
Interestingly, adrecizumab induced a rapid and profound dose‐dependent increase in its target peptide ADM, which is in line with results from animal studies and may be of importance for the underlying mechanism of action. It is well known from various in vitro 11, 12, 32, 33 and in vivo 13, 14, 34 studies that ADM is a strong endothelial barrier‐stabilizing peptide and that administration of ADM to animals with septic shock improves outcome. In fact, the adrecizumab‐induced increase in circulating ADM may well be responsible for the beneficial effects observed in preclinical studies, as a result of the non‐neutralizing nature of the antibody allowing ADM to bind with its receptors and activate second messengers 18. The proposed mechanism of action of adrecizumab and the role of the observed increase in ADM levels and resulting beneficial effects have recently been put forward 28. In short, because concentrations of the inactive peptide MR‐proADM (derived from the same precursor peptide) were not increased, the adrecizumab‐induced rise in ADM is probably not due to increased production. Hence, it appears plausible that two other mechanisms are responsible for the increase in ADM. First, the binding of ADM to the non‐neutralizing antibody adrecizumab prolongs its T½λ because ADM is normally subject to proteolytic degradation by proteases targeting its N‐terminus, which is now bound to adrecizumab 28. Second, ADM's distribution is shifted towards the blood. This is based upon the fact that ADM can normally diffuse freely across the blood barrier, whereas ADM–adrecizumab complexes cannot, as adrecizumab is a large IgG antibody with a low volume of distribution, indicating that it remains confined to the blood compartment. As a consequence, the binding of ADM to the antibody that remains in the circulation will drain ADM from the interstitium.
ADM has been described as a double‐edged sword in sepsis 35 because, on the one hand, it improves endothelial barrier stability and outcome in preclinical animal sepsis models but, on the other hand, it possesses vasodilatory effects which can cause hypotension at high concentrations 16, 36, 37. Therefore, it could be hypothesized that the adrecizumab‐induced increase in ADM would cause hypotension. However, our data clearly showed that blood pressure was not influenced by adrecizumab, and that adrecizumab did not influence the LPS‐induced decrease in blood pressure. Moreover, we also measured dopamine and catecholamines in our first‐in‐human study, to evaluate whether adrecizumab influenced circulating levels of these vasoactive hormones and therefore might have counteracted, and thereby masked, vasodilation. The finding that the circulating concentrations of these endogenous were not relevantly influenced indicates that the higher levels of (adrecizumab‐bound) ADM do not reach vascular smooth muscle cells and cause vasodilation.
No relevant effects of adrecizumab on mean arterial pressure, heart rate or inflammatory parameters were observed. In relation to the inflammatory response, it should be stressed that the endotoxaemia study was not designed primarily to evaluate this endpoint. Adrecizumab administration was initiated 1 h following the start of the LPS infusion. Therefore, it is not surprising that cytokine kinetics were not influenced by adrecizumab, as the inflammatory response had already been activated, to a large extent, prior to adrecizumab administration. Unfortunately, it was not possible to evaluate endpoints which were favourably influenced by adrecizumab in preclinical studies, such as reduced catecholamine demand, organ dysfunction and endothelial barrier leakage 18, 19, 20, as the transient and relatively mild human endotoxaemia model does not induce such effects. Nevertheless, adrecizumab did attenuate the sickness score in the highest dose group and tended to decrease it for the two lower doses. This is remarkable, considering the small number of subjects per group and the stringent method of analysis used, yielding limited statistical power to detect significant differences. As no immunomodulatory effects were detected in the present study, it is tempting to speculate that adrecizumab's effects on the sickness score are mediated through beneficial effects on vascular integrity. As alluded to earlier, our group has previously shown that human endotoxaemia does not induce overt microvascular leakage 38 but the analytical methods used may not have been sensitive enough to detect subtler differences in vascular integrity.
Conclusion
Intravenous administration of adrecizumab was safe and well tolerated in healthy human volunteers, both in non‐inflammatory conditions and during systemic inflammation induced by the administration of endotoxin. Moreover, adrecizumab induced a rapid and profound increase in its target peptide ADM in the blood circulation. This is consistent with previously reported beneficial effects in preclinical sepsis models. Our results pave the way for further investigation of adrecizumab in septic patients. A phase II proof‐of‐concept, precision medicine study in septic shock patients has recently started recruitment and is anticipated to enrol 300 patients (http://clinicaltrials.gov, NCT‐number: NCT03085758). Furthermore, as endothelial dysfunction is not restricted to sepsis/septic shock but also occurs in other diseases 39, 40, 41, adrecizumab might have broader applicability.
Competing Interests
The work presented here was funded by Adrenomed AG. The sponsor was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations. P.P.’s institution received a research grant from Adrenomed AG that was used to conduct the present studies. P.P. has received travel reimbursement and consultancy fees from Adrenomed AG. C.G., A.B. and A.M, have received travel reimbursement from Adrenomed AG. UMR‐S 942, Inserm received a research grant from Adrenomed AG that was used to conduct preclinical studies. D.v.L., R.P., B.t.E. and M.K. have no competing interests to declare. Adrenomed AG holds patent rights to ADM antibodies.
Contributors
M.K. and P.P. designed the studies. C.G., D.v.L., R.P. and B.t.E. performed the experiments. C.G., M.K. and P.P. analysed the data. C.G., A.B., A.M., M.K. and P.P. contributed to interpretation of the results. C.G. drafted the manuscript. A.B., A.M., M.K. and P.P. critically revised the manuscript. All authors read and approved the final version of the manuscript.
The authors would like to thank Jelle Gerretsen for support with laboratory analyses, Yvonne Kaspers for secretarial support, Pleun Hemelaar for data management, Marieke van der A, Hetty van der Eng, Chantal Luijten, Noortje Roovers and Hellen van Wezel for membership of the unblinded team, and Lucas van Eijk, Saskia Houterman, Quirijn de Mast, Gert Jan Scheffer and Frank van de Veerdonk for taking part in the data safety monitoring board.
Supporting information
Table S1 Summary of adverse events by system organ class for the first‐in‐human study (A) and the human endotoxaemia study (B). AE, adverse event
Figure S1 The dose proportionality of Cmax and AUC0–∞ of adrecizumab was assessed using one‐way analysis of variance followed by a Bonferroni post‐hoc test in case a P‐value <0.05 indicated nonproportionality. Data from both the first‐in‐human study (A) and the human endotoxaemia study (B) were analysed. Data are expressed as mean ± standard deviation. AUC0‐∞, area under the plasma concentration–time curve from time zero to infinity; Cmax, highest observed plasma concentration
Figure S2 Vital signs in the first‐in‐human study: mean arterial pressure (A), heart rate (B), temperature (C) and peripheral oxygen saturation (D). Data are expressed as mean ± standard error of the mean. The dark grey area indicates the study drug administration period. Differences between adrecizumab groups and placebo were evaluated using repeated measures two‐way analysis of variance, and interaction term P‐values are displayed. *P < 0.05
Figure S3 Plasma concentrations of dopamine (A), renin (B), noradrenaline (C), adrenaline (D) and endothelin 1 (E), measured in the first‐in‐human study. Data are presented as mean ± standard error of the mean. Differences between adrecizumab groups and placebo were evaluated using repeated measures two‐way analysis of variance, and interaction term P‐values are displayed. *P < 0.05
Geven, C. , van Lier, D. , Blet, A. , Peelen, R. , ten Elzen, B. , Mebazaa, A. , Kox, M. , and Pickkers, P. (2018) Safety, tolerability and pharmacokinetics/pharmacodynamics of the adrenomedullin antibody adrecizumab in a first‐in‐human study and during experimental human endotoxaemia in healthy subjects. Br J Clin Pharmacol, 84: 2129–2141. 10.1111/bcp.13655.
References
- 1. Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P, et al Assessment of global incidence and mortality of hospital‐treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med 2016; 193: 259–272. [DOI] [PubMed] [Google Scholar]
- 2. Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med 2013; 41: 1167–1174. [DOI] [PubMed] [Google Scholar]
- 3. Vincent JL, Marshall JC, Namendys‐Silva SA, Francois B, Martin‐Loeches I, Lipman J, et al Assessment of the worldwide burden of critical illness: the intensive care over nations (ICON) audit. Lancet Respir Med 2014; 2: 380–386. [DOI] [PubMed] [Google Scholar]
- 4. Singer M, Deutschman CS, Seymour CW, Shankar‐Hari M, Annane D, Bauer M, et al The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis‐3). JAMA 2016; 315: 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ 2016; 353: i1585. [DOI] [PubMed] [Google Scholar]
- 6. Garcia‐Ponce A, Chanez Paredes S, Castro Ochoa KF, Schnoor M. Regulation of endothelial and epithelial barrier functions by peptide hormones of the adrenomedullin family. Tissue Barriers 2016; 4: e1228439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Geven C, Kox M, Pickkers P. Adrenomedullin and adrenomedullin‐targeted therapy as treatment strategies relevant for sepsis. Front Immunol 2018; 9: 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Temmesfeld‐Wollbruck B, Hocke AC, Suttorp N, Hippenstiel S. Adrenomedullin and endothelial barrier function. Thromb Haemost 2007; 98: 944–951. [DOI] [PubMed] [Google Scholar]
- 9. Caironi P, Latini R, Struck J, Hartmann O, Bergmann A, Maggio G, et al Circulating biologically active adrenomedullin (bio‐ADM) predicts hemodynamic support requirement and mortality during sepsis. Chest 2017; 152: 312–320. [DOI] [PubMed] [Google Scholar]
- 10. Marino R, Struck J, Maisel AS, Magrini L, Bergmann A, Di Somma S. Plasma adrenomedullin is associated with short‐term mortality and vasopressor requirement in patients admitted with sepsis. Crit Care 2014; 18: R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brell B, Temmesfeld‐Wollbruck B, Altzschner I, Frisch E, Schmeck B, Hocke AC, et al Adrenomedullin reduces Staphylococcus aureus alpha‐toxin‐induced rat ileum microcirculatory damage. Crit Care Med 2005; 33: 819–826. [DOI] [PubMed] [Google Scholar]
- 12. Hippenstiel S, Witzenrath M, Schmeck B, Hocke A, Krisp M, Krüll M, et al Adrenomedullin reduces endothelial hyperpermeability. Circ Res 2002; 91: 618–625. [DOI] [PubMed] [Google Scholar]
- 13. Muller‐Redetzky HC, Will D, Hellwig K, Kummer W, Tschernig T, Pfeil U, et al Mechanical ventilation drives pneumococcal pneumonia into lung injury and sepsis in mice: protection by adrenomedullin. Crit Care 2014; 18: R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Temmesfeld‐Wollbruck B, Brell B, David I, Dorenberg M, Adolphs J, Schmeck B, et al Adrenomedullin reduces vascular hyperpermeability and improves survival in rat septic shock. Intensive Care Med 2007; 33: 703–710. [DOI] [PubMed] [Google Scholar]
- 15. Kita T, Suzuki Y, Kitamura K. Hemodynamic and hormonal effects of exogenous adrenomedullin administration in humans and relationship to insulin resistance. Hypertens Res 2010; 33: 314–319. [DOI] [PubMed] [Google Scholar]
- 16. Lainchbury JG, Troughton RW, Lewis LK, Yandle TG, Richards AM, Nicholls MG. Hemodynamic, hormonal, and renal effects of short‐term adrenomedullin infusion in healthy volunteers. J Clin Endocrinol Metab 2000; 85: 1016–1020. [DOI] [PubMed] [Google Scholar]
- 17. Nakamura M, Yoshida H, Makita S, Arakawa N, Niinuma H, Hiramori K. Potent and long‐lasting vasodilatory effects of adrenomedullin in humans. Comparisons between normal subjects and patients with chronic heart failure. Circulation 1997; 95: 1214–1221. [DOI] [PubMed] [Google Scholar]
- 18. Struck J, Hein F, Karasch S, Bergmann A. Epitope specificity of anti‐adrenomedullin antibodies determines efficacy of mortality reduction in a cecal ligation and puncture mouse model. Intensive Care Med Exp 2013; 1: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wagner K, Wachter U, Vogt JA, Scheuerle A, McCook O, Weber S, et al Adrenomedullin binding improves catecholamine responsiveness and kidney function in resuscitated murine septic shock. Intensive Care Med Exp 2013; 1: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Geven C, Peters E, Schroedter M, Struck J, Bergmann A, McCook O, et al Effects of the humanized anti‐adrenomedullin antibody adrecizumab (HAM8101) on vascular barrier function and survival in rodent models of systemic inflammation and sepsis. Shock 2018. 10.1097/SHK.0000000000001102. [DOI] [PubMed] [Google Scholar]
- 21. Bahador M, Cross AS. From therapy to experimental model: a hundred years of endotoxin administration to human subjects. J Endotoxin Res 2007; 13: 251–279. [DOI] [PubMed] [Google Scholar]
- 22. Kiers D, Koch RM, Hamers L, Gerretsen J, Thijs EJ, van Ede L, et al Characterization of a model of systemic inflammation in humans in vivo elicited by continuous infusion of endotoxin. Sci Rep 2017; 7: 40149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eijk LT, Pickkers P, Smits P, Bouw MP, van der Hoeven JG. Severe vagal response after endotoxin administration in humans. Intensive Care Med 2004; 30: 2279–2281. [DOI] [PubMed] [Google Scholar]
- 24. Weber J, Sachse J, Bergmann S, Sparwaßer A, Struck J, Bergmann A. Sandwich immunoassay for bioactive plasma adrenomedullin. J Appl Lab Med 2017; 2: 222–233. [DOI] [PubMed] [Google Scholar]
- 25. Willemsen JJ, Ross HA, Jacobs MC, Lenders JW, Thien T, Swinkels LM, et al Highly sensitive and specific HPLC with fluorometric detection for determination of plasma epinephrine and norepinephrine applied to kinetic studies in humans. Clin Chem 1995; 41: 1455–1460. [PubMed] [Google Scholar]
- 26. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guides to IMMUNOPHARMACOLOGY. Nucl Acid Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA, et al The Concise guide to Pharmacology 2017/18: G protein‐coupled receptors. Br J Pharmacol 2017; 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Geven C, Bergmann A, Kox M, Pickkers P. Vascular effects of adrenomedullin and the anti‐adrenomedullin antibody adrecizumab in sepsis. Shock 2018. 10.1097/SHK.0000000000001103. [DOI] [PubMed] [Google Scholar]
- 29. Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol 2017; 6: 576–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tabrizi M, Bornstein GG, Suria H. Biodistribution mechanisms of therapeutic monoclonal antibodies in health and disease. AAPS J 2010; 12: 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ornan DA, Chaudry IH, Wang P. Saturation of adrenomedullin receptors plays an important role in reducing pulmonary clearance of adrenomedullin during the late stage of sepsis. Biochim Biophys Acta 2002; 1586: 299–306. [DOI] [PubMed] [Google Scholar]
- 32. Dunworth WP, Fritz‐Six KL, Caron KM. Adrenomedullin stabilizes the lymphatic endothelial barrier in vitro and in vivo . Peptides 2008; 29: 2243–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Temmesfeld‐Wollbruck B, Brell B, zu Dohna C, Dorenberg M, Hocke AC, Martens H, et al Adrenomedullin reduces intestinal epithelial permeability in vivo and in vitro . Am J Physiol Gastrointest Liver Physiol 2009; 297: G43–G51. [DOI] [PubMed] [Google Scholar]
- 34. Itoh T, Obata H, Murakami S, Hamada K, Kangawa K, Kimura H, et al Adrenomedullin ameliorates lipopolysaccharide‐induced acute lung injury in rats. Am J Physiol Lung Cell Mol Physiol 2007; 293: L446–L452. [DOI] [PubMed] [Google Scholar]
- 35. Kox M, Pickkers P. Adrenomedullin: its double‐edged sword during sepsis slices yet again. Intensive Care Med Exp 2014; 2: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meeran K, O'Shea D, Upton PD, Small CJ, Ghatei MA, Byfield PH, et al Circulating adrenomedullin does not regulate systemic blood pressure but increases plasma prolactin after intravenous infusion in humans: a pharmacokinetic study. J Clin Endocrinol Metab 1997; 82: 95–100. [DOI] [PubMed] [Google Scholar]
- 37. Nakamura K, Toda H, Terasako K, Kakuyama M, Hatano Y, Mori K, et al Vasodilative effect of adrenomedullin in isolated arteries of the dog. Jpn J Pharmacol 1995; 67: 259–262. [DOI] [PubMed] [Google Scholar]
- 38. Van Eijk LT, Pickkers P, Smits P, van den Broek W, Bouw MP, van der Hoeven JG. Microvascular permeability during experimental human endotoxemia: an open intervention study. Crit Care 2005; 9: R157–R164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Johansson P, Stensballe J, Ostrowski S. Shock induced endotheliopathy (SHINE) in acute critical illness – a unifying pathophysiologic mechanism. Crit Care 2017; 21: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jozwiak M, Teboul JL, Monnet X. Extravascular lung water in critical care: recent advances and clinical applications. Ann Intensive Care 2015; 5: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rajendran P, Rengarajan T, Thangavel J, Nishigaki Y, Sakthisekaran D, Sethi G, et al The vascular endothelium and human diseases. Int J Biol Sci 2013; 9: 1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Summary of adverse events by system organ class for the first‐in‐human study (A) and the human endotoxaemia study (B). AE, adverse event
Figure S1 The dose proportionality of Cmax and AUC0–∞ of adrecizumab was assessed using one‐way analysis of variance followed by a Bonferroni post‐hoc test in case a P‐value <0.05 indicated nonproportionality. Data from both the first‐in‐human study (A) and the human endotoxaemia study (B) were analysed. Data are expressed as mean ± standard deviation. AUC0‐∞, area under the plasma concentration–time curve from time zero to infinity; Cmax, highest observed plasma concentration
Figure S2 Vital signs in the first‐in‐human study: mean arterial pressure (A), heart rate (B), temperature (C) and peripheral oxygen saturation (D). Data are expressed as mean ± standard error of the mean. The dark grey area indicates the study drug administration period. Differences between adrecizumab groups and placebo were evaluated using repeated measures two‐way analysis of variance, and interaction term P‐values are displayed. *P < 0.05
Figure S3 Plasma concentrations of dopamine (A), renin (B), noradrenaline (C), adrenaline (D) and endothelin 1 (E), measured in the first‐in‐human study. Data are presented as mean ± standard error of the mean. Differences between adrecizumab groups and placebo were evaluated using repeated measures two‐way analysis of variance, and interaction term P‐values are displayed. *P < 0.05